

Abstract
Autosomal-dominant brachydactyly type A2 (BDA2), a limb malformation characterized by hypoplastic middle phalanges of the second and fifth fingers, has been shown to be due to mutations in the Bone morphogenetic protein receptor 1B (BMPR1B) or in its ligand Growth and differentiation factor 5 (GDF5). A linkage analysis performed in a mutation-negative family identified a novel locus for BDA2 on chromosome 20p12.3 that incorporates the gene for Bone morphogenetic protein 2 (BMP2). No point mutation was identified in BMP2, so a high-density array CGH analysis covering the critical interval of ∼1.3 Mb was performed. A microduplication of ∼5.5 kb in a noncoding sequence ∼110 kb downstream of BMP2 was detected. Screening of other patients by qPCR revealed a similar duplication in a second family. The duplicated region contains evolutionary highly conserved sequences suggestive of a long-range regulator. By using a transgenic mouse model we can show that this sequence is able to drive expression of a X-Gal reporter construct in the limbs. The almost complete overlap with endogenous Bmp2 expression indicates that a limb-specific enhancer of Bmp2 is located within the identified duplication. Our results reveal an additional functional mechanism for the pathogenesis of BDA2, which is duplication of a regulatory element that affects the expression of BMP2 in the developing limb.
Main Text
Embryonic development depends on tight control of gene expression. In many instances, regulatory promoters located immediately upstream of the transcription start site contain sufficient information to direct correct gene expression. In many developmentally important genes, though, more complex regulatory mechanisms are needed to drive dynamic spatially and temporally controlled expression patterns. Such cis-regulatory elements can be located upstream or downstream or within introns of the transcription unit.1 They are frequently conserved among species and may be located as far as 1.5 Mb in either direction. Several studies have identified such elements as essential regulators of developmental gene expression, that have the potential to switch genes off and on in particular types of cells/tissues during certain developmental time points. Given the importance of gene regulation in development, it is to be expected that a large number of developmental defects is caused by mutations affecting such regulatory elements (for review see 2). However, because of the relative paucity of information regarding the basic mechanisms of gene regulation, only a few gene alterations affecting regulatory elements have been reported so far. Here we describe a tandem duplication of a ∼5.5 kb element 3′ of BMP2 (MIM ∗112261), which is associated with brachydactyly type A2 (BDA2 [MIM #112600]).
The brachydactylies are a related group of conditions, several of which are caused by mutations in genes that are linked to the bone morphogenetic protein (BMP) pathway. BMPs and the related growth and differentiation factors (GDFs) are phylogenetically conserved signaling proteins that belong to the transforming growth factor beta (TGF-β) superfamily. Originally identified for their ability to induce bone, they were subsequently shown to be involved in multiple aspects of body patterning and morphogenesis.3 BMPs initiate their signaling pathways through binding to two types of transmembrane receptors, the BMP type I and the type II receptor. Upon ligand binding, the receptors dimerize and activate Smad transcription factors that subsequently regulate gene expression in the nucleus. The activity of BMPs is regulated at multiple levels including extracellular inhibitors such as Noggin (MIM ∗602991). Mutations in components of this pathway are known to result in various types of brachydactylies (BDA2 [MIM #112600]; brachydactyly type C, BDC [MIM #113100]; brachydactyly type B2, BDB2 [MIM #611377]), symphalangism (SYM1 [MIM #185800]), and multiple synostosis syndrome (SYNS1 [MIM #186500]) but are also associated with more complex skeletal disorders such as the acromesomelic chondrodysplasias (MIM #200700, MIM #201250, MIM #228900) and fibrodysplasia ossificans progessiva (FOP [MIM #135100]).4
BDA2 refers to a genetically heterogeneous subset of brachydactylies characterized by hypoplastic or aplastic middle phalanges of the second and fifth finger. So far, dominant-negative mutations in the BMP-receptor BMPR1B (MIM ∗603248) and a specific missense mutation in GDF5 (MIM ∗601146) resulting in a loss of binding to BMPR1B are known to cause isolated BDA2.5,6 In addition, mutations that affect the GDF5 cleavage site were shown to result in a similar phenotype.38 On the contrary, mutations that lead to an activation of GDF5 or in a loss of function (LOF) of its inhibitor NOGGIN result in fusion of phalanges caused by a lack of joint formation.6,7 Thus, a loss of BMPR1B-mediated activity appears to be associated with a hypoplasia of phalanges whereas activation of the BMP pathway results in joint fusions (symphalangism).
In this article we describe a novel molecular basis for the BDA2 phenotype in two unrelated families. The investigated pedigrees are shown in Figure 1A. One of the pedigrees is part of a large Brazilian kindred of German origin originally published as a clinical description in 1980 (family 1).8 The smaller family is of European origin as well (family 2). Within the families, the phenotype appeared to be variable but penetrance was complete. The phenotypes of affected hands are shown in Figure 1B. In family 1, we examined 21 affected and 5 unaffected individuals. Hand and foot radiographs were obtained for 14 affected individuals. The predominant trait was in accordance with BDA2, i.e., shortening of the second mesophalanx associated with a medial deviation in the proximal interphalangeal joint (PIP) and atypical or even absent phalangeal flexion creases. On radiographs, the second mesophalanx often appeared triangular (Figure 1B, ∗). In addition, we observed ulnar deviation and limited passive and active movement of the PIP or distal interphalangeal joint (DIP) in the third, fourth, and fifth fingers in some affected individuals. The lower limb phenotype was generally milder with shortening of the second toe deviating radially in the metatarsal-phalangeal joint, eventually combined with hallux valgus of the big toe. In a single patient, the hands appeared normal, and shortening of the second toe bilaterally was the only observed pathology. The most severely affected individual displayed short malformed third proximal phalanges and mesophalanges, absent third and fourth DIP flexion creases, and a simian flexion crease in addition to a severe shortening and malformation of the second proximal phalanx and mesophalanx (Figure 1B, ∗∗). This phenotype has similarities to BDC. His feet showed hallux valgus and a short triangular mesophalanx on the second toe bilaterally. In family 2, only the mother in generation II and her child were investigated. Both presented with characteristic BDA2 (Figure 1B, ∗∗∗).
