

Abstract
Objective
Osteoarthritis is a degenerative joint disease whose molecular mechanism is currently unknown. Wnt/β-catenin signaling has been demonstrated to play a critical role in the development and function of articular chondrocytes. To determine the role of β-catenin signaling in articular chondrocyte function, we generated Col2a1-ICAT–transgenic mice to inhibit β-catenin signaling in chondrocytes.
Methods
The expression of the ICAT transgene was determined by immunostaining and Western blot analysis. Histologic analyses were performed to determine changes in articular cartilage structure and morphology. Cell apoptosis was determined by TUNEL staining and the immunostaining of cleaved caspase 3 and poly(ADP-ribose) polymerase (PARP) proteins. Expression of Bcl-2, Bcl-xL, and Bax proteins and caspase 9 and caspase 3/7 activities were examined in primary sternal chondrocytes isolated from 3-day-old neonatal Col2a1-ICAT–transgenic mice and their wild-type littermates and in primary chicken and porcine articular chondrocytes.
Results
Expression of the ICAT transgene was detected in articular chondrocytes of the transgenic mice. Associated with this, age-dependent articular cartilage destruction was observed in Col2a1-ICAT– transgenic mice. A significant increase in cell apoptosis in articular chondrocytes was identified by TUNEL staining and the immunostaining of cleaved caspase 3 and PARP proteins in these transgenic mice. Consistent with this, Bcl-2 and Bcl-xL expression were decreased and caspase 9 and caspase 3/7 activity were increased, suggesting that increased cell apoptosis may contribute significantly to the articular cartilage destruction observed in Col2a1-ICAT–transgenic mice.
Conclusion
Inhibition of β-catenin signaling in articular chondrocytes causes increased cell apoptosis and articular cartilage destruction in Col2a1-ICAT–transgenic mice.
Osteoarthritis (OA) is a degenerative joint disease. Numerous genetic and environmental factors have been proposed as contributors to the development of OA. The progression of the disease is slow and eventually results in degeneration and loss of the articular cartilage in various joints, including fingers, knees, hips, and spine. The disease process leads to limitation of joint movement, joint deformity, joint stiffness, inflammation, and severe pain. The mechanism of OA pathogenesis remains undefined.
OA is mediated by a dynamic interplay between the articular chondrocytes, their matrix, and synovial cells. Articular chondrocytes, the cell type present in articular cartilage, are responsible for producing and maintaining the extracellular matrix (ECM), which supports the appropriate biomechanical function of the tissue. Phenotypic changes seen in OA are often accompanied by ECM degradation, which eventually results in cartilage destruction (1). Synovial fibroblasts are mesenchyme-derived cells that form a thin lining of synovial tissue surrounding the fibrous capsule of the joint. The physiologic roles of synovial tissue are to produce synovial fluid that lubricates the joints and to supply nutrients to the articular chondrocytes.
Articular cartilage plays a crucial role in joint function during adulthood, and destruction of articular cartilage has severe consequences. While most cartilages are replaced by bone during development, only a few cartilages are permanent, including articular cartilage. Articular chondrocytes, which function to maintain the matrix, are unique among growth plate chondrocytes in that they exhibit very limited mitotic activity, have a slow rate of matrix synthesis and degradation, and do not progress fully to the terminally differentiated phenotype displayed by cells in the growth plate (2). Very little is known regarding the mechanisms involved in establishing and maintaining this articular chondrocyte phenotype.
The function of articular chondrocytes is regulated by a variety of growth factors, including Wnt/β-catenin signaling molecules, which play a critical role in chondrocyte proliferation, differentiation, and apoptosis. Beta-catenin is a key molecule in the canonical Wnt signaling pathway and plays a critical role in multiple steps during chondrocyte formation and maturation (3,4). Inhibitor of β-catenin and T cell factor (ICAT) is an 82-amino-acid small molecule (5) whose crystal structure reveals binding capacity to the armadillo repeats of β-catenin. This binding disrupts the complex formation of β-catenin with T cell factor (TCF)/lymphoid enhancer factor (LEF) (6,7) and thus leads to inhibition of signaling in this pathway. To investigate the role of β-catenin signaling in the maintenance of articular cartilage function under physiologic and pathophysiologic conditions, we generated Col2a1-ICAT–transgenic mice in which the ICAT transgene was targeted specifically to chondrocytes using the Col2a1 promoter. We found that the ICAT transgene was highly expressed in articular chondrocytes in 6-month-old transgenic mice, leading to inhibition of β-catenin signaling in transgenic mouse chondrocytes. Associated with this, severe articular cartilage destruction was observed in Col2a1-ICAT–transgenic mice. Articular chondrocyte apoptosis was significantly increased, providing novel evidence regarding the contribution of β-catenin signaling to the maintenance of normal articular cartilage function.
MATERIALS AND METHODS
Generation of transgenic mice
To generate the Col2a1-ICAT transgene, DNA fragments encoding ICAT were cloned into the Not I site of a Colα1(II) (Col2a1)–based expression vector, PKN185 (8). The resulting vector contains the FLAG-tagged ICAT (FLAG-ICAT) complementary DNA including the 5′ Nde I site Col2a1 promoter (nucleotide 1940–2971, GenBank accession no. M65161), β-globin intron cassette, SV40 poly(A), and Col2a1 enhancer (nucleotide 4930–5571, GenBank accession no. M65161). The FLAG-ICAT transgene was excised by Nde I and Hind III digestion. The ICAT transgene was then purified and injected into pronuclei of fertilized eggs from C57BL/6J mice. Positive transgenic founder mice were identified by Southern blot analysis, and mice were genotyped by polymerase chain reaction (PCR). The F3 generation of 2 separate lines of Col2a1-ICAT–transgenic mice and their wild-type (WT) littermates were used for phenotype analysis and cellular function studies.
Histology and histomorphometry
Initial radiographic and histologic analyses were performed in 2 lines of the transgenic mice (lines 5 and 7), and similar articular cartilage destruction was found. Analysis of time-dependent articular cartilage destruction was then performed in 1 line of these transgenic mice (line 5). Knee joints from 6-, 9-, 12-, and 15-month-old WT and Col2a1-ICAT–transgenic mice were dissected, fixed in 10% formalin, decalcified, and embedded in paraffin. Serial midsagittal sections (3-µm thick) of knee joints from 6-month-old mice were cut every 10 µm from both the medial and lateral compartments. The sections were stained with Alcian blue/hematoxylin and eosin (H&E). Articular cartilage was outlined on the tibial surface, and an area algorithm in the software package ImagePro 4.5 (Leeds Precision Instruments, Minneapolis, MN) was used to determine the pixel area of outlined articular cartilage from each section. Using this approach, average articular cartilage area was determined. Cells in each section were counted, and the ImagePro counting algorithm was used to determine the average number of chondrocytes per unit cartilage area. Six mice per group were used for histomorphometric measurements.
