

1. Introduction
Peptidases and proteases catalyze the hydrolysis of the amide bonds that link amino acids together in peptides and proteins, a process called proteolysis, and these enzymes serve diverse functions in biology.1 Proteases in the digestive system, such as pepsin and trypsin, break down ingested proteins, allowing the amino acid building blocks to be utilized by the body to make new proteins specific for the physiology of the organism. Such degradation also takes place intracellularly via the proteasome, which disassembles misfolded proteins that might otherwise compromise cell function. Proteases, including the proteasome, also cleave protein components involved in the cell cycle, which regulates cell growth and replication, and proteolysis is essential for certain signaling pathways from the cell surface, which is essential for integrating the activities of the various cells of the body. A proteolytic cascade also regulates blood clotting, an essential stage of wound healing that, if not properly controlled, may lead to thrombosis, embolism and stroke. Another set of proteolytic events controls the levels of peptides involved in blood pressure regulation, and agents that target these proteases are among the most widely prescribed medications. Still other proteases are responsible for generating peptide hormones such as insulin.
These illustrations are only a fraction of the proteolytic events known to be important in biology and medicine. Over 500 proteases are predicted to be encoded in the human genome (~2% of all encoded genes)2, and the functions of many of these proteases have yet to be determined. In addition, pathogen-specific proteases are often essential for infectivity and the life cycle of the organism or virus, and inhibitors of HIV protease are part of drug cocktails that have turned AIDS from a death sentence to a manageable long-term illness. A number of proteases have been targeted successfully with medications, and still many other are considered top targets for emerging therapeutics.3
1.1 Protease terminology
The residues of protease substrates are considered relative to the site of backbone cleavage (Fig. 1). Those residues N-terminal to the cleavage site are said to be on the P side, and those residues C-terminal to the cleavage site are said to be on the P′ side. Immediately N-terminal to the cleavage site is residue P1, and moving further N-terminally are residues P2, P3, P4 and so on. Immediately C-terminal to the cleavage site is residue P1′, and moving further C-terminally are residues P2′, P3′, P4′ and so on. Thus, the protease cleaves the amide bond that links residue P1 and P1′ (Fig. 1, red amide group).
Figure 1.

General substrate and substrate binding sites for proteases. The scissile amide functionality is in red. Moving left from the cleavage site are residues P1, P2, P3, P4, etc. Moving from the right of the cleavage site are residues P1′, P2′, P3′, P4′, etc. Corresponding binding pockets on the protease for these residues are S1, S2, S3, S4, etc. on the left side and S1′, S2′, S3′, S4′, etc. on the right side.
Proteases contain binding pockets for substrate residues (Fig. 1). Those pockets that bind to the P side of the substrate are called S pockets, and those pockets that bind to the P′ side of the substrate are called S′ pockets. The protease pocket that interacts directly with the P1 residue of the substrate is called S1, and protease pockets that bind to resides P2, P3, P4, etc. are called, respectively, S2, S3, S4, etc. The protease pockets that interacts directly with the P1′ residue of the substrate is called S1′, and protease pockets that bind to residues P2′, P3′, P4′, etc. are called, respectively, S2′, S3′, S4′, etc. Thus, the residues involved in catalysis are located between the S1 and S1′ pockets. The character of these pockets dictates the degree of specificity of the protease (i.e., whether it will cleave only one particular sequence of amino acid residues, a wide variety of substrates, or somewhere in between).
1.2 Protease mechanisms
The proteolytic process requires the concerted effort of key residues within the protease active site. These hydrolytic enzymes are classified into four general types based on their catalytic residues and mechanism of action: (A) serine/threonine proteases, (B) cysteine proteases, (C) aspartyl proteases, and (D) metalloproteases. Each of these four main protease categories contains hundreds of known examples and have representatives in all forms of life.1
Serine/threonine proteases employ a conserved serine (or, more rarely, threonine) in the active site, which is activated by a nearby histidine base, which is often in turn activated by an adjacent aspartate anion, setting up what is referred to as a catalytic triad (Fig. 2, A). The net result is that the serine hydroxyl group is more nucleophilic and therefore more capable of attacking the carbonyl carbon of an amide bond. The nucleophilic attack converts the planar carbonyl group into a tetrahedral carbon with a negatively charged oxygen (B), an intermediate that is stabilized by a pocket in the enzyme called the oxyanion hole. Re-formation of the carbonyl functionality takes place with the breaking of the carbon-nitrogen bond (i.e., cleavage of the peptide backbone) and release of the newly formed amino terminus (C). The serine is now part of an ester bond, which is in turn hydrolyzed by incoming water that is activated in essentially the same way as the serine hydroxyl was activated in the first step (D, E). Release of the second proteolytic product, the newly formed carboxy terminus (F), then frees up the enzyme for another round of catalysis. Cysteine proteases go through a similar mechanism, with the cysteine thiol serving as the nucleophile attacking the amide carbonyl, but in this case the thiol is much more acidic (pKa ~ 8) than the hydroxyl group (pKa ~ 14) and is therefore more easily activated. A catalytic dyad, with a properly positioned histidine adjacent to the cysteine thiol, is usually sufficient.
Figure 2.

General catalytic mechanism for serine proteases. Active site serine, histidine and (often) aspartate comprise a catalytic triad that work together first to activate the serine hydroxyl group for nucleophilic attack of the amide carbonyl. After release of the amino group, water is then activated for nucleophilic attack of the ester carbonyl of the protease-substrate covalent intermediate. The carboxylate is then released, and the enzyme is back in its original state and ready for another round of catalysis.
Aspartyl proteases utilize two adjacent aspartate residues in the active site. Aspartate is normally deprotonated and therefore negatively charged at biological pH, but in aspartyl proteases only one is charged, because two proximal anions are unfavorable. These aspartates interact with water and the carbonyl oxygen of the scissile amide bond to increase the nucleophilicity of the water and activate the carbonyl carbon for nucleophilic attack (Fig. 3, A). A tetrahedral intermediate results (B), but unlike what occurs with serine and cysteine proteases, no covalent enzyme-substrate intermediate forms. Re-formation of the carbonyl group coincides with the departure of the amino group, simultaneously producing the new N- and C-termini (C). Metalloprotease likewise activate water directly and do not form covalent enzyme-substrate intermediates. A zinc atom is typically coordinated to two histidines, an aspartate, and to a glutamate, and it is the coordinated glutamate that interacts with and activates the water for catalysis (see later discussion about S2P structure and mechanism).
Figure 3.

General catalytic mechanism for aspartyl proteases. Two active site aspartates activate water and the scissile amide bond of the substrate via hydrogen bonding. Nucleophilic attack of water on the amide carbonyl forms a tetrahedral gem-diol intermediate that fragments to the two cleavage products.
1.3 Membrane-associated proteases
Many important proteolytic events take place at cellular membranes at the cell surface or in intracellular organelles. Up until just over a decade ago, all known membrane-associated proteases were simply tethered to the membrane, anchored by a transmembrane domain or a type of glycolipid called glycophosphatidylinositol (GPI). The catalytic domains of these proteases are completely outside the membrane and are related, both in sequence and in structure, to water-soluble proteases. Examples include a subset of matrix metalloproteases (MMPs), which are tethered through a single transmembrane domain or a glycophospatidyl inositol-anchoring domain and cleave a variety of proteins at the cell surface, such as growth factors and growth factor receptors.4 Other examples are fungal aspartyl proteases called yapsins, which are tethered through a GPI-anchor and are thought to play a role in cell wall integrity and remodeling.5 Several other membrane-tethered proteases will be discussed below, in the context of setting the stage for intramembrane proteolysis.
Until recently, as described above, all identified proteases had been water-soluble enzymes: either the entire enzyme is normally found in an aqueous environment or a membrane anchor holds down an otherwise aqueous protease. Beginning in 1997, however, new proteases have been discovered that are apparently embedded within the hydrophobic environment of the lipid bilayer and somehow carry out hydrolysis on the transmembrane region of their substrates in the generally water-excluding environment of the membrane. Another unusual feature of this process are the substrates, which are typically folded into an α-helix, a conformation that makes the backbone amide bonds inaccessible to nucleophilic attack due to steric hindrance by the amino acid side chains. These intramembrane-cleaving proteases (I-CLiPs)6–8 must therefore create an environment for water and the hydrophilic residues needed for catalysis, and bend or unwind their substrates to make the amide bonds susceptible to hydrolysis. Supporting these mechanistic notions is the observation that these newly discovered I-CLiPs are apparently variations on familiar themes in protease biochemistry: despite of the novelty of being membrane-embedded and cleaving transmembrane domains, the residues essential for catalysis by these I-CLiPs are virtually the same as those found in aqueous proteases. In this review, the known I-CLiPs are discussed by mechanistic category (specifically, as metallo, aspartyl and serine proteases; no cysteine I-CLiPs have yet been identified), covering the literature from their initial discovery in 1997 to the present.