Figure 1.
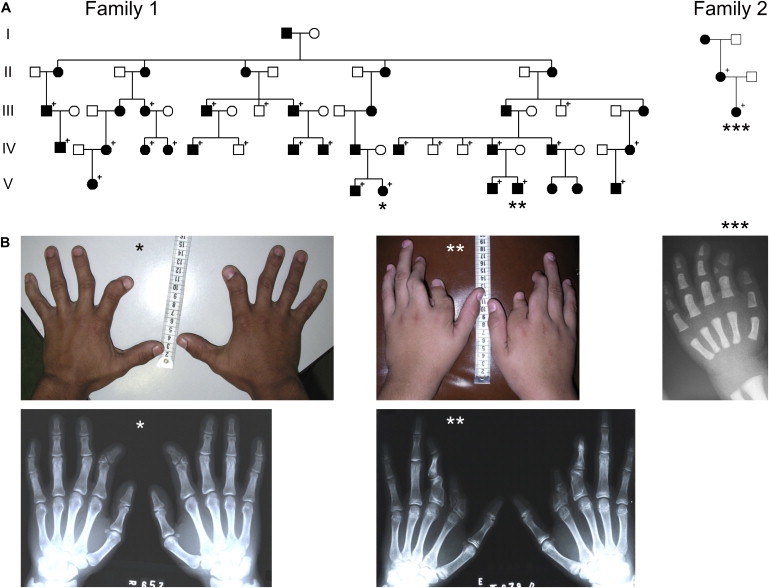
Pedigrees and Clinical Phenotype
(A) Pedigrees. Affected individuals are indicated by black symbols. Symbols with a + indicate individuals who were clinically examined and for whom further molecular analysis was performed.
(B) Clinical phenotype caused by the duplication. BDA2 with shortened and medially deviated second fingers caused by hypoplastic and triangular middle phalanges was observed in almost all of the affected individuals as demonstrated in the photograph and X-rays of an affected adult (∗) and an 18-month-old affected child (∗∗∗). One affected individual in family 1 (∗∗) showed a more severe phenotype, displaying considerable shortened and deviated second and third fingers resulting from hypoplastic and malformed proximal as well as middle phalanges showing similarities to BDC.
We obtained blood samples or buccal swabs from the family members indicated in the pedigrees and extracted DNA by standard methods. Selected affected individuals of the families described here were screened for mutations in BMPR1B, GDF5, as well as in other genes known to be associated with BD phenotypes such as IHH (MIM ∗600726), ROR2 (MIM ∗602337), and HOXD13 (MIM ∗142989), but no mutation was detected. All sequencing experiments were carried out by standard techniques as reported elsewhere.5,9–12 All participants gave their consent for molecular testing. The study was approved by the local ethics committee.
The molecular basis for BDA2 was unclear in family 1, so we genotyped DNA samples from 10 individuals of the family with the Affymetrix GeneChip Human Mapping 10K Array, version 2.0 (Affymetrix, Santa Clara, CA) and performed a genome-wide linkage analysis. Genotypes were called by the GeneChip DNA Analysis Software (GDAS v2.0; Affymetrix). Relationship errors were evaluated with the help of the program Graphical Relationship Representation (GRR).13 The program PedCheck was applied to detect Mendelian errors14 and data for SNPs with such errors were removed from the data set. Non-Mendelian errors were identified with the program MERLIN15 and unlikely genotypes for related samples were deleted. Parametric linkage analysis was performed with a modified version of the program GENEHUNTER 2.1.16,17 We used a sliding window with sets of 90 SNPs for calculation assuming autosomal-dominant inheritance with full penetrance and a disease allele frequency of 0.0001. Haplotypes were reconstructed with MERLIN and displayed graphically with HaploPainter.18 All data handling was performed with the graphical user interface ALOHOMORA.19
As a result, a further locus for brachydactyly was mapped to a ∼30 cM region between the telomer and marker rs953021 on chromosome 20p12.3 (lod score 3.31). Microsatellite markers were used for further fine mapping including all collected DNA samples. Because of an additional recombination in another family member, the critical region was narrowed down to a ∼1.3 Mb region between 5.68 and 7.01 Mb on chromosome 20 (positions according UCSC Human Genome July 2003). Searching for candidate genes in this interval, we first focused on BMP2, which is located at 6.696–6.708 Mb because a deregulation of BMP signaling is known to play a key role in the pathogenesis of BD. Sequencing of all exons, introns, 3′ UTR, and 5′ UTR as well as evolutionary highly conserved regions in the direct 3′ and 5′ flanking sequences of BMP2 revealed no mutation. Next, the coding regions of all annotated genes and transcripts lying in the confined interval were sequenced but no mutation was detected.