Immunostaining
Tissue sections were deparaffinized by immersing in xylene, then fixed with 4% paraformaldehyde for 15 minutes and treated with 0.5% Triton for 15 minutes followed by fixing with 4% paraformaldehyde for another 5 minutes. Sections were then incubated with a mouse anti-FLAG M2 monoclonal antibody (1:200 dilution; Sigma, St. Louis, MO), rabbit anti–cleaved caspase 3 monoclonal antibody (Asp175, 1:200 dilution; Cell Signaling Technology, Danvers, MA), and rabbit anti–poly(ADP-ribose) polymerase (anti-PARP) polyclonal antibody (1:50 dilution; Abcam, Cambridge, MA) overnight. Secondary incubations were performed with a fluorescence-conjugated secondary antibody for 60 minutes, and sections were mounted with Vectashield (LabVision, Fremont, CA). Slides were visualized under a fluorescence microscope.
Cell isolation and cell culture
Three-day-old neonatal mice were euthanized and genotyped using tail tissues obtained at the time of death. The anterior rib cage and sternum were harvested en bloc, washed with phosphate buffered saline (PBS), and then digested with Pronase (Roche Applied Science, Indianapolis, IN) dissolved in PBS (2 mg/ml) in a 37°C water bath with continuous shaking for 60 minutes. This was followed by incubation in a solution of collagenase D (3 mg/ml dissolved in serum-free Dulbecco’s modified Eagle’s medium [DMEM]; Roche Applied Science) for 90 minutes at 37°C. The soft tissue debris was thoroughly removed. The remaining sterna and costosternal junctions were further digested in a fresh collagenase D solution in petri dishes in a 37°C incubator for 5 hours with intermittent shaking. This step allows remnant fibroblasts to attach to the petri dish while the chondrocytes remain afloat in the medium. The digestion solution was filtered through Swinnex filter (Millipore, Bedford, MA) to remove all residual bone fragments. The solution was centrifuged, and the cells were resuspended in complete medium (DMEM with 10% fetal bovine serum [FBS], 1% penicillin/streptomycin, 100 mM L-glutamine, and 50 µg/ml ascorbic acid, pH 7.1). The cells were counted and plated at the appropriate density. To remove any remaining fibroblasts, 24-hour cultures were treated with 0.05% trypsin for 1 minute to lift the fibroblasts from the culture dish while allowing the chondrocytes to remain attached.
For chicken chondrocytes, 6-week-old chicks were euthanized and the femora and tibiae were carefully separated by removing the surrounding tissue at the knee joint. Articular cartilage was cut off and digested with trypsin (1 mg/ml; Sigma) on a rotator for 30 minutes. This was followed by incubation in a solution of hyaluronidase (1 mg/ml; Sigma) for 60 minutes. The remaining tissues were further digested in collagenase A (1 mg/ml; Roche Applied Science) on the rotator overnight. The digestion solution was filtered through Swinnex filter to remove all residual bone fragments. The solution was centrifuged, and the cells were resuspended in complete medium (DMEM with 5% FBS, 1% penicillin/streptomycin, and 50 µg/ml ascorbic acid, pH 7.1). Cells were counted and plated at the appropriate density.
Porcine articular chondrocytes were isolated from the articular cartilage of elbow and metatarsal joints of freshly slaughtered pigs. Briefly, articular cartilage was shaved from the articular surfaces and minced into 2 × 2–mm chunks. Articular chondrocytes were then released from the native ECM with a 1.5-hour treatment in 0.05% (weight/volume) Pronase (Calbiochem, San Diego, CA) followed by 16-hour digestion in 0.2% (w/v) collagenase (Worthington, Lakewood, NJ). Cells were then pelleted and resuspended in 20 ml of cell culture media containing DMEM, 1% penicillin/streptomycin, 10% FBS, and 50 µg/ml ascorbic acid before counting and plating.
Luciferase assay
Primary sternal chondrocytes were transfected with the Top-flash reporter construct (0.5 µg/well, 12-well plate) and control SV40 Ranilla luciferase construct (0.01 µg/well) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in the absence or presence of BIO (1 µM), the glycogen synthase kinase 3β (GSK-3β) inhibitor. Cell extracts were harvested, and the luciferase activity was measured 2 days after transfection by the Promega Dual Luciferase system (Promega, Madison, WI).
Western blot analysis
Primary chondrocytes were lysed on ice for 30 minutes in a buffer containing 50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P40, and 0.1% sodium dodecyl sulfate (SDS) supplemented with protease inhibitors (10 µg/ml leupeptin, 10 µg/ml pepstatin A, and 10 µg/ml aprotinin) and phosphatase inhibitors (1 mM NaF and 1 mM Na3VO4). Proteins were fractionated by SDS– polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and detected using anti–Bcl-2 and anti–Bcl-xL monoclonal antibodies (1:1,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) and an anti-Bax monoclonal antibody (1:1,000 dilution; Santa Cruz Biotechnology).
TUNEL staining
A TUNEL staining kit (DeadEnd Fluorometric TUNEL System; Promega) was used to assess programmed cell death by catalytically incorporating fluorescein-12-dUTP at 3′-OH DNA ends using the terminal deoxynucleotidyl transferase and recombinant enzyme. After deparaffinization, the tissue sections were placed in equilibrium buffer and then in a solution containing the equilibrium buffer, nucleotide mix, and terminal deoxynucleotidyl transferase and recombinant enzyme and incubated at 37°C for 1 hour. The reaction was stopped with 2× saline–sodium citrate. Hoechst 33342 was used to stain nuclei. Results were visualized under a fluorescence microscope. Articular cartilage area from the tidemark to the surface and from the anterior to posterior cartilage margins was outlined in each slide. Chondrocyte numbers within the outlined regions were counted on 4 nonconsecutive sections from each joint sample. The apoptotic cell rates were determined by counting the numbers of TUNEL staining–positive cells in the cartilage area divided by the total cell number. Six Col2a1-ICAT–transgenic mice and 6 WT littermates were analyzed.
Caspase activity assay
Caspase 3/7 activity in primary mouse sternal chondrocytes and chicken and porcine articular chondrocytes was measured by adding Apo-ONE Caspase-3/7 Reagent (Apo-ONE homogeneous caspase 3/7 assay; Promega) to each well of a black 96-well tissue culture plate that was either blank or that had cells in culture. After incubation for 2 hours, the fluorescence of each well was measured. Caspase 9 activity was measured by adding Caspase-Glo 9 Reagent (Promega) to each well of a white 96-well tissue culture plate that was either blank or that had cells in culture. After incubation for 2 hours, the luminescence of each well was measured. The caspase 8 activity was determined by adding the Caspase-Glo 8 Reagent (Promega) to the cell culture.