2. S2P Metalloproteases
The first discovery of an I-CLiP arose from studies on the regulation of sterol and fatty acid metabolism. Sterol regulatory element binding proteins (SREBPs) are transcription factors that promote the expression of genes involved in the synthesis of cholesterol and fatty acids.9 Coordinated gene expression is controlled through negative feedback inhibition by cholesterol to ensure that lipids and sterols are produced only when needed. SREBP is synthesized as a precursor protein containing three distinct domains: a domain exposed to the cytosol that binds DNA and activates transcription, two transmembrane regions, and a regulatory domain involved in the feedback control by cholesterol (Fig. 4). When cholesterol levels are high, the SREBP precursor is kept in the endoplasmic reticulum (ER) by a multi-pass membrane protein called SCAP (SREBP cleavage-activating protein)10,11 in complex with a small membrane protein called Insig.12 Reduced cholesterol levels result in dissociation of Insig from SCAP, allowing SCAP to shepherd SREBP to the Golgi apparatus. Proteolysis of SREBP in the Golgi results in release of the transcription factor and its translocation to the nucleus.
Figure 4.

S2P contains conserved HEXXH and LDG motifs found in metalloproteases. SREBP is first cleaved by S1P in the luminal loop. The regulatory domain (Reg) interacts with the cholesterol-sensing SCAP to ensure that S1P proteolysis only occurs when cholesterol levels are low. Subsequent intramembrane proteolysis releases this transcription factor for expression of genes essential to cholesterol and fatty acid synthesis.
2.1 Discovery of Mammalian S2P
Proteolytic release of SREBPs occurs in two steps (Fig. 4). First, the luminal loop between the two transmembrane regions is cleaved by a membrane-tethered serine protease called the Site-1 protease (S1P).13 Release of the transcription factor requires subsequent cleavage by the Site-2 protease (S2P), which performs a hydrolysis of an amide bond predicted to lie three residues within the transmembrane domain.14 The requirement for a prior proteolytic event is a common theme with I-CLiPs. Complementation cloning (that is, adding cDNA libraries to a cell line to find rescuers of a mutant phenotype) identified S2P as a multi-pass membrane protein containing a conserved HEXXH sequence characteristic of zinc metalloproteases.15 The two histidines and the glutamate are required for S2P activity, consistent with known metalloprotease biochemistry in which the two histidines coordinate with zinc and the zinc in turn activates the glutamate for interaction with the catalytic water. Further analysis led to the discovery of a conserved aspartate located ~300 residues from the HEXXH sequence that is likewise critical for S2P activity and thought to be a third residue involved in zinc coordination.16 Similar to SREBP, sequential processing by S1P and S2P of the otherwise membrane-associated transcription factor ATF6 are essential steps in the ER stress response.17
2.2 Bacterial S2P-like Proteases
Further support for the proteolytic function of S2P came from the discovery of a family of related proteins in bacteria.18 These prokaryotic proteins play an essential role in the proteolysis of otherwise membrane-bound transcription factors needed for sporulation. These factors control gene expression in the mother cell after engulfment of the forespore. Cleavage of pro-σk and release of the transcription factor requires the multi-pass membrane protein SpoIVFB in Bacillus subtilis (Fig. 5), and this protein likewise contains the HEXXH motif and a second conserved region with an aspartate, both of which are essential for proteolysis. Another bacterial S2P family member, YaeL (also called RseP) in Escherichia coli, similarly requires HEXXH and a conserved aspartate to play a role in coordinating cell growth and cell division, through intramembrane proteolysis of RseA, a factor critical for responding to extracytoplasmic stress.19 Interestingly, the membrane orientations of the mammalian substrate SREBP and the bacterial substrate σk are opposite to each other, correlating with that of their respective enzymes, S2P and SpoIVFB, which are similarly thought to have opposite orientations.18 This implies that the catalytic region must align with the peptide substrate with proper relative directionality.
Figure 5.

Bacillus subtilisS2P-like protease SpoIVFB and sporulation. Upon engulfment of the forespore by the mother cell, a signaling pathway involving the transcription factor σG is initiated in the forespore that triggers the synthesis of the IVB serine protease. This protease degrades SpoIVFA, which along with BofA serves to inhibit SpoIVFB. With the inhibition of the S2P-like protease released, SpoIVFB cleaves pro-σK, allowing this transcription factor to signal in the mother cell for further factors needed for spore maturation.
Although SpoIVFB and YaeL are both S2P-like bacterial enzymes that cleave transmembrane proteins, the regulation of this key intramembrane proteolytic event for these two I-CLiPs is quite different. For cleavage of RseA by YaeL/RseP, the regulation is similar to that in SREBP cleavage by S2P: intramembrane proteolysis requires a prior cleavage event outside the membrane by another (serine) protease called DegS.20 In contrast, SpoIVFB apparently does not require prior proteolysis, and regulation occurs more directly at the level of SpoIVFB. Two membrane proteins, BofA and SpoIVFA, serve to inhibit SpoIVFB activity, and this inhibition is released by proteolysis of SpoIVFA by the forspore-secreted serine protease IVB (Fig. 5).21,22 The roles of many other S2P metalloproteases in biology remain unknown; however, an S2P homolog encoded by the human pathogen Mycobacterium tuberculosis was found to regulate cell envelope composition as well as growth and persistence in vivo.23 These findings suggest that this protease may be an appropriate target for developing therapeutics for tuberculosis.
2.3 Substrate Recognition
As mentioned above, the α-helical conformation of the transmembrane substrate renders the amide bonds inaccessible to attack by a catalytic residue or water, requiring some bending or unwinding of the helix before proteolysis can occur. The SREBP substrate contains a conserved asparagine-proline (NP) sequence within its transmembrane region that is critical for proteolytic processing by S2P.24 These two residues have the lowest propensity to form α-helices, suggesting that the NP-containing SREBP transmembrane region may be metastable. After S1P cleavage and dissociation of the other transmembrane region, the NP sequence may facilitate unwinding of the residues immediately upstream, including the leucine-cysteine bond that gets cleaved. Unwinding was originally thought to possibly result in protrusion of this bond to the membrane surface and access by the active site residues of S2P. However, the recently solved crystal structure of an S2P-type protease shows that the active site is apparently well within the boundaries of the lipid bilayer (see below). Nevertheless, unwinding may be important for substrate recognition, entry into the active site, and accessibility of the scissile amide bond.
2.4 Structure of an S2P
The E. coli YaeL/RseP protease has been purified to homogeneity with preservation of proteolytic activity.25 Most recently, a high-resolution crystal structure of an S2P family member, from archaeabacteria species Methanocaldococcus jannaschii, MjS2P, has been reported,26 confirming the presence of zinc and its coordination with the key transmembrane histidines, glutamate, and aspartate residues (Fig. 6). The protease crystallized in two conformations, one in which the active site appears more accessible through lateral gating (“open”) and one in which it is less accessible (“closed”, shown in Fig. 6). Transmembrane domains (TMDs) 2–4 are highly conserved, contain the catalytic residues, and do not vary much between the two conformations. TMD 2–4 is thus thought to represent the core domain, while TMD 1, TMD 5 and TMD 6, which are less conserved and more conformationally flexible, are thought to be important for substrate gating from the hydrophobic lipid bilayer into the internal, water-containing active site. In the open conformation, TMD 1 and TMD 6 are spaced further apart, suggesting that substrate enters the active site by traversing between these two TMDs. This new and detailed structural information allows the generation of specific hypotheses about substrate recognition and processing that can be tested biochemically (e.g, through mutagenesis), as is already being done with the Rhomboid serine proteases (see below).
Figure 6.

Structure of archaeal MjS2P. (A) The six transmembrane domains (TMDs) are arranged as a helical bundle, with two histidines from TMD 2 and an aspartate from TMD 4 coordinated to the zinc atom (grey). A second crystallized conformation has TMD 1 and TMD 6 much further apart, suggesting that this is the site of lateral gating by which the transmembrane of the substrate accesses the internal active site. (B) Close up of the active site, explicitly showing another residue (a glutamate in TMD 2) coordinated with the zinc. This glutamate activates water for catalysis.
3. Presenilin-Type Aspartyl Proteases
A key step in the pathogenesis of Alzheimer’s disease is APP proteolysis resulting in the formation of the amyloid-β peptide (Aβ), the principle protein component of the characteristic cerebral plaques of the disease.27,28 The N-terminus of Aβ is produced from the amyloid β-protein precursor (APP) first by the action of β-secretase, a membrane-tethered enzyme that resembles pepsin and other water-soluble aspartyl proteases.29–33 This proteolysis leads to membrane shedding of the large luminal/extracellular APP domain (Fig. 7). The 99-residue membrane-bound remnant is then cleaved in the middle of its transmembrane region by γ-secretase, releasing Aβ and again near the inner leaflet at the ε site to release the APP intracellular domain (AICD).34 Rare mutations in the APP gene, found in and around the Aβ region, cause familial early-onset Alzheimer’s disease, and these mutations alter the production of Aβ or its aggregation properties, important evidence for the amyloid hypothesis of Alzheimer pathogenesis.27,28 Alzheimer-causing mutations also occur in one of the subunits of γ-secretase (see below). Also described below, chemical probes played important roles in the characterization, identification, purification and mechanistic understanding of the I-CLiP that is now known as the γ-secretase complex.
Figure 7.