BMP2 is embedded in a “gene desert,” based on the lack of annotated genes ∼600 kb upstream and ∼1.1 Mb downstream of BMP2. Large gene deserts containing conserved sequences flanking developmental genes such as BMP2 argue for the possibility of noncoding regulatory elements, which can be located hundreds of kb away of the gene itself. In other genes, e.g., SHH (MIM ∗600725), SHOX (MIM ∗312865), PAX6 (MIM ∗607108), or SOX9 (MIM ∗608160), it was already shown that such elements are involved in the regulation of tissue-specific gene expression and also control target gene expression in defined temporal processes during embryonic development.2,20–23 Because genomic aberrations are a possible cause of hand and foot malformations,21,22 we subsequently performed array-based comparative genomic hybridization (array CGH) analysis to screen for submicroscopic chromosomal aberrations in the family 1 mapped to 20p12.3. Previous karyotyping of GTG-banded chromosomes from lymphocytes in an affected family member did not reveal any chromosomal abnormalities. To assess imbalances at the BMP2 locus, we designed a custom array (Roche NimbleGen Inc., Madison WI) for array CGH analysis that covers the critical region between 5.68 Mb and 7.01 Mb on human chromosome 20 at high density (average probe spacing 8 bp). Array CGH analysis was performed as a service at Roche NimbleGen, Iceland, according to manufacturer's protocol. In brief, 2 μg of genomic DNA were sonicated, yielding 500–2000 bp fragments. After fluorescent labeling, the test and reference DNA were hybridized for 15 to 20 hr on a MAUI Station. The array was washed and consecutively scanned on a GenePix 4000B scanner (Axon Instruments). Scanned images were analyzed with the NimbleScan software. Circular binary segmentation algorithm24 was employed for aberration detection. The genomic profile was visualized by the SignalMap software (SignalMap v1.9.0.03, NimbleGen Systems Inc.). With this custom array, a small duplication of about 5.5 kb ranging from approximately 6,808,500 to 6,814,000 bp was identified in an affected family member (Figure 2A). The duplication is located ∼110 kb downstream of BMP2 in a noncoding sequence.
Figure 2.
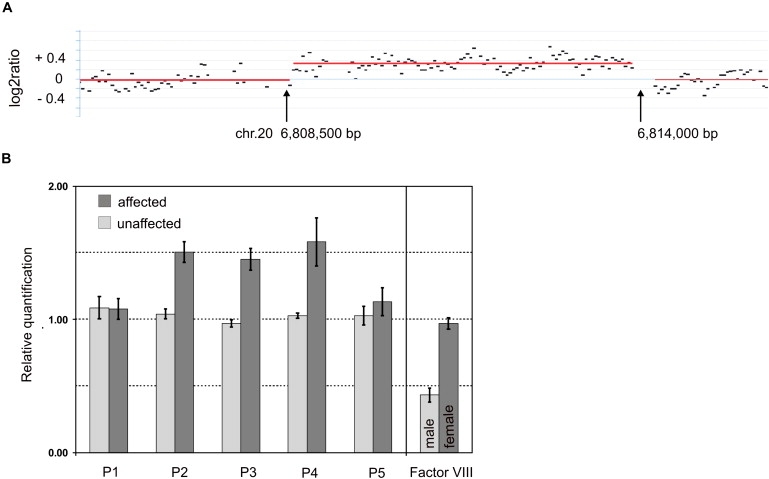
Microduplication on 20p12.3
(A) Genomic profile of the microduplication as detected on NimbleGen custom array. The detected breakpoints are indicated by arrows. The duplication comprises ∼5.5 kb. The gray horizontal lines indicate the segments as calculated by the CBS algorithm with 50 bp segmentation. x axis shows genomic positions on chromosome 20; y axis shows log2ratio.
(B) Microduplication confirmed by quantitative real-time PCR. The mean values for relative quantification were exported from the 7900 SDS software. For 4 affected (dark gray bars) and 4 nonaffected (light gray bars) individuals, mean values and standard deviations (error bars) for each target amplicon relative to Albumin as a two-copy reference gene were calculated. For gender determination, mean values and SDs for factor VIII on the X chromosome were calculated relative to Albumin as an autosomal two-copy reference gene. Results were calibrated to the mean value determined for a healthy female control. P1–P5 refers to primers shown in Table S1. One duplicated allele plus one normal allele results in three copies for the amplicons of primer pairs P2, P3, and P4 within the duplicated region in the affected individuals and therefore in a ratio of 1.5 relative to the two copies of the healthy female control. Localization of qPCR amplicons is illustrated in Figure 3C.
With quantitative real-time PCR (qPCR), the duplication was confirmed in 4 affected individuals of pedigree 1 and excluded in 4 unaffected members of the family (Figure 2B). The dark gray bars in Figure 2B show the mean value of the investigated affected individuals and the light gray bars the mean value of unaffected individuals. The mean values for relative quantification were exported from the 7900 SDS software. Five primer pairs covered the respective proximal and distal breakpoint regions, with three primer pairs within the duplicated region and two within the flanking sequences as proposed by the custom array results. Primer sequences are given in Table S1 available online and genomic positions are illustrated in Figure 3C. With a qPCR and breakpoint-PCR screening, the microduplication was not detected in more than 200 DNA samples of clinically asymptomatic control individuals excluding a common copy number variant (CNV) (data not shown). The qPCR experiments were performed as described previously.21
Figure 3.