RESULTS
Generation of Col2a1-ICAT–transgenic mice and verification of signaling phenotype
Col2a1-ICAT–transgenic mice were engineered to overexpress ICAT in chondrocytes under control of the mouse Col2a1 promoter. As mentioned, ICAT is a small peptide which selectively binds to the armadillo repeats of β-catenin and disrupts the interaction between β-catenin and TCF. Overexpression of ICAT specifically inhibits β-catenin signaling without disturbing cell adhesion (9). This unique feature of ICAT makes it an ideal molecule for the selective blockade of β-catenin signaling in target cells. In the present studies, we first examined the expression of the ICAT transgene in articular cartilage by immunostaining using an anti-FLAG antibody and found that the FLAG-ICAT transgene was specifically expressed in articular chondrocytes in 2-week-old and 6-month-old Col2a1-ICAT–transgenic mice (Figures 1a and b). To determine whether β-catenin signaling is blocked in Col2a1-ICAT–transgenic mouse chondrocytes, we performed β-catenin reporter assays using primary sternal chondrocytes that were transfected with the Top-flash reporter construct and treated with the GSK-3β inhibitor, BIO. We found that BIO-induced reporter activity was completely inhibited in chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 1c).
Figure 1. Generation of Col2a1-ICAT–transgenic mice and verification of signaling phenotype.
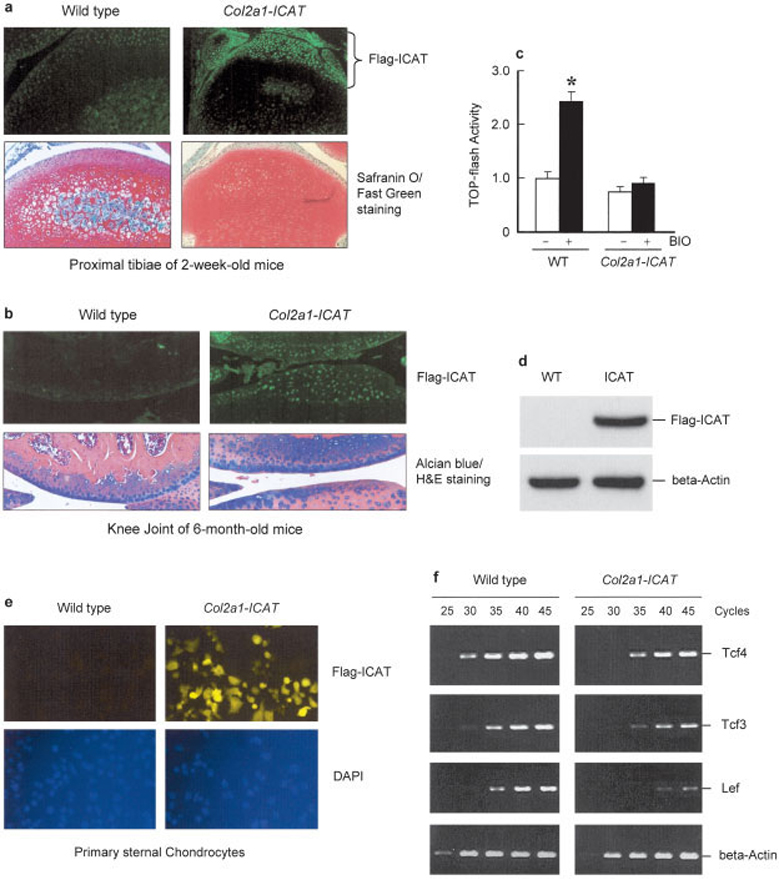
Two lines of Col2a1-ICAT–transgenic mice were established. The expression of the FLAG-ICAT transgene and the effect of inhibitor of β-catenin and T cell factor (ICAT) on β-catenin signaling in chondrocytes were examined. a and b, Immunofluorescence staining was performed using tissue sections from long bones of 2-week-old mice (a) and 6-month-old mice (b). Safranin O–fast green–stained histologic sections from 2-week-old mice and Alcian blue/hematoxylin and eosin (H&E)–stained histologic sections from 6-month-old mice were used as controls. Using the anti-FLAG M2 antibody, the expression of the FLAG-ICAT transgene was detected in articular chondrocytes of Col2a1-ICAT–transgenic mice but not in those of their wild-type (WT) littermates. c, To determine changes in β-catenin signaling in Col2a1-ICAT–transgenic mice, primary sternal chondrocytes isolated from 3-day-old Col2a1-ICAT–transgenic mice and their WT littermates were transfected with Top-flash reporter construct and treated for 48 hours with glycogen synthase kinase 3β inhibitor, BIO (1 µM). Values are the mean and SEM. BIO stimulated Top-flash reporter activity in WT chondrocytes (* = P < 0.05 versus unstimulated WT chondrocytes, by unpaired t-test). In transgenic mouse chondrocytes, the basal reporter activity was reduced and the stimulatory effect of BIO was completely inhibited. d and e, To further demonstrate FLAG-ICAT transgene expression in vitro, we performed Western blot (d) and immunostaining (e) assays using primary sternal chondrocytes derived from 3-day-old Col2a1-ICAT–transgenic mice and their WT littermates. Western blot and immunostaining data showed that FLAG-ICAT was detected only in chondrocytes derived from Col2a1-ICAT–transgenic mice and not in those from their WT littermates. f, The expression of β-catenin signaling downstream genes, Tcf3, Tcf4, and Lef, was examined by reverse transcriptase–polymerase chain reaction assays. The mRNA expression of Tcf3, Tcf4, and Lef was reduced in chondrocytes derived from Col2a1-ICAT–transgenic mice. (Original magnification × 10 in a; × 20 in b and e.) DAPI = 4′, 6-diamidino-2-phenylindole.
To further demonstrate the expression of the ICAT transgene in vitro, we examined FLAG-ICAT expression in isolated sternal chondrocytes via Western blotting using the anti-FLAG antibody. The results showed that the expression of ICAT protein could be detected in sternal chondrocytes derived from Col2a1-ICAT–transgenic mice but not in cells from the WT littermates (Figure 1d). The results from in vitro immunostaining assays further showed the expression of the FLAG-ICAT transgene in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 1e). To determine changes in β-catenin downstream genes, we examined the expression of TCFs and LEF and found that the messenger RNA expression of Tcf3, Tcf4, and Lef was significantly reduced in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 1f). The results demonstrate that we have established a transgenic mouse model with overexpression of ICAT in chondrocytes and the associated inhibition of β-catenin signaling specifically in chondrocytic cells.