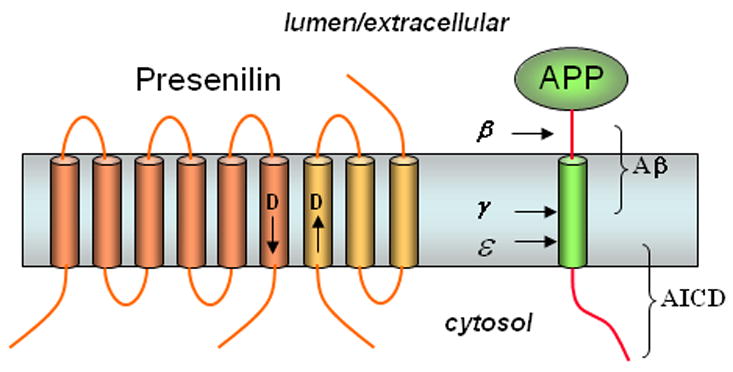
Presenilin, the γ-secretase complex, and the proteolysis of APP to Aβ. Presenilin is processes into two pieces, an N-terminal fragment (NTF, dark portion) and a C-terminal fragment (CTF, light portion) that remain associated. Each fragment donates one aspartate essential for γ-secretase activity (arrows near these aspartates denote N- to C-terminal directionality of the protein sequence3). APP is first cleaved in the extracellular domain by β-secretase, and the remnant is cleaved twice within the membrane by γ-secretase to produce the Aβ peptide of Alzheimer’s disease (secreted) and the intracellular domain (AICD, freed into the cytosol).
3.1 Discovery of Presenilin as a Protease
Several more or less contemporaneous observations provided critical clues for the identification of the elusive γ-secretase, a subject of intense interest as a potential therapeutic target. First, genes encoding the multi-pass membrane proteins presenilin-1 and presenilin-2 were discovered in a search to identify other genes associated with familial, early-onset Alzheimer’s disease.35–37 The disease-causing missense mutations were soon found to alter how γ-secretase cuts APP, leading to increased proportions of longer, more aggregation-prone forms of Aβ.38–41 Second, knockout of presenilin genes eliminated γ-secretase cleavage of APP.42–44 Third, the types of compounds that could inhibit γ-secretase contained moieties typically found in aspartyl protease inhibitors.45,46 These findings led to the identification of two conserved transmembrane aspartates in the multi-pass presenilin that are critical for γ-secretase cleavage of APP (Fig. 7), suggesting that presenilins might be the responsible aspartyl proteases.47–49
Presenilin is cut into two pieces, an N-terminal fragment (NTF) and a C-terminal fragment (CTF),50,51 the formation of which is regulated by limiting cellular factor(s).52 NTF and CTF remain physically associated in a high-molecular weight complex and are metabolically stable.50,51,53–55 These and other results suggested that the NTF-CTF heterodimer is the biologically active form.48 Intriguingly, the NTF and CTF each contribute one of the essential and conserved aspartates, suggesting that the γ-secretase active site might be at the interface between these two presenilin fragments. In strong support of this hypothesis, transition-state analogue inhibitors of γ-secretase, compounds designed to interact with the active site of the protease (e.g., compound 1, Fig. 10), were found to bind directly to presenilin NTF and CTF.56,57 However, presenilins are apparently part of a larger multi-protein complex which constitutes γ̃-secretase (see below).
Figure 10.
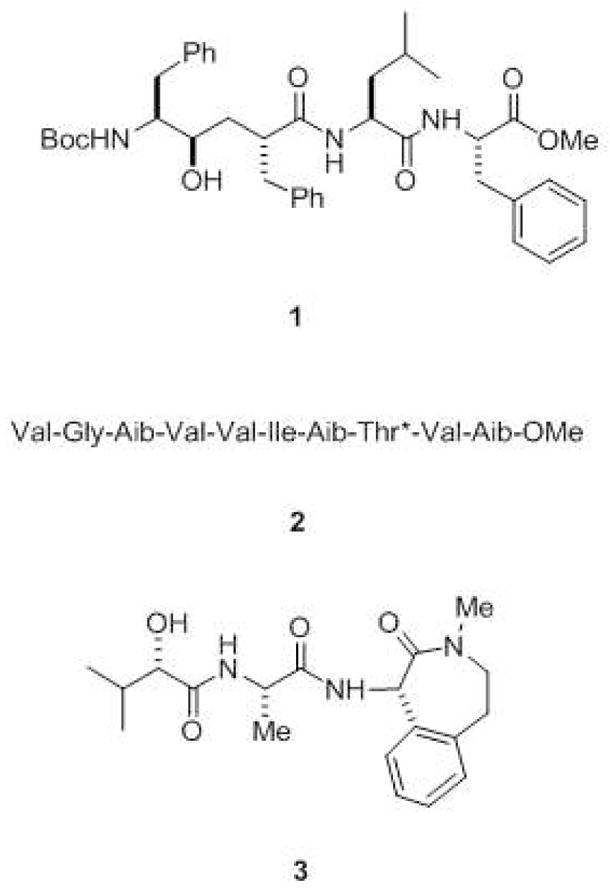
Inhibitors of γ-secretase. Transition-state analogue inhibitors such as 1 include hydroxyl-containing moieties that interact with the catalytic aspartates of aspartyl proteases. Helical peptide inhibitors include α-aminoisobutyric acid (Aib)-containing substrate mimics such as 2 (*denotes that the threonine residue contains an O-benzyl group). These helical peptides mimic the APP transmembrane domain and interact with the substrate docking site on the protease. Also shown is the potent benzodiazepine inhibitor 3 (LY-450,139), which is in late-stage clinical trials for the treatment of Alzheimer’s disease.
3.2 The Notch Receptor and Other Substrates
At the same time presenilins were discovered as susceptibility loci for Alzheimer’s disease, they were also shown to be required for Notch signaling,58 a pathway essential for cell differentiation during development and beyond.59 After Notch is synthesized in the ER, the receptor is cleaved in its extracellular domain during its passage through the secretory pathway, and the two pieces so generated remain associated.60 Upon interaction with a cognate ligand, Notch becomes susceptible to a second extracellular proteolysis, by a membrane-tethered metalloprotease, near the membrane.61,62 The membrane-associated remnant is then cleaved within its transmembrane domain by a presenilin-dependent γ-secretase-like protease,63 releasing the Notch intracellular domain (NICD). NICD translocates to the nucleus and activates transcription after associating with the nuclear partner CSL (CBP/RBPjk, Su(H), Lag-1).64 Knock-in of a Notch-1 transmembrane mutation that greatly reduces presenilin-mediated proteolysis leads to a lethal phenotype in mice similar that seen in Notch-1 knockout mice, indicating that efficient γ-secretase cleavage is essential for Notch signaling during development.65
Since the discovery that Notch is cleaved by γ-secretase, a plethora of other substrates have been identified, including Erb-B4, E- and N-cadherins, CD44, the low density lipoprotein receptor, Nectin-1, and the Notch ligands Delta and Jagged.66,67 Knowledge of the cellular functions of these proteolytic events vary, but in the case of N-cadherin, the produced intracellular domain associates with the transcriptional activator CBP (CREB binding protein) and promotes its migration to the cytosol and degradation by the proteasome.68 Also, neuregulin-1-triggered cleavage of ErbB4 inhibits astrocyte differentiation by interacting with repressors of astrocyte gene expression.69 While cellular function can be ascribed in some cases, the ability of γ-secretase to cleave so many different substrates and its apparently poor sequence specificity raises the question of whether a major role of this enzyme is to serve as a general degrading protease for membrane-bound protein remnants.70 Indeed, γ-secretase appears to be unique among intramembrane proteases in its ability to process so many different substrates. The broad substrate recognition by γ-secretase is likely related to the fact that, unlike the other intramembrane proteases, the enzyme apparently does not require helix-breaking residues near the cleavage sites within the substrates.
3.3 Other Subunits of the γ-Secretase Complex
The highly conserved role of γ-secretase in Notch signaling and its importance in development made possible genetic screens in worms (specifically Caenorhabditis elegans) that identified two Notch modifiers, a single-pass membrane protein APH-2 (nicastrin) and a multi-pass protein APH-1.71–73 Nicastrin was independently isolated biochemically as a presenilin-associated protein and found to be essential for γ-secretase processing of both APP and Notch.74 A saturation screen in C. elegans for presenilin modifiers netted all these proteins and added Pen-2. All four proteins (presenilin, nicastrin, Aph-1, and Pen-2) associate with one another75,76 and with an immobilized γ-secretase inhibitor.76,77 Moreover, their coexpression increased γ-secretase activity in both Drosophila and mammalian cells75,76 and reconstituted activity in yeast.78 Because yeast have no such protease activity and contain no apparent orthologs of these metazoan proteins, these findings strongly suggest that this quartet of proteins is necessary and sufficient for γ-secretase activity. This was subsequently confirmed through purification of the protease complex to virtual homogeneity.79
γ-Secretase is so far unique among intramembrane proteases in being composed of several different proteins: all the others apparently work alone as single proteins. Coexpression, RNA interference, and the identification of assembly intermediates suggest the order in which these four subunits come together,75,80,81 and partial dissociation of the protease complex with detergent offers a model for how these subunits interact (Fig. 8).80 Nicastrin and Aph-1 together can stabilize full-length presenilin, and final addition of Pen-2 apparently triggers presenilin endoproteolysis and γ-secretase activity.75 Pen-2 is also required to stabilize the presenilin subunits.82 The specific biochemical functions of these presenilin cofactors have been mostly enigmatic; however, nicastrin has been suggested to play a role in substrate recognition (see section 3.4 below).