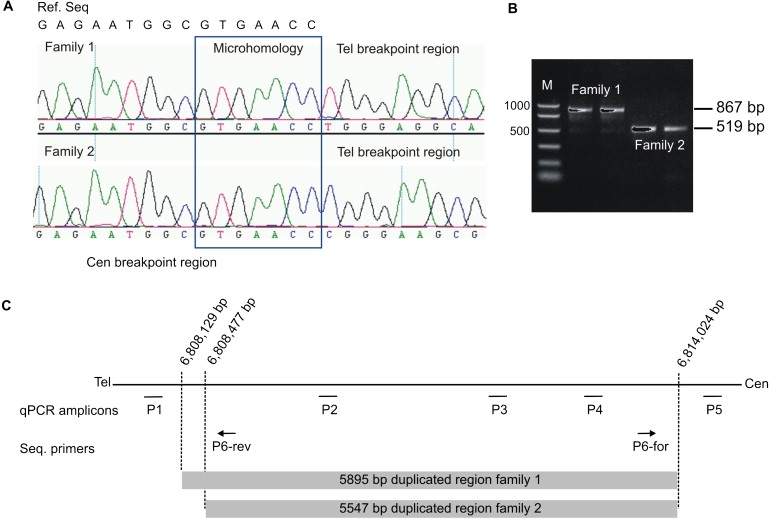
Breakpoint Identification by Sequence Analysis
(A) Sequence analysis of the junction fragment revealed a tandem duplication of 5,895 bp in family 1 and a tandem duplication of 5,547 bp in family 2. The top line on the left displays the centromeric reference sequence (ref. seq.). Below are the electropherograms of one affected individual of each family. The centromeric breakpoint is identical in both families. Note the differences in the telomeric sequence after the seven homologous nucleotides (surrounded by the blue box) of the junction fragment between the two families. The breakpoints are located within this homologous sequence or at the adjacent nucleotides.
(B) PCR products obtained with primers P6-forward and P6-reverse result in an 867 bp fragment in family 1 and in a 519 bp fragment in family 2.
(C) Illustration of qPCR amplicons and sequencing primer positions as well as localization of the duplicated regions and breakpoints in family 1 and 2 on chromosome 20.
Analysis by PCR with primers P6-forward and P6-reverse (Table S1; Figure 3C) on genomic DNA level allowed the identification of a junction fragment that included the transition site between the telomeric and centromeric breakpoints in all affected family members' DNA of pedigree 1 (Figures 3A and 3B). This fragment of 867 bp was undetectable in nonaffected blood relatives as well as in the in-laws of the family (data not shown), showing cosegregation of the aberration with the BDA2 phenotype. By the following direct sequencing of this fragment, we were able to identify a microhomology of seven nucleotides GTGAACC beginning at the telomeric position 6,808,129 bp and at the centromeric side on position 6,814,024 bp (positions according to UCSC Human Genome March 2006). Thus, the two breakpoints are apparently located within the microhomology of seven nucleotide on either side. Based on these findings, we conclude that the fragment was duplicated in tandem and had a size of 5,895 bp (Figure 3C).
Furthermore, we performed a screening of 22 unrelated affected individuals with well-characterized BDA2 or BDC phenotypes without mutations in the so far known brachydactyly genes with qPCR with all five primer pairs mentioned above. With this approach, a similar duplication was identified in one affected mother and her affected daughter (family 2) (Figures 3A and 3B). Breakpoint analysis in these individuals revealed an identical centromeric breakpoint region as detected in family 1 but a different telomeric breakpoint region subsequently located between 6,808,476 and 6,808,477 bp or within the following seven nucleotides (positions according to UCSC Human Genome March 2006), resulting in a slightly smaller tandem duplication of 5,547 bp (Figure 3C). DNA samples of the remaining 21 patients were also screened for single nucleotide changes in the conserved elements of the duplicated region but no pathogenic alterations were detected. Point mutations in the coding sequence of BMP2 were also excluded in these patients.
As conceivable mechanisms that lead to the identified genomic duplications, different possibilities have to be considered. The telomeric breakpoints in both families as well as the common centromeric breakpoint are located within regions of short interspersed elements (SINEs) of the Alu gene family. Because of their repetitive sequence, Alu elements can mediate unequal homologous recombination that is estimated to be the underlying mechanism for approximately 0.3% of all human genetic diseases.25 In addition, the breakpoints described here are located within a 7 nucleotide microhomology sequence that contains a recognition sequence (5′-GTT-3′) for topoisomerase I. Thus, nonallelic homologous recombination mediated by topoisomerases has to be considered as an alternative mechanism.26 For illegitimate recombination, microhomology of two to six nucleotides is necessary after the cleavage site. In our case, the cleavage site is followed by the three nucleotides 5′-CAC-3′ (note that Figure 3A shows the sequences in reverse orientation; recognition sequence in underlined, i.e., 3′-GTGAACC-5′). A replication-based model called fork stalling and template switching (FoSTeS) described by Lee et al. should also be taken into consideration.27 The hypothesis states that during the DNA replication process the replication fork can stall at DNA regions of genomic instability, e.g., at repetitive sequences, leading as a consequence to a switch of the lagging strand to another active replication fork in physical proximity. Microhomology between some base pairs, as identified in the breakpoints described here, is sufficient for that template switching. Thus, the DNA sequence can be replicated once again, which results in a duplication.