Articular cartilage destruction in Col2a1-ICAT–transgenic mice
The joint/articular cartilage phenotype was initially evaluated by radiographic and histologic analyses of 2 lines (lines 5 and 7) of 6- and 9-month-old Col2a1-ICAT–transgenic mice. Based on these analyses, both lines were found to possess a similar articular cartilage phenotype. Detailed time-dependent assessment of the articular cartilage destruction was performed using histologic and histomorphometric analyses in 1 line of these transgenic mice (line 5). Alcian blue/H&E staining was performed. General evaluation of the histologic specimens revealed a progressive loss of the smooth surface of knee articular cartilage in Col2a1-ICAT–transgenic mice. At age 6 months, mild degeneration was observed and the number of articular chondrocytes was reduced, with the cartilage in weight-bearing areas completely lacking cells (Figure 2a). Interestingly, remaining chondrocytes were grouped into clusters, suggesting cell cloning (Figure 2a). Histologic and histomorphometric analyses showed that the articular cartilage area and articular chondrocyte numbers were significantly reduced in Col2a1-ICAT–transgenic mice (Figures 2b–d).
Figure 2. Articular cartilage destruction in 6-month-old Col2a1-ICAT–transgenic mice.
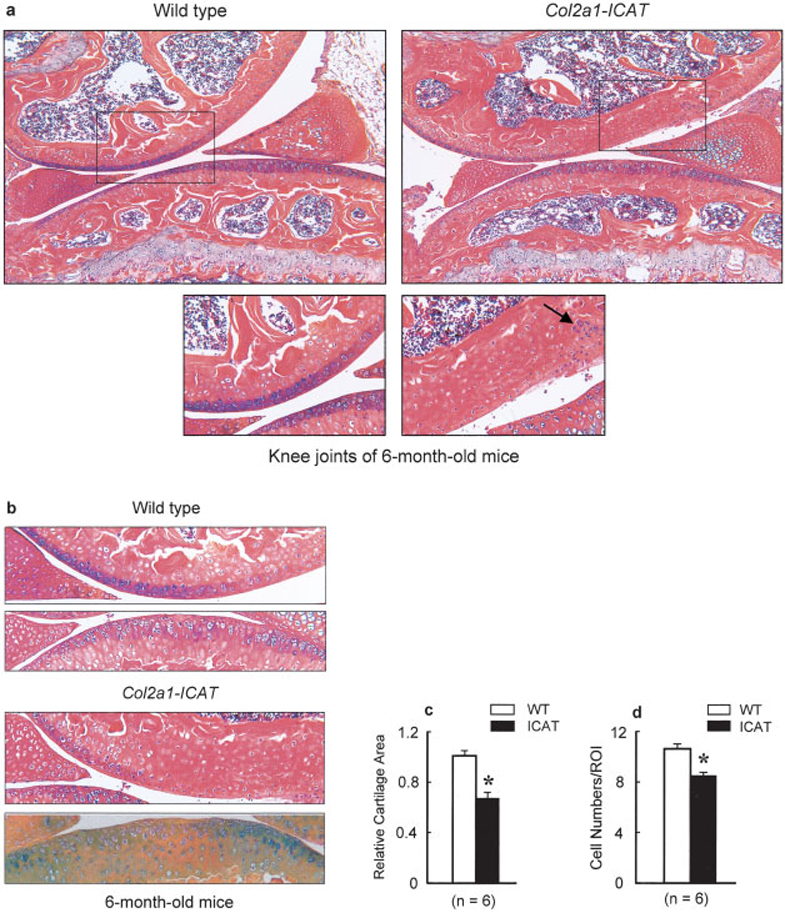
a, Changes in knee joint structure of Col2a1-ICAT–transgenic mice were analyzed by histology using Alcian blue/H&E and orange G staining. Mild cartilage degeneration was observed at the articular surface of knee joints in 7 of 10 6-month-old Col2a1-ICAT–transgenic mice. In the weight-bearing area, the articular layer was thinner or even completely missing in transgenic mice. Chondrocyte clusters (arrow) were found in Col2a1-ICAT–transgenic mice. No articular cartilage damage was found in 6-month-old WT mice. Boxed areas in upper panels are shown at higher magnification in lower panels. b–d, Histologic and histomorphometric analyses showed that articular area (b and c) and articular chondrocyte numbers (d) were significantly reduced in Col2a1-ICAT–transgenic mice. Values are the mean and SEM. * = P < 0.05 versus WT mice, by unpaired t-test (n = 6 mice per group). (Original magnification × 10.) ROI = region of interest (see Figure 1 for other definitions).
At age 9 months, more severe destruction of the articular cartilage was observed (Figure 3a), and by age 12 months, the progressive destruction of the articular cartilage resulted in severe loss of the entire articular cartilage surface in Col2a1-ICAT–transgenic mice. Detached fragments of bone covered with cartilage were also observed (Figure 3b). Histologic analysis showed that 7 of 10 (70%), 6 of 9 (67%), and 6 of 11 (55%) Col2a1-ICAT–transgenic mice at ages 6, 9, and 12 months, respectively, had articular cartilage destruction. In contrast, no articular cartilage damage was found in 6-month-old WT mice, and only minor articular cartilage damage was found in <10% of age-matched 9- and 12-month-old WT mice (10 mice were analyzed).
Figure 3. Progressive destruction of articular cartilage in 9- and 12-month-old Col2a1-ICAT–transgenic mice.
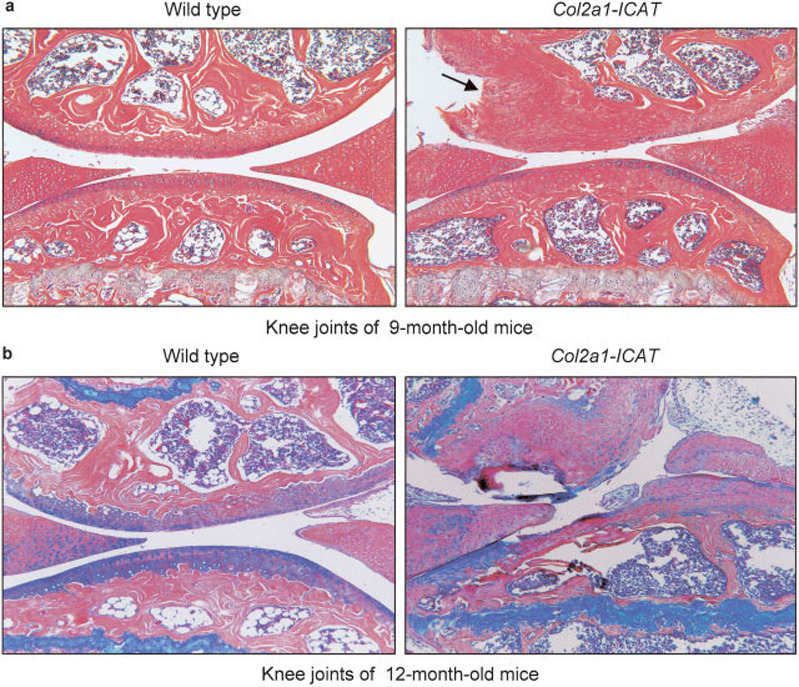
a, Histologic analysis showed moderate destruction of articular cartilage tissue, and defects were extended to the calcified cartilage area in 6 of 9 9-month-old Col2a1-ICAT–transgenic mice (arrow). b, Severe destruction of articular cartilage and subchondral bone was detected in 6 of 11 12-month-old Col2a1-ICAT–transgenic mice. In contrast, only minor articular cartilage damage was found in <10% of 9- and 12-month-old wild-type mice (10 mice were analyzed). (Original magnification × 10.)