Figure 8.

Presenilin interacts with three other membrane proteins, nicastrin, Aph-1, and Pen-2, whereupon presenilin is cleaved into NTF and CTF to form active γ-secretase.
As for the stoichiometry of the γ-secretase complex, this has been a matter of some controversy, with discrepancies in the reported size of the complex and in the number of presenilin molecules per complex. Sizes of 100–150 KDa to 2 MDa have been reported,53,54,76,83–85 and several studies suggested that two presenilins reside at the catalytic core of the protease complex.86–88 Most recently, however, rigorous biochemical and biophysical experiments have shown that isolated, active complexes contain only one of each component 89 and, consistent with this stoichiometry, that the size of the purified enzyme is ~230 kDa, as determined by scanning electron microscopy.90
3.4 Substrate Recognition
Among the more intriguing questions about the entire emerging family of I-CLiPs is how they handle substrates and cleave within their TMDs. Because it presumably contains water and uses hydrophilic residues, the membrane-embedded active site should be sequestered from the hydrophobic environment of the surrounding lipid tails. Thus, the active site might be envisioned to be part of a pore or channel that could allow entry of water.6 However, the substrate passes through the membrane and cannot enter such a pore or channel directly; docking on the outer surface of the protease, with lateral gating to bring the substrate into the internal active site, might be required.6 Initial evidence for such a mechanism came from isolation of the γ-secretase complex with an immobilized transition-state analogue inhibitor.77 Detergent-solubilized membranes from a cultured human cell line were passed through this affinity matrix, resulting in copurification of γ-secretase complex members and an endogenous membrane-bound APP stub found in these cells. This stub results from alternative processing of APP by membrane-tethered α-secretases, and like the stub produced by β-secretase, it is also a γ-secretase substrate. Thus, an endogenous substrate copurified with the γ-secretase complex while the protease active site was blocked by the immobilized transition-state analogue inhibitor, suggesting the existence of a separate substrate binding site. Substrate bound to this special type of exosite, dubbed the “docking site”, could copurify without being subject to proteolysis.
3.4.1 Helical Peptide Substrate Mimics
Designed peptides based on the transmembrane domain of APP and constrained in a helical conformation (e.g., compound 2, Fig. 10) can potently inhibit γ-secretase, apparently by interacting with this docking site.91 Conversion of these helical peptide inhibitors into affinity labeling reagents led to the localization of the substrate docking site to the presenilin NTF/CTF interface.92 Transition-state analogue inhibitors also bind directly to the NTF/CTF interface, but at a site distinct from that of helical peptide inhibitors. These findings suggest a pathway for γ-secretase substrate from docking site to active site: upon binding to the outer surface of presenilin at the NTF/CTF interface, the substrate can pass, either in whole or in part, between these two presenilin subunits to access the internal active site (Fig. 9). Interestingly, extension of a ten-residue helical peptide inhibitor by just three additional residues resulted in a potent inhibitor93 apparently capable of binding both docking site and active site,92 suggesting that these two substrate binding sites are relatively close.
Figure 9.

Model for how substrates interact with presenilin. Substrate is light green, presenilin NTF is copper, presenilin CTF is gold, Pen-2 is red, Aph-1 is blue, and nicastrin is bright green. The active site (represented by the blue star in stage 1), containing water and two aspartates, is thought to be sequestered away from the hydrophobic environment of the lipid bilayer, necessitating an external docking site for the transmembrane domain of the substrate. Studies with chemical probes (helical peptides and transition-state analogues) support this model (see text). The transmembrane domain of the substrate interacts with the docking site, while the N-terminus of the substrate putatively interacts with the Nicastrin ectodomain (stage 2). The transmembrane domain of the substrate then passes either in whole or in part into the active site for proteolysis (stage 3).
3.4.2 Nicastrin in Substrate Recognition
Up until recently, all the action seemed to be taking place on presenilin. However, one study has suggested that nicastrin also plays a critical role in substrate recognition.94 The ectodomain of nicastrin bears sequence resemblance to aminopeptidases, although certain catalytic residues are not conserved. Nevertheless, nicastrin may recognize the N-terminus of γ-secretase substrates derived from APP and Notch (Fig. 9), and consistent with this notion, mutation of the aminopeptidase domain was reported to prevent this interaction. One conserved glutamate was noted to be especially important, perhaps because this residue forms an ion pair with the amino terminus of the substrate. The sequence of the substrate N-terminus is apparently not critical for the interaction, but a free amino group is. Indeed, simple formylation of the substrate N-terminus was enough to prevent effective substrate interaction and proteolytic processing. Thus, nicastrin may be a kind of gatekeeper for the γ-secretase complex: type I membrane proteins that have not shed their ectodomains cannot interact properly with nicastrin and do not gain access to the active site. However, a new study contradicts this view, with evidence that mutation of the aminopeptidase domain can interfe re with the maturation of the γ-secretase complex, not with the activity of the mature complex.95
3.5 Inhibitors and Modulators
γ-Secretase has in many ways been an attractive target for Alzheimer therapeutics,96 with one inhibitor now in advanced clinical trials97 (compound 3, Fig. 10). However, interference with Notch processing and signaling may lead to toxicities that preclude clinical use of such inhibitors. Long-term treatment with γ-secretase inhibitors causes severe gastrointestinal toxicity and interferes with the maturation of B- and T-lympocytes in mice, effects that are indeed due to inhibition of Notch processing and signaling.98,99 However, compounds that can modulate the enzyme to alter or block Aβ production with little or no effect on Notch would bypass this potential roadblock to therapeutics. Recent studies suggest that certain compounds can alter substrate selectivity and the sites of substrate proteolysis, both in cells and in purified biochemical systems.
Certain non-steroidal anti-inflammatory drugs (NSAIDs; e.g., ibuprofen, indomethacin, and sulindac sulfide) can reduce the production of the highly aggregation-prone Aβ42 peptide and increase a 38-residue form of Aβ, a pharmacological property independent of inhibition of cyclooxygenase.100 The alteration of the proteolytic cleavage site is observed with isolated or purified γ-secretase,79,101,102 indicating that the compounds can interact directly with the protease complex to exert these effects. Enzyme kinetic studies and displacement experiments suggest the selective NSAIDs can be noncompetitive with respective to APP substrate102 and to a transition-state analogue inhibitor, suggesting interaction with a site distinct from the active site.103 The site of cleavage within the Notch transmembrane domain may be similarly affected, but this subtle change does not inhibit the release of the intracellular domain and thus does not affect Notch signaling.104 For this reason, these agents may be safer as Alzheimer therapeutics than inhibitors that block the active site or the docking site. Indeed, one compound, R-flurbiprofen (also called tarenflurbil, compound 4, Fig. 11), is in late-stage clinical trials.105 Surprisingly, the site of proteolytic cleavage by the presenilin homologue signal peptide peptidase (SPP; see section 3.7) can also be modulated by the same NSAIDs that affect γ-secretase. Because SPP apparently does not require other protein cofactors, these findings suggest that presenilin is the site of NSAID binding within the γ-secretase complex and that SPP and presenilin share a conserved drug binding site for allosteric modulation of substrate cleavage sites.106 However, a new study suggests that the substrate itself is the target, specifically in the region of APP substrate at the extracellular/membrane junction.107 A much weaker interaction was observed with a comparable substrate based on Notch, suggesting an alternative explanation for the lack of effect on Notch signaling.
Figure 11.
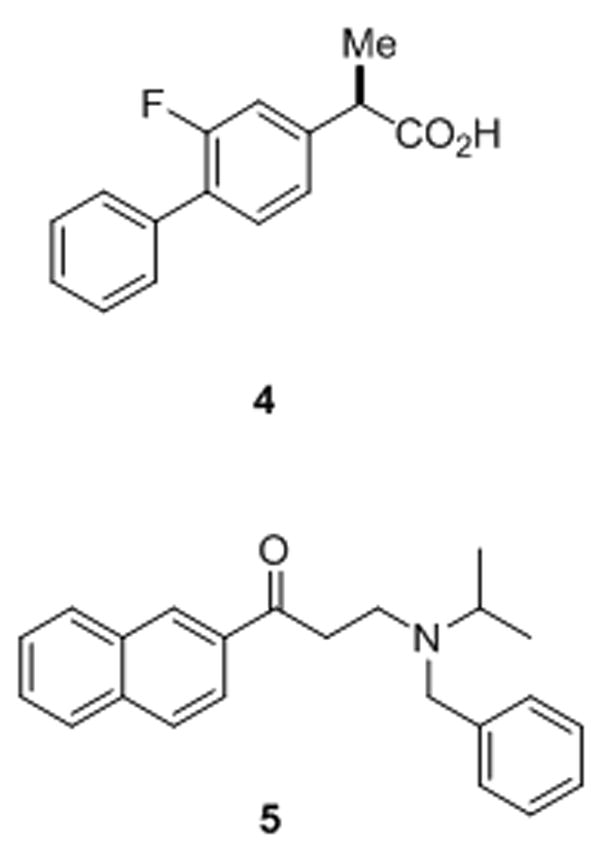
Modulators of γ-secretase. NSAID-like modulator 4 (R-Flurbiprofen or tarenflurbil), which is in late-stage clinical trials for Alzheimer’s disease, shifts where γ-secretase cuts APP, reducing the aggregation-prone Aβ42 and elevating more soluble Aβ38. In contrast, naphthyl ketone 5 inhibits total Aβ production without interfering with the ability of γ-secretase to cleave Notch receptor substrates.