We identified two blocks of highly conserved noncoding sequences within the duplication (Figure 4). It is known that regulatory elements are located with highly conserved regions, so we hypothesized that the microduplication contains a potential regulator for Bmp2. To test this hypothesis, we investigated the effect of the duplicated WT region in a transgenic mouse model. According to the smaller duplication in family 2, the homologous mouse sequence on chromosome 2 was identified to range from 133,355,678 to 133,360,193 bp (UCSC Mouse Genome, February 2006), comprising 4,515 bp (Figure 4). This region was cloned with the mouse BAC clone RP24-82A15 obtained from Children's Hospital Oakland Research Institute. The following primers containing NotI restriction sites (/) were used: forward gactgcggccgc/GCCATGGCATTAATCAGACA and reverse gactgcggccgc/TTCAGCACACCGTGCTTATC. This fragment is subsequently called mouse Bmp2 homolog-enhancer region (mBmp2-h-ER). mBmp2-h-ER was ligated into pSfi-Hsp68lacZ (a kind gift of Douglas P. Mortlock) in forward orientation.20,23,28 The mBmp2-h-ER_pSfi-Hsp promoter X-Gal transgene construct was verified by direct sequencing. For the generation of a transgenic mouse model, the mBmp2-h-ER_pSfi-Hsp X-Gal plasmid was used for pronuclear injection of C57BL6 embryos. Plasmid DNA was linearized with SalI and further purified by gelelectrophoresis with the omni pure kit from Omni life science (Hamburg, Germany). Pronuclear injections and oviduct transfers were approved by the competent local authority (LAGeSo) and performed according to standard protocols. Transgenic embryos were created, collected at E11.5–E14.5, and subjected to whole-mount X-Gal staining as previously described.29 The retained yolk sacs were used for genotyping. These analyses revealed a specific X-Gal staining in the limb buds and the developing phalanges but not in other parts of the embryos (Figures 5A and 5B).
Figure 4.
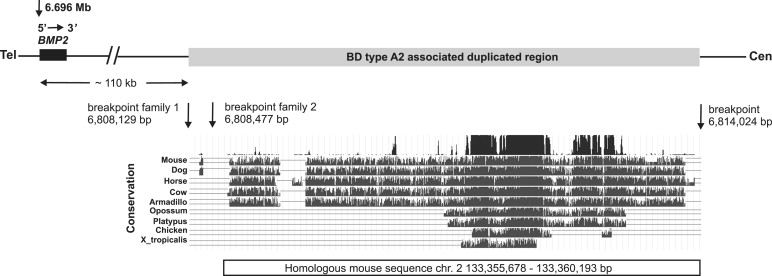
Schematic Representation of the Critical Region on 20p12.3
BMP2 and the 3′ distant duplicated region containing a limb-specific regulatory element that lies about 110 kb downstream of the gene itself are shown. The duplicated region (gray box) in family 1 described here starts at nucleotide 6,808,129 bp and ends at nucleotide 6,814,024 bp on chromosome 20. In family 2, the duplication starts at nucleotide 6,808,477 bp and ends similar as in family 1 at nucleotide 6,808,024 bp (positions according to UCSC Human Genome March 2006). The conservation of the duplicated sequence in different species is depicted in the UCSC genome browser plot. Within the sequence here are two highly conserved regions between mammals and chicken. The positions of the homologous mouse sequence on chromosome 2 are shown under the alignments ranging from 133,355,678 to 133,360,193 bp (UCSC Mouse Genome February 2006). This sequence was cloned into the mBmp2-h-ER_pSfi-Hsp promoter X-Gal transgene construct.
Figure 5.
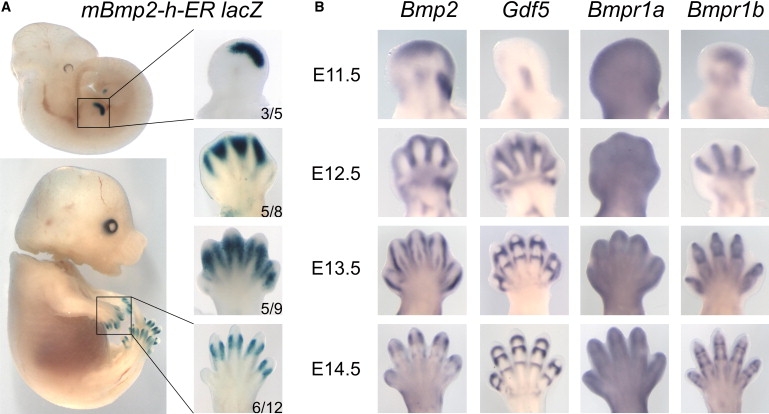
Expression Profiles of X-Gal in Transgenic Mice Compared to Bmp2, Gdf5, and Their Type I Receptors
(A) X-Gal staining of transgenic mice carrying the BDA2-associated duplicated region (mBmp2-h-ER). Left panel shows the entire embryo, right panel magnification of fore limb. Developmental stage is given on right side. The number of embryos with specific X-gal staining in the limbs out of the total number of transgene-positive embryos is shown in the right lower corner. Note that staining is present exclusively in the limb autopod. At E11.5, X-Gal staining is present in a distinct region of the distal autopod. Coincidental with the formation of the digit anlagen (E12.5), the staining moves toward the interdigital space. At E13.5 and E14.5, staining becomes restricted to the distal interphalangeal joint region.