At age 15 months, radiography showed ectopic bone formation in Col2a1-ICAT–transgenic mice (Figure 4a). Histologic analyses revealed surface fibrillation and vertical cleft development at the knee articular surface in Col2a1-ICAT–transgenic mice (Figure 4b). Complete loss of subchondral bone, which was replaced by the formation of new woven bone secondary to the articular destruction, was found in transgenic mice (Figure 4b). Overall, these phenotypic changes resembled the clinical features commonly observed in OA patients. The modified Chamber’s scoring system reported by Glasson et al (10) was used to determine that the average histologic grade in Col2a1-ICAT–transgenic mice was 4 of 6 (i.e., moderate loss of ~40–60% of the articular cartilage).
Figure 4. Ectopic and new bone formation in articular cartilage area of Col2a1-ICAT–transgenic mice.
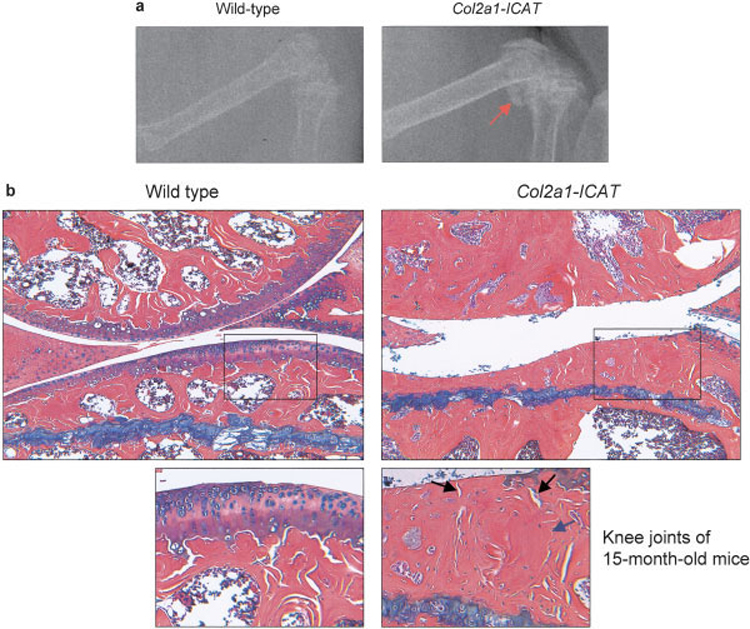
a, Radiography shows ectopic bone formation (arrow) in the knee joint of a 15-month-old Col2a1-ICAT–transgenic mouse. b, Histologic analysis reveals the complete loss of articular tissue at the weight-bearing area in a 15-month-old transgenic mouse. The destruction extends to the subchondral bone region. Boxed areas in upper panels are shown at higher magnification in lower panels. The high-magnification picture shows the deep cleft (black arrows) and new woven bone formation (blue arrow) in the knee joint area of a 15-month-old Col2a1-ICAT–transgenic mouse. (Original magnification × 10.)
Increase in chondrocyte apoptosis in Col2a1-ICAT–transgenic mice
We investigated the underlying mechanism of the pathogenesis of the articular cartilage destruction in Col2a1-ICAT–transgenic mice. No significant changes in cell proliferation were observed in articular chondrocytes of transgenic mice, as assessed by Ki-67 staining (data not shown). We then analyzed the expression of matrix genes via real-time reverse transcriptase–PCR using primary sternal chondrocytes isolated from neonatal WT and Col2a1-ICAT–transgenic mice and found that the expression of type X collagen was reduced in primary chondrocytes from transgenic mice (data not shown), suggesting that the chondrocyte maturation process was delayed. Increased expression of matrix metalloproteinase 13 (MMP-13) is often associated with OA (11,12). However, in Col2a1-ICAT–transgenic mice, MMP-13 expression was also reduced (data not shown). These results suggest that articular cartilage destruction observed in Col2a1-ICAT–transgenic mice may not be caused by the premature acceleration of articular chondrocyte differentiation.
We then analyzed cell apoptosis by TUNEL staining and found that the number of apoptotic cells in the articular cartilage was significantly increased in Col2a1-ICAT–transgenic mice (Figures 5a and b). Quantitative analysis showed a 75% increase in the number of apoptotic cells detected in the knee articular cartilage of the transgenic mice compared with that of the WT mice (Figure 5b). To further verify changes in cell apoptosis, we performed immunostaining using antibodies against the cleaved caspase 3 and PARP in the articular cartilage using knee joint sections derived from 6-month-old Col2a1-ICAT–transgenic mice and littermate controls. We found that staining of both cleaved caspase 3 and PARP was increased in Col2a1-ICAT–transgenic mice (Figures 5c and d). These results are consistent with the TUNEL staining results and indicate that apoptosis of articular chondrocytes is indeed increased in transgenic mice, suggesting that articular cartilage destruction in these mice may be caused by an increase in articular chondrocyte apoptosis.
Figure 5. Increased cell apoptosis in articular cartilage of Col2a1-ICAT–transgenic mice.