Another type of allosteric modulator is compounds that resemble kinase inhibitors and interact with a nucleotide binding site on the γ-secretase complex. The discovery that ATP can increase Aβ production in membrane preparations prompted the testing of a variety of compounds that interact with ATP binding sites on other proteins.108 In this focused screen, the Abl kinase inhibitor Gleevec emerged as a selective inhibitor of Aβ production in cells without affecting the proteolysis of Notch. In light of these findings, ATP and other nucleotides were tested for effects on purified γ-secretase preparations and found to selectively increase the proteolytic processing of a purified recombinant APP-based substrate without affecting the proteolysis of a Notch counterpart.109 Furthermore, certain compounds known to interact with ATP binding sites were found to selectively inhibit APP processing vis-à-vis Notch in purified protease preparations (e.g., compound 5, Fig. 11). These and other results suggest that the γ-secretase complex contains a nucleotide binding site and that this site allows allosteric regulation of γ-secretase processing of APP with respect to Notch. Whether this regulation is physiologically important is unclear, but the pharmacological relevance is profound and may lead to new therapeutic candidates for Alzheimer’s disease.
3.6 Towards the Structure of γ-Secretase
The purification of the γ-secretase complex79 has allowed the first glimpse into its structure. Electron microscopy and single particle analysis reveals that the complex has a globular structure that at low resolution (10–15 Ǻ) appears rather amorphous110 (Fig. 12). [Another structure, elucidated in a similar manner but of much poorer resolution (~45 Ǻ), has also been reported.111] Nevertheless, two important features can be gleaned. The first is a rather large low-density interior cavity of ~ 20 Ǻ diameter that is presumably where the active site resides, a characteristic reminiscent of the proteasome. The second is the presence of two small openings that may be the site of entry for water.
Figure 12.

Structure of the γ-secretase complex as deduced by electron microscopy. (A) Although the resolution is relatively low (~15 Ǻ) and the structure is globular, (B) a central cavity of low density suggests the location of the active site, and two ports, H1 and H2, suggest site of water entry. Reprinted with permission from reference 110. Copyright 2006 National Academy of Science.
Other structural features have been revealed by cysteine mutagenesis with crosslinking of chemical probes.112,113 The generation of a cysteine-less version of presenilin that retains the ability to assemble with other complex members, undergo endoproteolysis to NTF and CTF, and process APP allowed incorporation of single cysteine resides at various sites near the key aspartates. Disulfide formation with thiol-containing reagents then provided information about the relative accessibility of these sites from the aqueous milieu, allowing the construction of a model in which water can funnel down to where the aspartates reside. Furthermore, simultaneous mutation of the two conserved transmembrane aspartates to cysteine and apparent intramolecular crosslinking provided the first evidence that these two aspartates are indeed in close proximity,112 which is required for them to coordinate and serve catalytic function. Using this same approach (cysteine mutagenesis and crosslinking), two recent studies suggests that TMD 9 serves as a gatekeeper for lateral entry of the substrate TMD.114,115 Still another cysteine-crosslinking study suggests that TMD1 is in direct contact with TMD8.116 More detailed information will likely require a crystal structure of presenilin or a presenilin homologue (see below).
3.7 Signal Peptide Peptidases
The concept of presenilin as the catalytic component for γ-secretase was considerably strengthened when signal peptide peptidase (SPP) was found to be a similar intramembrane aspartyl protease. SPP clears remnant signal peptides from the membrane after their production by signal peptidase (Fig. 13). However, this process apparently also plays a role in immune surveillance, in which signal peptides from the major histocompatibility complex (MHC) type I are cleaved by SPP and the peptide products presented onto the cell surface as an indication to natural killer cells whether MHC synthesis is proceeding normally.117 In addition, SPP is exploited by the hepatitis C virus for the maturation of its core protein, suggesting that this protease may be a suitable target for antiviral therapy.118 SPP was identified by affinity labeling with a peptidomimetic inhibitor, and the protein sequence displayed intriguing parallels with presenilin.119 SPP contains two conserved aspartates, each predicted to lie in the middle of a transmembrane domain (Fig. 13), and the aspartate-containing sequences resemble those found in presenilins. The predicted topology of SPP also resembles that of presenilins, placing the key aspartates in the same relative position to each other in the membrane. As with the metalloprotease S2P compared with its bacterial relatives, the orientation of the aspartate-containing transmembrane domains of SPP is apparently opposite that of presenilins, again in correlation with the orientation of SPP substrates, which is opposite that of γ-secretase substrates. Interestingly, prior to the identification of SPP, a computational search for presenilin-like proteins netted an entire family of so-called presenilin homologs (PSHs);120 however, it is not yet clear if all of these proteins have catalytic activity. Two homologs, SPP-like proteases, SPPL2a and SPPL2b, have recently been found to cleave tumor necrosis factor α 121 and the dementia-associated Bri2 protein,122 although the biological roles of these proteolytic events are unknown.
Figure 13.
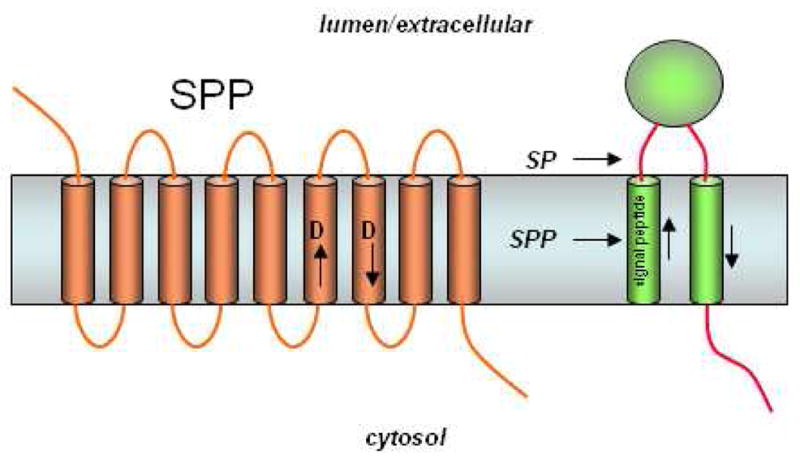
The presenilin homolog signal peptide peptidase (SPP). Signal peptides are removed from membrane proteins via signal peptidase (SP), and these peptides are released from the membrane by SPP-mediated intramembrane proteolysis. SPP, like presenilin, contains two aspartates essential for protease activity, but the conserved aspartate-containing motifs are in the opposite orientation compared with their presenilin counterparts (cf. Fig. 7). Consistent with the flipped orientation of SPP vis-à-vis presenilin, the substrates of these two proteases also run in the opposite direction. Unlike presenilin, SPP apparently does not require other protein cofactors or cleavage into two subunits for proteolytic activity.
SPP appears to be less complicated than γ-secretase. Expression of human SPP in yeast reconstituted the protease activity, suggesting that the protein has activity on its own and does not require other mammalian protein cofactors.119 This has recently been confirmed by the expression of various SPP orthologs in E. coli and purification of active enzyme to homogeneity.123 Moreover, unlike presenilins, SPP is not processed into two pieces. Thus, SPP may be a more tractable enzyme for understanding this type of intramembrane aspartyl protease and may shed light on γ-secretase structure and function. Indeed, the catalytic sites of the two proteases appear remarkably similar: their activities are inhibited by some of the same active site directed peptidomimetics124,125 and helical peptides,106 and activity can be modulated by the same NSAIDs that affect γ-secretase.106 SPP forms a homodimer very rapidly in cells, and this dimer is stable enough to allow isolation and analysis.126 Moreover, this dimer can be specifically labeled by a transition-state analog inhibitor, suggesting that the dimer is catalytically active. The functional importance of this dimer, however, is unclear; dimerization is not necessary for proteolytic activity, as determined from purification of recombinant SPP from E. coli.123 In terms of substrate recognition, however, SPP does display an important difference with γ-secretase: the apparent requirement for helix-breaking residues that should facilitate the ability of the enzyme to access the site of hydrolysis.127
4. Rhomboid Serine Proteases
The study of a conserved growth factor signaling pathway also led to intramembrane proteolysis. Epidermal growth factor (EGF) receptor ligands are synthesized as single-pass membrane proteins, but signaling requires proteolytic release and secretion of the ligand for interaction with its cognate receptor. In vertebrates, this is accomplished by membrane-tethered metalloproteases. Genetic analysis in Drosophila, however, identified two essential players, dubbed Star and Rhomboid-1, in the proteolysis of an EGF ortholog Spitz. No other components are apparently required. Full-length Spitz remains in the ER until it is ushered by Star to the Golgi apparatus, where it encounters Rhomboid-1.128 Rhomboid-mediated proteolysis in the Golgi is then followed by secretion for intercellular communication. But how does Rhomboid allow cleavage of Spitz?