(B) Whole-mount in situ hybridization of Bmp2, Gdf5, and their receptors Bmpr1a and Bmpr1b. Note overlapping but not identical staining pattern between Bmp2 and X-Gal. Gdf5 expression marks the developing joints. Bmpr1a is expressed ubiquitously throughout the limb, whereas Bmpr1b is restricted to the digit anlagen and the interphalangeal joints.
To compare the transgene expression pattern to the typical expression pattern for Bmp2 and other genes known to play an important role in the pathogenesis of BDA2 and BDC, additional in situ hybridizations on WT mouse embryos were performed for Bmp2, Gdf5, and the receptors Bmpr1a and Bmpr1b on limb stages E11.5 to E14.5. Whole-mount in situ hybridization was performed as previously described with minor modifications.30–32 The results show that the endogenous expression of Bmp2 in the limb is very similar to the X-Gal expression of the transgene. Other sites of staining observed for Bmp2 such as the whiskers, the somites, and the kidneys28 were not seen in X-Gal-stained embryos, indicating that mBmp2-h-ER drives expression exclusively in the limbs. At E11.5, Bmp2 is known to be expressed in the apical ectodermal ridge (AER), the underlying mesenchyme, and at the posterior side of the limb. The latter domain, as well as the AER expression, was not seen in the X-Gal-stained animals. At E12.5, expression was located mainly between the newly formed digits as well as in an area corresponding to the posterior side of the wrist-forming region, in the wrist itself, and in the distal joints of radius and ulna. At this stage, X-Gal staining was also present in the interdigital mesenchyme but not in the more proximal domains. At E13.5, Bmp2 expression was confined to a layer of cells surrounding the cartilage condensation and the dorsal tendons. At E14.5, Bmp2 expression was found mainly around the joint regions of the middle and proximal phalanges. X-Gal staining was very similar at these stages flanking the joint regions of the distal interphalangeal joints.
Our results suggest that Bmp2 expression in the limb is regulated by a 3′ distant cis-acting enhancer that is located within the duplication. Chandler and colleagues reported that a BAC spanning the duplicated enhancer is able to drive expression in the limb digits, whereas a BAC with a deletion of this region showed no digit expression. Furthermore, they identified two highly conserved regions, named ECR-1 and ECR-2, located about +156 kb and +160 kb 3′ from Bmp2. One of these elements, ECR-1, was shown to function as distant enhancers regulating Bmp2 expression in differentiating osteoblasts.28 It is to be expected that multiple other regulatory elements are present in the vicinity of Bmp2 that regulate expression in various tissues and stages of development.
Proteins of the BMP family are secreted signaling molecules that are involved in numerous developmental processes including patterning and organ development. The role of Bmp2 during embryogenesis has been investigated in detail before. It was shown that Bmp2 is expressed in several embryonic regions, e.g., the kidneys, hair follicles, tooth buds, and the gut epithelium. In addition, it is expressed in hypertrophic chondrocytes of the growth plate, in the osteogenic perichondrium, and in osteoblasts.33–35 During development of the interphalangeal joints, Bmp2 is first expressed in a region surrounding the future joints. At E14.5, Bmp2 expression is also found in the joint interzone.6
Functional studies of specific mutations resulting in BDA2 or BDC indicate that both phenotypes are the result of a deregulation within the BMP pathway that alters the equilibrium between the ligand GDF5 and the BMP type 1 receptors leading to a decreased GDF5-BMPR1B signaling.5,6,36,37 The brachydactyly phenotype in the families described here is similar to the phenotype caused by mutations in GDF5 or BMPR1B, so we hypothesize that the identified duplication of the BMP2 enhancer region disturbs the functional balance of BMPR1B signaling in a similar manner. Different mechanisms have to be taken into consideration for how this pathogenic effect could take place.
One possible explanation for the phenotype is a misexpression of BMP2. For example, an inversion involving Shh was shown to result in ectopic expression of Shh in the digits producing a BDA-like phenotype in mice.38 Duplication of the ZRS, a distant locus control region regulating the expression of Shh in the limb, results in preaxial polydactyly and syndactyly.21 The same phenotype can be caused by point mutations in the ZRS.39 Interestingly, similar mutations have been described in mice and cats with polydactyly and these mutations were shown to result in ectopic expression of Shh at the anterior margin of the limb bud.40,41 Based on the assumption that similar phenotypes are caused by similar pathogenetic mechanisms, the ZRS duplication reported by Klopocki and colleagues is thus likely to result in Shh misexpression.21 In analogy, the duplication described here may lead to misexpression of Bmp2 and thus a deregulation of BMP signaling during digit development.
Theoretically, the tandem duplication of a single regulatory element could impair its normal function by positional effects resulting in decreased BMP2 expression. However, conditional ablation of Bmp2 in the mouse limb did not cause any limb phenotype, showing that, at least in the mouse, loss of Bmp2 is dispensable for limb formation.42 A partial downregulation by this heterozygous mutation is thus unlikely to have an effect. A more likely scenario is an increase in BMP2 expression. Two patients with large duplication of chromosome 20 encompassing BMP2 and the duplicated regulator sequence described here were reported to have a BDA2 phenotype in association with other malformations.43,44 It is to be expected that a duplication of the entire BMP2 gene including the regulatory regions results in an increase in BMP2 expression. Assuming that the isolated BDA2 phenotype is caused by the same mechanism, a positional effect of the duplication reported here seems thus unlikely. Thus, the duplication most likely results in an increase in expression which, in contrast to the chromosomal duplications, is restricted to the developing limb.