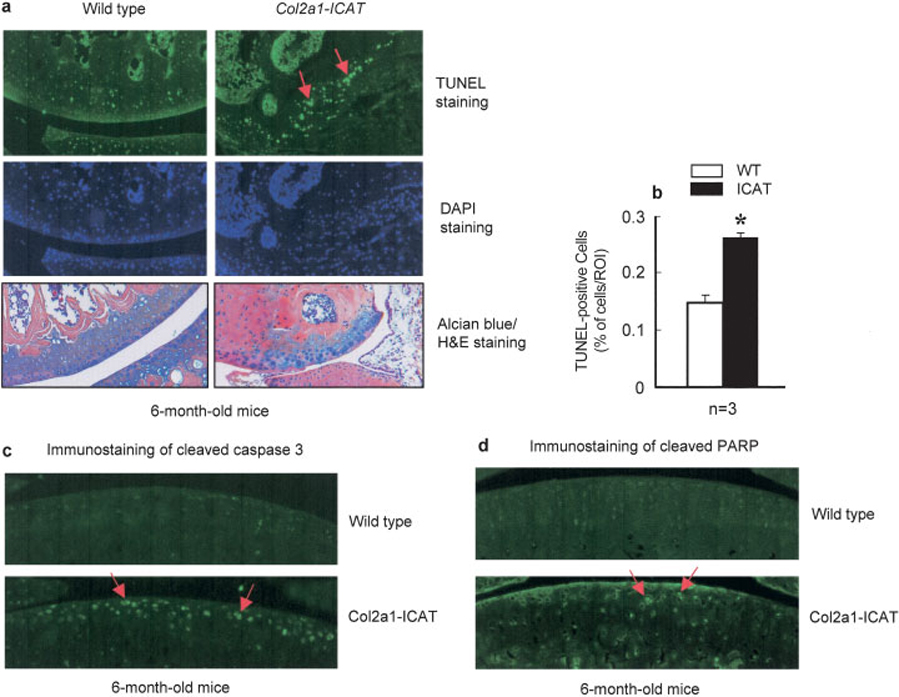
a, Cell apoptosis was determined by TUNEL staining. A significant increase in numbers of apoptotic cells was found in articular chondrocytes of 6-month-old Col2a1-ICAT–transgenic mice (arrows). DAPI staining was used as a positive control. The Alcian blue/H&E–stained histologic pictures from the same tissue blot are also shown. b, Quantification of apoptotic cells showed a 75% increase in the number of apoptotic cells in Col2a1-ICAT–transgenic mice. Values are the mean and SEM. * = P < 0.05 versus WT mice, by unpaired t-test (n = 3 mice per group). c and d, The immunostaining of cleaved caspase 3 (c) and poly(ADP-ribose) polymerase (PARP) (d) proteins was performed using knee joint sections derived from 6-month-old WT and Col2a1-ICAT–transgenic mice. The immunostaining of both cleaved caspase 3 and PARP was significantly increased in articular cartilage of Col2a1-ICAT–transgenic mice (arrows). (Original magnification × 20.) ROI = region of interest (see Figure 1 for other definitions).
It has been reported that the expression of Bcl-2 and Bcl-xL in human articular chondrocytes protects cells from nitric oxide–induced apoptosis (13). In the present study, we found that the expression of Bcl-2 and Bcl-xL proteins was decreased in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 6a). In contrast, the expression of Bax protein was increased in these cells (Figure 6a). Caspase 9 activation occurs downstream of Bcl-2/cytochrome c in the cytokine-induced activation of apoptosis. Thus, we examined changes in caspase 9 activity and found that it was increased in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 6b). In contrast, no changes were found in caspase 8 activity (data not shown). Caspases 3 and 7 are downstream of caspase 9 and have been shown to be involved in chondrocyte apoptosis and development of OA (14,15). In the present studies, we found that caspase 3/7 activity was also significantly increased in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (Figure 6b).
Figure 6. Changes in Bcl-2, Bcl-xL, and Bax expression and caspase 9 and caspase 3/7 activities in primary sternal chondrocytes of Col2a1-ICAT–transgenic mice and in primary chicken and porcine articular chondrocytes.
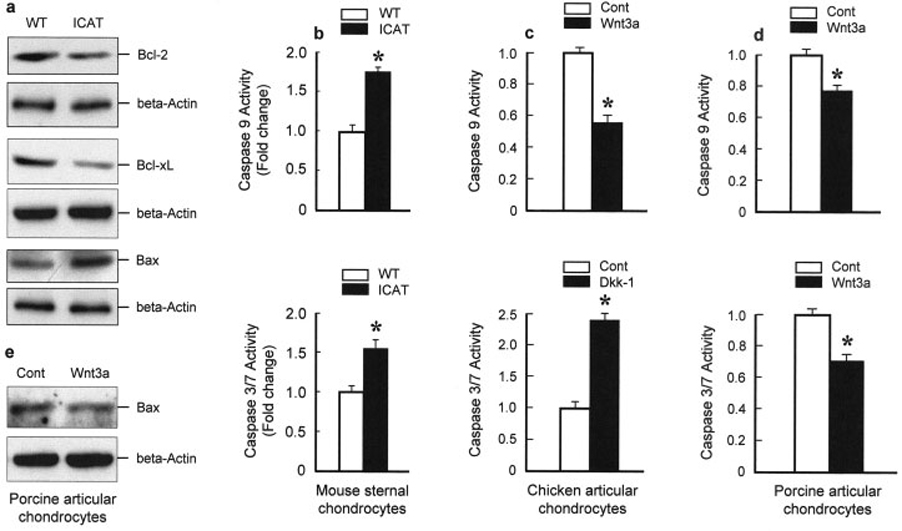
a, Primary sternal chondrocytes were isolated from 3-day-old Col2a1-ICAT–transgenic mice and their WT littermates. The expression of Bcl-2, Bcl-xL, and Bax proteins was detected by Western blot analysis. The expression levels of Bcl-2 and Bcl-xL proteins were decreased in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice, while the expression level of Bax protein was increased. b, Caspase 9 and caspase 3/7 activities were measured and found to be significantly increased in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice. c and d, Caspase 9 and caspase 3/7 activities were also measured in primary chicken (c) and porcine (d) articular chondrocytes. Wnt-3A (100 ng/ml) significantly inhibited caspase 9 activity and Dkk-1 (1 µg/ml) stimulated caspase 3/7 activity in chicken articular chondrocytes after 48-hour treatment. In contrast, Wnt-3A (100 ng/ml, 48-hour treatment) inhibited both caspase 9 and caspase 3/7 activities in porcine articular chondrocytes. Values in b–d are the mean and SEM. * = P < 0.05 versus WT mice or versus control (Cont), by unpaired t-test (n = 4 wells per group). e, Bax protein expression was reduced by 48-hour treatment with Wnt-3A (100 ng/ml) in primary porcine articular chondrocytes. See Figure 1 for other definitions.
To further determine the regulatory role of β-catenin signaling in caspase activities in articular chondrocytes, we also measured caspase 9 and caspase 3/7 activities in primary chicken and porcine articular chondrocytes following activation or inhibition of canonical Wnt signaling. Consistent with the results from Col2a1-ICAT–transgenic mice, we found that Wnt-3A (100 ng/ml) significantly inhibited caspase 9 activity in chicken articular chondrocytes, while Dkk-1 (1 µg/ml) stimulated caspase 3/7 activity (Figure 6c). In contrast, Wnt-3A (100 ng/ml) inhibited both caspase 9 and caspase 3/7 activities in porcine articular chondrocytes (Figure 6d). In addition, we found that Bax protein expression was reduced by treatment with Wnt-3A (100 ng/ml) in primary porcine articular chondrocytes (Figure 6e).
To determine the potential contribution of the ERK and phosphatidylinositol 3-kinase/Akt pathways to the apoptosis observed in Col2a1-ICAT–transgenic mice, we examined levels of phosphorylated ERK-1/2 and Akt in WT and transgenic mouse chondrocytes. No changes in either phosphorylated ERK-1/2 or Akt levels were observed in primary sternal chondrocytes derived from Col2a1-ICAT–transgenic mice (data not shown), suggesting that the ERK and Akt pathways are not involved in Col2a1-ICAT–induced cell apoptosis. Overall, these results suggest that increased cell apoptosis is mediated by the Bcl-2/caspase 9/caspase 3/7 pathway.