4.1 Discovery of Rhomboid as a Protease
Mutational analysis of conserved non-glycine residues revealed a tantalizing requirement for a serine, a histidine, and an asparagine, which together might serve as a catalytic triad typically found in serine proteases129 (although subsequent studies support a Ser-His dyad130) (Fig. 14). These three residues were predicted to reside about the same depth within the membrane and thus have the potential to interact with each other. Consistent with this idea, the cleavage site of Spitz was estimated to be at an equivalent depth in the transmembrane region, and Spitz cleavage was sensitive only to serine protease inhibitors. Moreover, a careful analysis of concentration dependence revealed that expression of catalytic amounts of Rhomboid-1 still allowed Spitz proteolysis. Taken together, Rhomboid-1 is apparently a novel intramembrane serine protease.
Figure 14.

Rhomboid serine proteases. Rhomboids contain a conserved serine and histidine, which comprise a putative catalytic dyad of a serine protease. Rhomboid-1 cleaves within the transmembrane region of the Drosophila EGF-like growth factor Spitz.
4.2 Substrate Recognition and Rhomboid Regulation
What determines Rhomboid substrate specificity, and how is this proteolytic event regulated? Most of the Spitz transmembrane region could be swapped with that of a nonsubstrate protein without affecting cleavage by Rhomboid; however, the N-terminal quarter of the transmembrane region was critical for substrate recognition.131 Indeed, incorporation of this substrate motif into Delta allowed this Notch ligand to be processed by Rhomboid. Further examination of the substrate motif led to the tentative identification of a critical glycine-alanine, suggesting that, as with S2P and SPP, Rhomboid seems to require helix-destabilizing residues within the transmembrane domain of its substrates. Rhomboid activity is distinguished from that of most of the other I-CLiPs because Rhomboid does not require prior substrate cleavage by another protease. Rhomboid regulation apparently occurs mainly by translocation of the substrate from the ER to the Golgi (mediated by Star) and spatial control of Rhomboid transcription.
4.3 Other Rhomboids in Biology
Like S2P, Rhomboid genes have been conserved throughout evolution. Surprisingly, in spite of overall low homology with Rhomboids from multicellular organisms, a number of unicellular Rhomboids were capable of cleaving Drosophila Rhomboid substrates, and mutation of the putative catalytic residues abolished protease activity, illustrating the evolutionary conservation of the serine protease function of Rhomboid.132 The natural substrates for microbial Rhomboids are unknown, with two notable exceptions: (A) Providencia stuartii Rhomboid protease AarA cleaves a protein called TatA as part of a quorum-sensing signal;133 and (B) a Rhomboid from a parasitic amoeba sheds cell surface receptors as a means of evading the immune system.134 As for substrates of eukaryotic Rhomboid-1 homologues, two mitochondrial membrane proteins have been identified as substrates for yeast Rhomboid RBD1.135–137 RBD1-mediated release of one of these substrate (dynamin-like GTPase Mgm1p) is essential for remodeling of the mitochondrial membrane, and the human ortholog of RBD1, PARL, could restore substrate proteolysis and proper growth rates and mitochondrial morphology in a yeast RBD1 mutant,136 suggesting that the role of these Rhomboids in mitochrondrial function has been evolutionarily conserved. Indeed, a recent study identified a mitochondrial protein OPA1 as a likely substrate for PARL, the cleavage of this substrate being critical to cristae remodeling and cytochrome c release during apoptosis.138 In Toxoplasma, TgROM5, one of five nonmitochondrial Rhomboids in these parasites, cleaves a cell surface adhesion protein as a key step in cell invasion, and similar findings in the related Plasmodium falciparum, the malarial parasite, have recently been reported,139 suggesting that Rhomboids are potential targets for treating infections by these deadly pathogens.
4.4. Structure of Rhomboid
Rhomboid provided the first crystal structures of an I-CLiP, with four reports on the E. coli Rhomboid GlpG140–143 and one on the Haemophilus influenza Rhomboid (HiGlpG).144 These structures show remarkable similarities and important differences that provide insight into how this class of membrane-embedded protease carries out hydrolysis in the lipid bilayer. The structures all reveal that the key serine and histidine implicated as the catalytic dyad are indeed coordinated with each other and lie at a depth within the membrane consistent with where Rhomboids cleave their transmembrane substrates (Fig. 15A). A cavity is open to the periplasmic space, with the catalytic dyad at the bottom of this opening, and this cavity contains multiple water molecules.
Figure 15.

Structure of E. coli Rhomboid GlpG. (A) The serine in transmembrane domain 4 and the histidine in transmembrane domain 6 are coordinated in a manner consistent with known serine proteases and at a depth within the membrane consistent with the site of proteolysis of Rhomboid substrates. (B) Close up view of the active site from a crystal structure with a bound phospholipid. The interaction of the phosphate group with the backbone NH of serine 102 and with the side chain of asparagines 154 suggests the site of the oxyanion hole.
How substrate enters this cavity is not entirely clear, but the position of transmembrane domain 5 varies in the different structures, and movement of this domain can provide a space through which substrate may reach the catalytic dyad. Indeed, one of the reported structures contains a bound lipid in this space,142 with the phosphate group residing near the Ser-His dyad and a key Asn residue that may contribute to the oxyanion hole that stabilizes intermediates and transition-states during serine protease catalysis (Fig. 15B). Furthermore, mutational analysis revealed that altering residues predicted to disrupt TMD 5’s role as a gate led to increased proteolytic activity,145 providing further validation for the site of lateral entry of the substrate. These structural findings validate the molecular and biochemical studies on Rhomboids and suggest that such approaches have been providing true mechanistic insight into the workings of other I-CLiPs. As with the structure of an S2P metalloprotease (see above), these structures offer details that inspire specific hypotheses about how Rhomboids handle substrates to hydrolyze transmembrane domains.
5. Conclusions and Perspective
I-CLiPs are membrane-embedded enzymes that hydrolyze transmembrane substrates, and the residues essential to catalysis reside within the boundaries of the lipid bilayer. These proteases appear to recapitulate the mechanisms of soluble proteases, and the crystal structures of Rhomboids and an S2P support this notion, at least for the serine and metallo I-CLiPs. All I-CLiPs would be predicted to contain an initial substrate docking site, but to date evidence for such a docking site has only been provided for γ-secretase. The I-CLiPs discovered so far each play critical roles in biology and are closely regulated, but the means of control vary. They are all involved in cell signaling, but do so in a variety of ways. Membrane topology seems to dictate the types of substrates that can be cleaved, but this concept remains speculative. Most I-CLiPs appear to require helix-breaking residues near the cleavage sites of their substrates, although γ-secretase may be a notable exception.
Critical remaining issues include the identity of substrates for the I-CLiP family members whose roles are unknown. For instance, although an entire family of PSHs and Rhomboids have been discovered, natural substrates are only known for a handful of these proteins, and very little is known about the natural substrates of microbial and parasitic I-CLiPs. The conservation of putative catalytic residues implies conservation of proteolytic function, but the search for substrates is far from trivial. A computational approach for sequence motifs that are apparently required for substrate proteolysis by Rhomboids led to identification of adhesion proteins in Toxoplasma as potential substrates.131 Most typically, genetic and cell biological studies suggest a connection between protease and putative substrate, with follow up molecular and biochemical studies for validation.
Another key issue is understanding the specific mechanisms of these proteases This includes elucidating conformational changes that take place in both enzyme and substrate during proteolysis and identifying enzyme residues that directly interact with substrate. Structural biology is clearly the emerging frontier in the study of I-CLiPs, with Rhomboid and S2P providing the first fruits of such endeavors. Detailed structural understanding should provide clearer appreciation for how these remarkable enzymes work. The development of small molecule tools should ultimately dovetail with these structural studies, allowing cocrystal structures that offer further insight into mechanism and that pave the way for structure-based design in cases where the target has high therapeutic relevance.
Acknowledgments
The author’s work on intramembrane proteases is supported from NIH R01 grants AG17574, NS41355 and GM79555 (to M.S.W.) and P01 grant AG15379 (to Dennis J. Selkoe). Figure 15A was rendered by Raquel L. Lieberman.
Biography
 Michael S. Wolfe is a Professor of Neurology at Harvard Medical School and Brigham and Women’s Hospital. He received his B.S. in chemistry in 1984 from the Philadelphia College of Pharmacy and Science and earned his Ph.D. in medicinal chemistry in 1990 from the University of Kansas. After postdoctoral stints at the University of Kansas (medicinal chemistry) and the NIH (cell biology), he joined the faculty of the University of Tennessee in Memphis in 1994. In 1999, he moved to Harvard Medical School, where his work has focused on understanding the molecular basis of Alzheimer’s disease and identifying effective approaches for pharmacological intervention. In 2006, Dr. Wolfe founded the Laboratory for Experimental Alzheimer Drugs (LEAD) at Harvard Medical School.
Michael S. Wolfe is a Professor of Neurology at Harvard Medical School and Brigham and Women’s Hospital. He received his B.S. in chemistry in 1984 from the Philadelphia College of Pharmacy and Science and earned his Ph.D. in medicinal chemistry in 1990 from the University of Kansas. After postdoctoral stints at the University of Kansas (medicinal chemistry) and the NIH (cell biology), he joined the faculty of the University of Tennessee in Memphis in 1994. In 1999, he moved to Harvard Medical School, where his work has focused on understanding the molecular basis of Alzheimer’s disease and identifying effective approaches for pharmacological intervention. In 2006, Dr. Wolfe founded the Laboratory for Experimental Alzheimer Drugs (LEAD) at Harvard Medical School.