As shown in previous studies, BMP signaling appears to be extremely dosage dependent, in particular during development of the phalanges and their joints. In contrast to Gdf5, which signals primarily through its high-affinity receptor BMPR1B, Bmp2 binds with high affinity to BMPR1A and BMPR1B.45 Too much Bmp2 would thus result in a deregulation of the fine-tuned BMP pathway by increasing the BMPR1A signal and, consequently, in a relative decrease of BMPR1B signaling. Furthermore, it is conceivable that BMP2 interferes with the binding of GDF5 to BMPR1B because of a competitive effect between the two ligands, further shifting the equilibrium of BMP type 1 receptor signaling toward BMPR1A. Such a reduction in BMPR1B signaling is compatible with the previously proposed molecular pathology of BDA2.
In summary, we demonstrate that a 5.5 kb tandem duplication downstream of BMP2 is associated with BDA2. The duplication contains highly conserved sequences that are likely to function as a cis-regulatory element regulating BMP2 expression in the limb. Duplications of regulatory elements can be considered as a mutational mechanism for developmental defects.
Acknowledgments
We thank Eleidi A. Chautard-Freire-Maia for helping to retrace the family originally published by her husband, Newton Freire-Maia. We thank the patients and their family members for participating in this study. We thank Douglas P. Mortlock for generously providing us with the vector pSfi-Hsp68lacZ. We acknowledge the technical help of Randi Koll, Fabienne Trotier, and Ingo Voigt. This work was funded by the DFG to K.D. (LE1851/1-2) and SFB 760 to P.S. and S.M. WJC is established by the Danish National Research Foundation. The authors declare no conflict of interest.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Decipher, https://decipher.sanger.ac.uk/ (Patient numbers: family 1, BER00248957; family 2, BER00248969)
Ensembl, http://www.ensembl.org/index.html
GenBank, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Bioinformatics, http://genome.ucsc.edu/
References
- 1.Howard M.L., Davidson E.H. cis-Regulatory control circuits in development. Dev. Biol. 2004;271:109–118. doi: 10.1016/j.ydbio.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 2.Kleinjan D.A., van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kishigami S., Mishina Y. BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev. 2005;16:265–278. doi: 10.1016/j.cytogfr.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Seemann P., Mundlos S., Lehmann K. Alterations of bone morphogenetic protein signaling pathway(s) in skeletal diseases. In: Vukicevic S., Sampath K.T., editors. Bone Morphogenetic Proteins: From Local to Systemic Therapeutics. Birkhäuser Verlag AG; Basel: 2008. pp. 141–159. [Google Scholar]
- 5.Lehmann K., Seemann P., Stricker S., Sammar M., Meyer B., Suring K., Majewski F., Tinschert S., Grzeschik K.H., Muller D. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc. Natl. Acad. Sci. USA. 2003;100:12277–12282. doi: 10.1073/pnas.2133476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seemann P., Schwappacher R., Kjaer K.W., Krakow D., Lehmann K., Dawson K., Stricker S., Pohl J., Ploger F., Staub E. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 2005;115:2373–2381. doi: 10.1172/JCI25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong Y., Krakow D., Marcelino J., Wilkin D., Chitayat D., Babul-Hirji R., Hudgins L., Cremers C.W., Cremers F.P., Brunner H.G. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat. Genet. 1999;21:302–304. doi: 10.1038/6821. [DOI] [PubMed] [Google Scholar]
- 8.Freire-Maia N., Maia N.A., Pacheco C.N. Mohr-Wriedt (A2) brachydactyly: analysis of a large Brazilian kindred. Hum. Hered. 1980;30:225–231. doi: 10.1159/000153133. [DOI] [PubMed] [Google Scholar]
- 9.Schwabe G.C., Turkmen S., Leschik G., Palanduz S., Stover B., Goecke T.O., Mundlos S. Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. Am. J. Med. Genet. A. 2004;124:356–363. doi: 10.1002/ajmg.a.20349. [DOI] [PubMed] [Google Scholar]
- 10.Schwabe G.C., Tinschert S., Buschow C., Meinecke P., Wolff G., Gillessen-Kaesbach G., Oldridge M., Wilkie A.O., Komec R., Mundlos S. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am. J. Hum. Genet. 2000;67:822–831. doi: 10.1086/303084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson D., Kan S.H., Oldridge M., Trembath R.C., Roche P., Esnouf R.M., Giele H., Wilkie A.O. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am. J. Hum. Genet. 2003;72:984–997. doi: 10.1086/374721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao B., Guo J., She C., Shu A., Yang M., Tan Z., Yang X., Guo S., Feng G., He L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 2001;28:386–388. doi: 10.1038/ng577. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 14.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 16.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 17.Strauch K., Fimmers R., Kurz T., Deichmann K.A., Wienker T.F., Baur M.P. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am. J. Hum. Genet. 2000;66:1945–1957. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiele H., Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 19.Ruschendorf F., Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 20.DiLeone R.J., Russell L.B., Kingsley D.M. An extensive 3′ regulatory region controls expression of Bmp5 in specific anatomical structures of the mouse embryo. Genetics. 1998;148:401–408. doi: 10.1093/genetics/148.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klopocki E., Ott C.E., Benatar N., Ullmann R., Mundlos S., Lehmann K. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J. Med. Genet. 2008;45:370–375. doi: 10.1136/jmg.2007.055699. [DOI] [PubMed] [Google Scholar]