DISCUSSION
In the present studies, we selectively blocked β-catenin signaling in chondrocytes by generating Col2a1-ICAT–transgenic mice. Specific expression of the ICAT transgene in articular chondrocytes of 6-month-old transgenic mice was observed, and severe articular cartilage destruction was identified. We found that increased cell apoptosis in articular chondrocytes may contribute to the articular cartilage destruction in these transgenic mice.
Seven transgenic founder mice were initially generated. We could not establish transgenic lines for 2 of the founders since newborn mice were extremely small and did not survive past age 1 month. Ultimately, 2 transgenic lines (lines 5 and 7) with moderate phenotype were established and analyzed. Although severe articular cartilage destruction was observed, articular cartilage defects were not detected in 100% of the Col2a1-ICAT–transgenic mice, especially in older animals. Since ICAT may not bind to β-catenin in an irreversible manner, it is possible that β-catenin signaling may not have been completely blocked in some of the transgenic mice.
The phenotypic stability and survival of the chondrocytes are essential for the maintenance of a proper cartilage matrix. It has been reported that activation of Wnt/β-catenin signaling inhibits cell apoptosis (16–18). In contrast, inhibition of Wnt proteins promotes programmed cell death in different types of cancer cells (19–23). The Wnt antagonist secreted Frizzled-related protein 1 (sFRP-1) promotes osteoblast and osteocyte apoptosis (24), and Wnt-7A inhibits apoptosis in articular chondrocytes (25). Serum withdrawal–induced apoptosis can be prevented by the canonical and the noncanonical Wnt proteins in osteoblasts (26). Furthermore, in human OA cartilage, FrzB-2 (encoding sFRP-4 protein) is highly expressed and is associated with chondrocyte apoptosis (27). These observations are consistent with our present finding that in Col2a1-ICAT–transgenic mice, cell apoptosis is significantly increased in articular chondrocytes. These findings indicate that Wnt/β-catenin signaling proteins are involved in the antiapoptosis signaling pathway.
FRZB encodes sFRP-3, a glycoprotein that antagonizes the signaling of Wnt ligands through Frizzled membrane-bound receptors (28). In vitro transfection assays demonstrated that sFRP-3 can inhibit β-catenin nuclear translocation and TCF/LEF-dependent transcriptional activation (29). The mutation of sFRP-3 involving the Arg324Gly substitution causes sFRP-3 to lose its ability to antagonize Wnt signaling and may predispose humans to OA by altering the development or stability of cartilage in weight-bearing joints (29,30). These findings suggest that activation of Wnt/β-catenin signaling may also be associated with the development of OA and further demonstrate that Wnt/β-catenin signaling is critical for joint development, maintenance, and OA pathogenesis.
Beta-catenin plays an important role in articular chondrocyte function as well as in embryonic cartilage development (3). Although Col2a1-ICAT–transgenic mice show severe cartilage destruction, the possibility that phenotypic changes are due to defects in epiphyseal subchondral bone that occurred during early cartilage development cannot be ruled out in the present studies. Due to the limitation of the transgenic approach in this study, we will not be able to separate the effect of ICAT in articular chondrocytes from its effect on growth plate chondrocytes. To determine the specific role of β-catenin in articular chondrocytes in a cell-autonomous manner in adult mice, cell-specific deletion of the β-catenin gene in an inducible manner in articular chondrocytes in adult mice is required. Since tissue-specific knockout of the β-catenin gene (targeted by Col2a1-Cre) results in perinatal lethality (3), we have recently generated Col2a1-CreER–transgenic mice (31,32), and we are planning to generate mice with deletion of the β-catenin gene specifically in adult articular cartilage using this tamoxifen-inducible Cre/loxP system. These mouse models will provide more information about the interrelationship or exclusivity of the growth plate and articular cartilage effects when there are alterations in β-catenin signaling.
In summary, in this study we provide novel evidence that selective inhibition of β-catenin signaling in articular chondrocytes causes articular cartilage destruction in Col2a1-ICAT–transgenic mice. The findings not only suggest that increased cell apoptosis may contribute to the articular cartilage damage in these mice, they also establish the idea that therapeutic approaches to OA involving inhibition of β-catenin signaling might not be helpful.
ACKNOWLEDGMENTS
We would like to thank Dr. Tetsu Akiyama (University of Tokyo) for providing us the ICAT expression plasmid. We would also like to acknowledge Dr. Tian-fang Li, Dr. Tzongjen Sheu, Erica Dussmann, and Tony Chen for their technical assistance.
Dr. Di Chen’s work was supported by NIH grants AR-051189, AR-052411, and AR-054465. Dr. O’Keefe’s work was supported by NIH grant AR-053717.
Footnotes
AUTHOR CONTRIBUTIONS
Dr. D. Chen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design. D. Chen.
Acquisition of data. Zhu, M. Chen, Zuscik, Wu, Wang.
Analysis and interpretation of data. Zhu, M. Chen, Rosier, O’Keefe, D. Chen.
Manuscript preparation. Zuscik, D. Chen.
Statistical analysis. Zhu.
Contributor Information
Mei Zhu, current address: Shanghai University of Chinese Medicine, Shanghai, China.
Mo Chen, current address: Shanghai University of Chinese Medicine, Shanghai, China.
Michael Zuscik, current address: Shanghai University of Chinese Medicine, Shanghai, China.
Qiuqian Wu, current address: Shanghai University of Chinese Medicine, Shanghai, China.
Yong-Jun Wang, current address: Shanghai University of Chinese Medicine, Shanghai, China.
Randy N. Rosier, University of Rochester, Rochester, New York..
Regis J. O’Keefe, University of Rochester, Rochester, New York..
Di Chen, University of Rochester, Rochester, New York..