References
- 1.Barrett AJ, Rawlings ND, Woessner JF, editors. The Handbook of Proteolytic Enzymes. Academic Press, Ltd.; Amsterdam: 2004. [Google Scholar]
- 2.Rawlings ND, Tolle DP, Barrett AJ. Nucleic Acids Res. 2004;32:D160. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turk B. Nat Rev Drug Discov. 2006;5:785. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 4.Sternlicht MD, Werb Z. Annu Rev Cell Dev Biol. 2001;17:463. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krysan DJ, Ting EL, Abeijon C, Kroos L, Fuller RS. Eukaryot Cell. 2005;4:1364. doi: 10.1128/EC.4.8.1364-1374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfe MS, De Los Angeles J, Miller DD, Xia W, Selkoe DJ. Biochemistry. 1999;38:11223. doi: 10.1021/bi991080q. [DOI] [PubMed] [Google Scholar]
- 7.Weihofen A, Martoglio B. Trends Cell Biol. 2003;13:71. doi: 10.1016/s0962-8924(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 8.Wolfe MS, Kopan R. Science. 2004;305:1119. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 9.Brown MS, Goldstein JL. Cell. 1997;89:331. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 10.Hua X, Nohturfft A, Goldstein JL, Brown MS. Cell. 1996;87:415. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- 11.Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Proc Natl Acad Sci U S A. 1999;96:11235. doi: 10.1073/pnas.96.20.11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawson RB. Nat Rev Mol Cell Biol. 2003;4:631. doi: 10.1038/nrm1174. [DOI] [PubMed] [Google Scholar]
- 13.Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, Brown MS. Mol Cell. 1998;2:505. doi: 10.1016/s1097-2765(00)80150-1. [DOI] [PubMed] [Google Scholar]
- 14.Duncan EA, Dave UP, Sakai J, Goldstein JL, Brown MS. J Biol Chem. 1998;273:17801. doi: 10.1074/jbc.273.28.17801. [DOI] [PubMed] [Google Scholar]
- 15.Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Mol Cell. 1997;1:47. doi: 10.1016/s1097-2765(00)80006-4. [DOI] [PubMed] [Google Scholar]
- 16.Zelenski NG, Rawson RB, Brown MS, Goldstein JL. J Biol Chem. 1999;274:21973. doi: 10.1074/jbc.274.31.21973. [DOI] [PubMed] [Google Scholar]
- 17.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. Mol Cell. 2000;6:1355. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 18.Rudner DZ, Fawcett P, Losick R. Proc Natl Acad Sci U S A. 1999;96:14765. doi: 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehara K, Akiyama Y, Ito K. Gene. 2001;281:71–9. doi: 10.1016/s0378-1119(01)00823-x. [DOI] [PubMed] [Google Scholar]
- 20.Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. Genes Dev. 2002;16:2156. doi: 10.1101/gad.1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou R, Kroos L. Mol Microbiol. 2005;58:835. doi: 10.1111/j.1365-2958.2005.04870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campo N, Rudner DZ. Mol Cell. 2006;23:25. doi: 10.1016/j.molcel.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 23.Makinoshima H, Glickman MS. Nature. 2005;436:406. doi: 10.1038/nature03713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS. Proc Natl Acad Sci U S A. 2000;97:5123. doi: 10.1073/pnas.97.10.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akiyama Y, Kanehara K, Ito K. EMBO J. 2004;23:4434. doi: 10.1038/sj.emboj.7600449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. Science. 2007;318:1608. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- 27.Hardy J, Selkoe DJ. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 28.Tanzi RE, Bertram L. Cell. 2005;120:545. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Science. 1999;286:735. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 30.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, John V. Nature. 1999;402:537. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 31.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Nature. 1999;402:533. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 32.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Mol Cell Neurosci. 1999;14:419. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 33.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Proc Natl Acad Sci U S A. 2000;97:1456. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G. Biochemistry. 2002;41:2825–2835. doi: 10.1021/bi015794o. [DOI] [PubMed] [Google Scholar]
- 35.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, StGeorge-Hyslop PH. Nature. 1995;375:754. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 36.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Science. 1995;269:973. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 37.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Placentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, StGeorge-Hyslop PH. Nature. 1995;376:775. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 38.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Nat Med. 1996;2:864. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 39.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Nature. 1996;383:710. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 40.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ, Arango JC. Nat Med. 1996;2:1146. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 41.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Nat Med. 1997;3:67. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 42.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Nature. 1998;391:387. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 43.Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Nat Cell Biol. 2000;2:461. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA. Nat Cell Biol. 2000;2:463. doi: 10.1038/35017108. [DOI] [PubMed] [Google Scholar]
- 45.Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Donkor IO, Selkoe DJ. Biochemistry. 1999;38:4720. doi: 10.1021/bi982562p. [DOI] [PubMed] [Google Scholar]
- 46.Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL. Biochemistry. 2000;39:8698. doi: 10.1021/bi0005456. [DOI] [PubMed] [Google Scholar]
- 47.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Nature. 1999;398:513. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 48.Laudon H, Mathews PM, Karlstrom H, Bergman A, Farmery MR, Nixon RA, Winblad B, Gandy SE, Lendahl U, Lundkvist J, Naslund J. J Neurochem. 2004;89:44. doi: 10.1046/j.1471-4159.2003.02298.x. [DOI] [PubMed] [Google Scholar]
- 49.Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. J Biol Chem. 1999;274:28669. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- 50.Ratovitski T, Slunt HH, Thinakaran G, Price DL, Sisodia SS, Borchelt DR. J Biol Chem. 1997;272:24536. doi: 10.1074/jbc.272.39.24536. [DOI] [PubMed] [Google Scholar]
- 51.Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, Koo EH, Seubert P, StGeorge-Hyslop P, Teplow DB, Selkoe DJ. Neurobiol Dis. 1997;3:325. doi: 10.1006/nbdi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 52.Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. J Biol Chem. 1997;272:28415. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- 53.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE. J Biol Chem. 1998;273:16470. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- 54.Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C. J Biol Chem. 1998;273:3205. doi: 10.1074/jbc.273.6.3205. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. J Biol Chem. 1998;273:12436. doi: 10.1074/jbc.273.20.12436. [DOI] [PubMed] [Google Scholar]
- 56.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Nature. 2000;405:689. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 57.Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, Wolfe MS. Nat Cell Biol. 2000;2:428. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- 58.Levitan D, Greenwald I. Nature. 1995;377:351. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 59.Selkoe D, Kopan R. Annu Rev Neurosci. 2003;26:565. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- 60.Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A. Proc Natl Acad Sci U S A. 1998;95:8108. doi: 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israël A. Mol Cell. 2000;5:207. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 62.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. Mol Cell. 2000;5:197. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 63.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. Nature. 1999;398:518. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 64.Schroeter EH, Kisslinger JA, Kopan R. Nature. 1998;393:382. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 65.Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Nature. 2000;405:966. doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- 66.De Strooper B. Neuron. 2003;38:9. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 67.Beel AJ, Sanders CR. Cell Mol Life Sci. 2008;65:1311. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. Cell. 2003;114:635. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 69.Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Cell. 2006;127:185. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 70.Kopan R, Ilagan MX. Nat Rev Mol Cell Biol. 2004;5:499. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 71.Goutte C, Hepler W, Mickey KM, Priess JR. Development. 2000;127:2481. doi: 10.1242/dev.127.11.2481. [DOI] [PubMed] [Google Scholar]
- 72.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D. Dev Cell. 2002;3:85. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 73.Goutte C, Tsunozaki M, Hale VA, Priess JR. Proc Natl Acad Sci U S A. 2002;99:775. doi: 10.1073/pnas.022523499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P. Nature. 2000;407:48. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 75.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. Nature. 2003;422:438. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- 76.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Proc Natl Acad Sci U S A. 2003;100:6382. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS. Proc Natl Acad Sci U S A. 2002;99:2720. doi: 10.1073/pnas.052436599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Nat Cell Biol. 2003;5:486. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 79.Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, Van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS. Biochemistry. 2004;43:9774. doi: 10.1021/bi0494976. [DOI] [PubMed] [Google Scholar]