- 22.Klopocki E., Schulze H., Strauss G., Ott C.E., Hall J., Trotier F., Fleischhauer S., Greenhalgh L., Newbury-Ecob R.A., Neumann L.M. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortlock D.P., Guenther C., Kingsley D.M. A general approach for identifying distant regulatory elements applied to the Gdf6 gene. Genome Res. 2003;13:2069–2081. doi: 10.1101/gr.1306003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olshen A.B., Venkatraman E.S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 25.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J., Schiestl R.H. Topoisomerase I involvement in illegitimate recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996;16:1805–1812. doi: 10.1128/mcb.16.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 28.Chandler R.L., Chandler K.J., McFarland K.A., Mortlock D.P. Bmp2 transcription in osteoblast progenitors is regulated by a distant 3′ enhancer located 156.3 kilobases from the promoter. Mol. Cell. Biol. 2007;27:2934–2951. doi: 10.1128/MCB.01609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lobe C.G., Koop K.E., Kreppner W., Lomeli H., Gertsenstein M., Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev. Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 30.Stricker S., Fundele R., Vortkamp A., Mundlos S. Role of Runx genes in chondrocyte differentiation. Dev. Biol. 2002;245:95–108. doi: 10.1006/dbio.2002.0640. [DOI] [PubMed] [Google Scholar]
- 31.Albrecht A.N., Schwabe G.C., Stricker S., Boddrich A., Wanker E.E., Mundlos S. The synpolydactyly homolog (spdh) mutation in the mouse–a defect in patterning and growth of limb cartilage elements. Mech. Dev. 2002;112:53–67. doi: 10.1016/s0925-4773(01)00639-6. [DOI] [PubMed] [Google Scholar]
- 32.Zou H., Wieser R., Massague J., Niswander L. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev. 1997;11:2191–2203. doi: 10.1101/gad.11.17.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyons K.M., Pelton R.W., Hogan B.L. Organogenesis and pattern formation in the mouse: RNA distribution patterns suggest a role for bone morphogenetic protein-2A (BMP-2A) Development. 1990;109:833–844. doi: 10.1242/dev.109.4.833. [DOI] [PubMed] [Google Scholar]
- 34.Kulessa H., Turk G., Hogan B.L. Inhibition of Bmp signaling affects growth and differentiation in the anagen hair follicle. EMBO J. 2000;19:6664–6674. doi: 10.1093/emboj/19.24.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pathi S., Rutenberg J.B., Johnson R.L., Vortkamp A. Interaction of Ihh and BMP/Noggin signaling during cartilage differentiation. Dev. Biol. 1999;209:239–253. doi: 10.1006/dbio.1998.9181. [DOI] [PubMed] [Google Scholar]
- 36.Ploger F., Seemann P., Schmidt-von Kegler M., Lehmann K., Seidel J., Kjaer K.W., Pohl J., Mundlos S. Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum. Mol. Genet. 2008;17:1222–1233. doi: 10.1093/hmg/ddn012. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann K., Seemann P., Boergermann J., Morin G., Reif S., Knaus P., Mundlos S. A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type C/symphalangism-like phenotype or brachydactyly type A2. Eur. J. Hum. Genet. 2006;14:1248–1254. doi: 10.1038/sj.ejhg.5201708. [DOI] [PubMed] [Google Scholar]
- 38.Niedermaier M., Schwabe G.C., Fees S., Helmrich A., Brieske N., Seemann P., Hecht J., Seitz V., Stricker S., Leschik G. An inversion involving the mouse Shh locus results in brachydactyly through dysregulation of Shh expression. J. Clin. Invest. 2005;115:900–909. doi: 10.1172/JCI200523675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lettice L.A., Horikoshi T., Heaney S.J., van Baren M.J., van der Linde H.C., Breedveld G.J., Joosse M., Akarsu N., Oostra B.A., Endo N. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl. Acad. Sci. USA. 2002;99:7548–7553. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill R.E. How to make a zone of polarizing activity: insights into limb development via the abnormality preaxial polydactyly. Dev. Growth Differ. 2007;49:439–448. doi: 10.1111/j.1440-169X.2007.00943.x. [DOI] [PubMed] [Google Scholar]
- 41.Lettice L.A., Hill A.E., Devenney P.S., Hill R.E. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 2008;17:978–985. doi: 10.1093/hmg/ddm370. [DOI] [PubMed] [Google Scholar]
- 42.Bandyopadhyay A., Tsuji K., Cox K., Harfe B.D., Rosen V., Tabin C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lucas J., Le Mee F., Le Marec B., Pluquailec K., Journel H., Picard F. Trisomy 20p derived from a maternal inversion and brachymesophalangy of the index finger. Ann. Genet. 1985;28:167–171. [PubMed] [Google Scholar]
- 44.Pfeiffer R.A., Kandler C., Sieber E., Rauch A., Trautmann U. Brachydactyly in a child with duplication-deficiency subsequent to t(15;20)(q25.2;p12.2)mat. Candidate regions on one or both chromosomes? Clin. Genet. 1997;51:357–360. doi: 10.1111/j.1399-0004.1997.tb02489.x. [DOI] [PubMed] [Google Scholar]
- 45.Kotzsch A., Nickel J., Seher A., Heinecke K., van Geersdaele L., Herrmann T., Sebald W., Mueller T.D. Structure analysis of bone morphogenetic protein-2 type I receptor complexes reveals a mechanism of receptor inactivation in juvenile polyposis syndrome. J. Biol. Chem. 2008;283:5876–5887. doi: 10.1074/jbc.M706029200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.