REFERENCES
- 1.Kuettner KE, Goldberg VM. Introduction. In: Kuettner KE, Goldberg VM, editors. Osteoarthritic disorders. Rosemont (IL): American Academy of Orthopaedic Surgeons; 1995. pp. xxi–xxv. [Google Scholar]
- 2.Kuettner KE, Aydelotte MB, Thonar EJ. Articular cartilage matrix and structure: a minireview [review] J Rheumatol Suppl. 1991;27:46–48. [PubMed] [Google Scholar]
- 3.Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, et al. Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 5.Tago K, Nakamura T, Nishita M, Hyodo J, Nagai S, Murata Y, et al. Inhibition of Wnt signaling by ICAT, a novel β-catenin-interacting protein. Genes Dev. 2000;14:1741–1749. [PMC free article] [PubMed] [Google Scholar]
- 6.Graham TA, Clements WK, Kimelman D, Xu W. The crystal structure of the β-catenin/ICAT complex reveals the inhibitory mechanism of ICAT. Mol Cell. 2002;10:563–571. doi: 10.1016/s1097-2765(02)00637-8. [DOI] [PubMed] [Google Scholar]
- 7.Daniels DL, Weis WI. ICAT inhibits β-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol Cell. 2002;10:573–584. doi: 10.1016/s1097-2765(02)00631-7. [DOI] [PubMed] [Google Scholar]
- 8.Tsuda M, Takahashi S, Takahashi Y, Asahara H. Transcriptional co-activators CREB-binding protein and p300 regulate chondrocyte-specific gene expression via association with Sox9. J Biol Chem. 2003;278:27224–27229. doi: 10.1074/jbc.M303471200. [DOI] [PubMed] [Google Scholar]
- 9.Chen M, Zhu M, Awad H, Sheu T, Boyce BF, Chen D, et al. Inhibition of β-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 2008 doi: 10.1242/jcs.020362. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- 11.Moldovan F, Pelletier JP, Hambor J, Cloutier JM, Martel-Pelletier J. Collagenase-3 (matrix metalloprotease 13) is preferentially localized in the deep layer of human arthritic cartilage in situ: in vitro mimicking effect by transforming growth factor β. Arthritis Rheum. 1997;40:1653–1661. doi: 10.1002/art.1780400915. [DOI] [PubMed] [Google Scholar]
- 12.Tardif G, Pelletier JP, Dupuis M, Geng C, Cloutier JM, Martel-Pelletier J. Collagenase 3 production by human osteoarthritic chondrocytes in response to growth factors and cytokines is a function of the physiologic state of the cells. Arthritis Rheum. 1999;42:1147–1158. doi: 10.1002/1529-0131(199906)42:6<1147::AID-ANR11>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 13.Surendran S, Kim SH, Jee BK, Ahn SH, Gopinathan P, Han CW. Anti-apoptotic Bcl-2 gene transfection of human articular chondrocytes protects against nitric oxide-induced apoptosis. J Bone Joint Surg Br. 2006;88:1660–1665. doi: 10.1302/0301-620X.88B12.17717. [DOI] [PubMed] [Google Scholar]
- 14.Watrin-Pinzano A, Etienne S, Grossin L, Gaborit N, Cournil-Henrionnet C, Mainard D, et al. Increased apoptosis in rat osteoarthritic cartilage corresponds to degenerative chondral lesions and concomitant expression of caspase-3. Biorheology. 2006;43:403–412. [PubMed] [Google Scholar]
- 15.D’Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54:1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- 16.Longo KA, Kennell JA, Ochocinska MJ, Ross SE, Wright WS, MacDougald OA. Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J Biol Chem. 2002;277:38239–38244. doi: 10.1074/jbc.M206402200. [DOI] [PubMed] [Google Scholar]
- 17.Ueda Y, Hijikata M, Takagi S, Takada R, Takada S, Chiba T, et al. Wnt/β-catenin signaling suppresses apoptosis in low serum medium and induces morphologic change in rodent fibroblasts. Int J Cancer. 2002;99:681–688. doi: 10.1002/ijc.10418. [DOI] [PubMed] [Google Scholar]
- 18.Yang F, Zeng Q, Yu G, Li S, Wang CY. Wnt/β-catenin signaling inhibits death receptor-mediated apoptosis and promotes invasive growth of HNSCC. Cell Signal. 2006;18:679–687. doi: 10.1016/j.cellsig.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 19.You L, He B, Xu Z, Uematsu K, Mazieres J, Fujii N, et al. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res. 2004;64:5385–5389. doi: 10.1158/0008-5472.CAN-04-1227. [DOI] [PubMed] [Google Scholar]
- 20.You L, He B, Xu Z, Uematsu K, Mazieres J, Mikami I, et al. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene. 2004;23:6170–6174. doi: 10.1038/sj.onc.1207844. [DOI] [PubMed] [Google Scholar]
- 21.You L, He B, Uematsu K, Xu Z, Mazieres J, Lee A, et al. Inhibition of Wnt-1 signaling induces apoptosis in β-catenin-deficient mesothelioma cells. Cancer Re. 2004;64:3474–3478. doi: 10.1158/0008-5472.CAN-04-0115. [DOI] [PubMed] [Google Scholar]
- 22.He B, You L, Uematsu K, Xu Z, Lee AY, Matsangou M, et al. A monoclonal antibody against Wnt-1 induces apoptosis in human cancer cells. Neoplasia. 2004;6:7–14. doi: 10.1016/s1476-5586(04)80048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY, et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene. 2005;24:3054–3058. doi: 10.1038/sj.onc.1208511. [DOI] [PubMed] [Google Scholar]
- 24.Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, McLarney S, Mangine A, et al. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J Cell Biochem. 2005;96:1212–1230. doi: 10.1002/jcb.20599. [DOI] [PubMed] [Google Scholar]
- 25.Hwang SG, Ryu JH, Kim IC, Jho EH, Jung HC, Kim K, et al. Wnt-7a causes loss of differentiated phenotype and inhibits apoptosis of articular chondrocytes via different mechanisms. J Biol Chem. 2004;279:26597–26604. doi: 10.1074/jbc.M401401200. [DOI] [PubMed] [Google Scholar]
- 26.Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by β-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem. 2005;280:41342–41351. doi: 10.1074/jbc.M502168200. [DOI] [PubMed] [Google Scholar]
- 27.James IE, Kumar S, Barnes MR, Gress CJ, Hand AT, Dodds RA, et al. FrzB-2: a human secreted frizzled-related protein with a potential role in chondrocyte apoptosis. Osteoarthritis Cartilage. 2000;8:452–463. doi: 10.1053/joca.1999.0321. [DOI] [PubMed] [Google Scholar]
- 28.Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns [review] Bioessays. 2002;24:811–820. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- 29.Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, et al. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A. 2004;101:9757–9762. doi: 10.1073/pnas.0403456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min JL, Meulenbelt I, Riyazi N, Kloppenburg M, Houwing-Duistermaat JJ, Seymour AB, et al. Association of the Frizzled-related protein gene with symptomatic osteoarthritis at multiple sites. Arthritis Rheum. 2005;52:1077–1080. doi: 10.1002/art.20993. [DOI] [PubMed] [Google Scholar]
- 31.Chen M, Lichtler AC, Sheu T, Xie C, Zhang X, O’Keefe RJ, et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis. 2007;45:44–50. doi: 10.1002/dvg.20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu M, Chen M, Lichtler AC, O’Keefe RJ, Chen D. Tamoxifen-inducible Cre-recombination in articular chondrocytes of adult Col2a1-CreERT2 transgenic mice. Osteoarthritis Cartilage. 2008;16:129–130. doi: 10.1016/j.joca.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]