- 80.LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ. J Biol Chem. 2003;278:37213. doi: 10.1074/jbc.M303941200. [DOI] [PubMed] [Google Scholar]
- 81.Hu Y, Fortini ME. J Cell Biol. 2003;161:685. doi: 10.1083/jcb.200304014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H. J Biol Chem. 2004;279:23255. doi: 10.1074/jbc.M401789200. [DOI] [PubMed] [Google Scholar]
- 83.Edbauer D, Winkler E, Haass C, Steiner H. Proc Natl Acad Sci U S A. 2002;99:8666. doi: 10.1073/pnas.132277899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Evin G, Canterford LD, Hoke DE, Sharples RA, Culvenor JG, Masters CL. Biochemistry. 2005;44:4332. doi: 10.1021/bi0481702. [DOI] [PubMed] [Google Scholar]
- 85.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Proc Natl Acad Sci U S A. 2000;97:6138. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R. Proc Natl Acad Sci U S A. 2003;100:13075. doi: 10.1073/pnas.1735338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cervantes S, Saura CA, Pomares E, Gonzalez-Duarte R, Marfany G. J Biol Chem. 2004;279:36519. doi: 10.1074/jbc.M404832200. [DOI] [PubMed] [Google Scholar]
- 88.Clarke EE, Churcher I, Ellis S, Wrigley JD, Lewis HD, Harrison T, Shearman MS, Beher D. J Biol Chem. 2006;281:31279. doi: 10.1074/jbc.M605051200. [DOI] [PubMed] [Google Scholar]
- 89.Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS. J Biol Chem. 2007;282:33985. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- 90.Osenkowski P, Li H, Ye W, Li D, Aeschbach L, Fraering PC, Wolfe MS, Selkoe DJ, Li H. J Mol Biol. 2009;385:642. doi: 10.1016/j.jmb.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Das C, Berezovska O, Diehl TS, Genet C, Buldyrev I, Tsai JY, Hyman BT, Wolfe MS. J Am Chem Soc. 2003;125:11794. doi: 10.1021/ja037131v. [DOI] [PubMed] [Google Scholar]
- 92.Kornilova AY, Bihel F, Das C, Wolfe MS. Proc Natl Acad Sci U S A. 2005;102:3230. doi: 10.1073/pnas.0407640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bihel F, Das C, Bowman MJ, Wolfe MS. J Med Chem. 2004;47:3931. doi: 10.1021/jm049788c. [DOI] [PubMed] [Google Scholar]
- 94.Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, 3rd, Sudhof T, Yu G. Cell. 2005;122:435. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 95.Chavez-Gutierrez L, Tolia A, Maes E, Li T, Wong PC, de Strooper B. J Biol Chem. 2008;283:20096. doi: 10.1074/jbc.M803040200. [DOI] [PubMed] [Google Scholar]
- 96.Wolfe MS. Curr Top Med Chem. 2008;8:2. doi: 10.2174/156802608783334024. [DOI] [PubMed] [Google Scholar]
- 97.Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Clin Neuropharmacol. 2007;30:317. doi: 10.1097/WNF.0b013e31805b7660. [DOI] [PubMed] [Google Scholar]
- 98.Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. J Biol Chem. 2003;278:46107. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- 99.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, Zhang L, Higgins GA, Parker EM. J Biol Chem. 2004;279:12876. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 100.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. Nature. 2001;414:212. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 101.Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. J Biol Chem. 2003;278:31831. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- 102.Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T. J Biol Chem. 2003;278:18664. doi: 10.1074/jbc.M301619200. [DOI] [PubMed] [Google Scholar]
- 103.Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, Shearman MS. J Biol Chem. 2004;279:43419. doi: 10.1074/jbc.M404937200. [DOI] [PubMed] [Google Scholar]
- 104.Okochi M, Fukumori A, Jiang J, Itoh N, Kimura R, Steiner H, Haass C, Tagami S, Takeda M. J Biol Chem. 2006;281:7890. doi: 10.1074/jbc.M513250200. [DOI] [PubMed] [Google Scholar]
- 105.Galasko DR, Graff-Radford N, May S, Hendrix S, Cottrell BA, Sagi SA, Mather G, Laughlin M, Zavitz KH, Swabb E, Golde TE, Murphy MP, Koo EH. Alzheimer Dis Assoc Disord. 2007;21:292. doi: 10.1097/WAD.0b013e31815d1048. [DOI] [PubMed] [Google Scholar]
- 106.Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. Biochemistry. 2006;45:8649. doi: 10.1021/bi060597g. [DOI] [PubMed] [Google Scholar]
- 107.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE. Nature. 2008;453:925. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Proc Natl Acad Sci U S A. 2003;100:12444. doi: 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fraering PC, Ye W, Lavoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. J Biol Chem. 2005;280:41987. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H. Proc Natl Acad Sci U S A. 2006;103:6889. doi: 10.1073/pnas.0602321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ogura T, Mio K, Hayashi I, Miyashita H, Fukuda R, Kopan R, Kodama T, Hamakubo T, Iwatsubo T, Tomita T, Sato C. Biochem Biophys Res Commun. 2006;343:525. doi: 10.1016/j.bbrc.2006.02.158. [DOI] [PubMed] [Google Scholar]
- 112.Tolia A, Chavez-Gutierrez L, De Strooper B. J Biol Chem. 2006;281:27633. doi: 10.1074/jbc.M604997200. [DOI] [PubMed] [Google Scholar]
- 113.Sato C, Morohashi Y, Tomita T, Iwatsubo T. J Neurosci. 2006;26:12081. doi: 10.1523/JNEUROSCI.3614-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tolia A, Horre K, De Strooper B. J Biol Chem. 2008;15:15. doi: 10.1074/jbc.M802461200. [DOI] [PubMed] [Google Scholar]
- 115.Sato C, Takagi S, Tomita T, Iwatsubo T. J Neurosci. 2008;28:6264–71. doi: 10.1523/JNEUROSCI.1163-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kornilova AY, Kim J, Laudon H, Wolfe MS. Biochemistry. 2006;45:7598. doi: 10.1021/bi060107k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. J Immunol. 2001;167:6441. doi: 10.4049/jimmunol.167.11.6441. [DOI] [PubMed] [Google Scholar]
- 118.McLauchlan J, Lemberg MK, Hope G, Martoglio B. EMBO J. 2002;21:3980. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Science. 2002;296:2215. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- 120.Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, Golde TE. Hum Mol Genet. 2002;11:1037. doi: 10.1093/hmg/11.9.1037. [DOI] [PubMed] [Google Scholar]
- 121.Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, Bohland C, Imhof A, Martoglio B, Teplow DB, Haass C. Nat Cell Biol. 2006;8:894. doi: 10.1038/ncb1450. [DOI] [PubMed] [Google Scholar]
- 122.Martin L, Fluhrer R, Reiss K, Kremmer E, Saftig P, Haass C. J Biol Chem. 2008;283:1644. doi: 10.1074/jbc.M706661200. [DOI] [PubMed] [Google Scholar]
- 123.Narayanan S, Sato T, Wolfe MS. J Biol Chem. 2007;282:20172. doi: 10.1074/jbc.M701536200. [DOI] [PubMed] [Google Scholar]
- 124.Kornilova AY, Das C, Wolfe MS. J Biol Chem. 2003;278:16470. doi: 10.1074/jbc.C300019200. [DOI] [PubMed] [Google Scholar]
- 125.Weihofen A, Lemberg MK, Friedmann E, Rueeger H, Schmitz A, Paganetti P, Rovelli G, Martoglio B. J Biol Chem. 2003;278:16528. doi: 10.1074/jbc.M301372200. [DOI] [PubMed] [Google Scholar]
- 126.Nyborg AC, Kornilova AY, Jansen K, Ladd TB, Wolfe MS, Golde TE. J Biol Chem. 2004;279:15153. doi: 10.1074/jbc.M309305200. [DOI] [PubMed] [Google Scholar]
- 127.Lemberg MK, Martoglio B. Mol Cell. 2002;10:735. doi: 10.1016/s1097-2765(02)00655-x. [DOI] [PubMed] [Google Scholar]
- 128.Lee JR, Urban S, Garvey CF, Freeman M. Cell. 2001;107:161. doi: 10.1016/s0092-8674(01)00526-8. [DOI] [PubMed] [Google Scholar]
- 129.Urban S, Lee JR, Freeman M. Cell. 2001;107:173. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- 130.Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. EMBO J. 2005;24:464. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Urban S, Freeman M. Mol Cell. 2003;11:1425. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- 132.Urban S, Schlieper D, Freeman M. Curr Biol. 2002;12:1507. doi: 10.1016/s0960-9822(02)01092-8. [DOI] [PubMed] [Google Scholar]
- 133.Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Proc Natl Acad Sci U S A. 2007;104:1003. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baxt LA, Baker RP, Singh U, Urban S. Genes Dev. 2008;22:1636. doi: 10.1101/gad.1667708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Esser K, Tursun B, Ingenhoven M, Michaelis G, Pratje E. J Mol Biol. 2002;323:835. doi: 10.1016/s0022-2836(02)01000-8. [DOI] [PubMed] [Google Scholar]
- 136.McQuibban GA, Saurya S, Freeman M. Nature. 2003;423:537. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- 137.Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS. J Biol Chem. 2003;278:27781. doi: 10.1074/jbc.M211311200. [DOI] [PubMed] [Google Scholar]
- 138.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. Cell. 2006;126:177. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 139.Baker RP, Wijetilaka R, Urban S. PLoS Pathog. 2006;2:e113. doi: 10.1371/journal.ppat.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Y, Zhang Y, Ha Y. Nature. 2006;444:179. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 141.Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Nat Struct Mol Biol. 2006;13:1084. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 142.Ben-Shem A, Fass D, Bibi E. Proc Natl Acad Sci U S A. 2007;104:462. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Y, Ha Y. Proc Natl Acad Sci U S A. 2007;104:2098. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. Proc Natl Acad Sci U S A. 2007;104:750. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baker RP, Young K, Feng L, Shi Y, Urban S. Proc Natl Acad Sci U S A. 2007;104:8257. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]