

Abstract
Parkinson disease (PD) is a complex, multifactorial neurodegenerative disease with substantial evidence for genetic risk factors. We conducted a genome-wide linkage screen of 5824 single-nucleotide polymorphisms in 278 families of European, non-Hispanic descent to localize regions that harbor susceptibility loci for PD. By using parametric and nonparametric linkage analyses and allowing for genetic heterogeneity among families, we found two loci for PD. Significant evidence for linkage was detected on chromosome 18q11 (maximum lod score [MLOD] = 4.1) and suggestive evidence for linkage was obtained on chromosome 3q25 (MLOD = 2.5). These results were strongest in families not previously screened for linkage, and simulation studies suggest that these findings are likely due to locus heterogeneity rather than random statistical error. The finding of two loci (one highly statistically significant) suggests that additional PD susceptibility genes might be identified through targeted candidate gene studies in these regions.
Main Text
Parkinson disease (PD [MIM 168600]) is a neurodegenerative disorder affecting more than one million people in the United States.1 It is characterized by progressive depletion of dopaminergic neurons within the substantia nigra, which results in the clinical symptoms of tremor, rigidity, and bradykinesia. Despite inconsistent results from studies aiming to establish evidence for a genetic component to the etiology of PD,2 many genes have been identified in which mutations cause a Mendelian form of PD (PARK2 [MIM 602544], PINK1 [MIM 608309], DJ1 [MIM 602533], LRRK2 [MIM 609007], SNCA [MIM 163890], UCHL1 [MIM 191342]).3 Additionally, studies of the more common, late-onset form of PD have identified associations between variants in several genes (MAPT4 [MIM 157140], GBA5 [MIM 606463], FGF206,7 [MIM 605558], mitochondrial haplogroups8 [MIM 556500], and MAOB9–12 [MIM 309860]) and risk of PD. More complex patterns of interaction among risk factors, including gene-gene13,14 and gene-environment interactions,15,16 have been established. Common to all studies is the observation that clinical and genetic heterogeneity likely underlies these effects.3
Many genome-wide screens for loci linked or associated with PD have been performed to date.17–27 Most of the linkage studies in PD to date are based on microsatellite marker sets. However, the development of modern genotyping methods allows more efficient genotyping and increased information extraction via dense maps of single-nucleotide polymorphisms (SNPs). In order to further identify candidate regions that harbor the genes for PD, we extended our initial linkage screen of microsatellite markers in 174 families17 with a genome-wide linkage screen (GWLS) of 5824 SNPs in an expanded data set of 320 families.
For this study, participants from families with multiple individuals affected with PD were ascertained for genetic studies by 13 clinical sites. Clinical evaluations were conducted by neurologists or by clinical staff trained and supervised by a neurologist. Affected individuals possessed at least two of the cardinal signs of PD (resting tremor, rigidity, bradykinesia); unaffected individuals had no clinical signs of PD; and unclear individuals showed only one cardinal sign and/or atypical clinical signs. DNA was extracted from whole blood samples with the PUREGENE DNA purification kit by Gentra (Minneapolis, MN). All participants provided written consent for participation in the study and were enrolled under protocols approved by the institutional review boards at each participating site.
We conducted a GWLS with the Illumina Linkage Panel IVb (containing 6008 SNPs) in 1546 individuals from 320 families. A genotyping efficiency of 95% was required to retain a SNP for analysis, and unreliable markers were identified by including four duplicated quality control (QC) samples per 96-well PCR plate; markers with mismatched QC samples were removed from analysis.17 After removing 160 markers with low-efficiency or mismatched QC samples, 16 Y markers, and 8 pseudoautosomal XY markers, 5824 SNPs (average spacing 0.62 cM) were analyzed. Hardy-Weinberg equilibrium was evaluated in one affected and one unaffected individual per family via the Genetic Data Analysis program, and two SNPs with significant deviation from HWE in both groups (indicative of potential genotyping errors) were removed from the analysis. Linkage maps provided by Illumina established the location of each SNP by extrapolating from the physical map location (NCBI Build 35) and the deCODE linkage map (in cM). Family relationships were checked with RELPAIR software.28 Nine individuals were removed from analysis because of inconsistencies with the established pedigree structure, but no families were removed.
After eliminating 42 families carrying known causative mutations in the SNCA, PARK2, or LRRK2 genes, the final overall data set included 278 families of European, non-Hispanic descent with two or more sampled individuals with PD. This final data set included 158 families from our original screen and 120 new families. These families are described in more detail in Table 1. The total number of individuals with DNA samples available in the final data set was 1339 (an average of 4.8 individuals per family). There were 607 individuals with PD, 609 unaffected individuals, and 123 individuals with unclear status. The mean age at onset in individuals with PD was 60.2 years. The data set included 228 affected sibpairs and 147 other affected relative pairs. There were no differences in the 158 original families or 120 new families by mean age at onset or mean number of individuals with PD per family. However, a greater proportion of the new families (44%) contained affected relative pairs compared to the original screen families (22%), and there were differences in the numbers of families contributed by site.
Table 1.
Description of 278 Families Included in the Genome-wide Linkage Screen
| Original Screen Families (N = 158) | New Families (N = 120) | Total Families (N = 278) | |
|---|---|---|---|
| Individuals affected with PD sampled | 353 | 254 | 607 |
| Unaffected individuals sampled | 369 | 240 | 609 |
| Individuals with unclear diagnosis sampled | 74 | 49 | 123 |
| Mean age at onset (standard error) in sampled, affected individuals | 59.9 (12.7) | 60.8 (13.0) | 60.2 (12.8) |
| Number of sampled affected sibpairs | 150 | 78 | 228 |
| Number of sampled affected relative pairs | 76 | 72 | 147 |
| Mean number (range) of sampled affected individuals per family | 2.3 (2-5) | 2.2 (2-5) | 2.3 (2-5) |
| Proportion of families with sampled affected relative pairs | 22% | 44% | 31% |
| Number of families included by clinical site | |||
| Baylor College of Medicine | 7 | 6 | 13 |
| Charlotte Neurologic Center | 7 | 2 | 9 |
| Duke University Medical Center | 7 | 44 | 51 |
| Emory University | 22 | 9 | 31 |
| University of Pennsylvania | 12 | 5 | 17 |
| Marshfield Clinic | 12 | 1 | 13 |
| Ohio State University | 25 | 7 | 32 |
| Rush-Presbyterian-St. Luke's Hospital | 8 | 2 | 10 |
| University of California, Los Angeles | 2 | 3 | 5 |
| University of Kansas | 16 | 1 | 17 |
| University of Minnesota | 34 | 24 | 58 |
| University of Western Australia | 4 | 0 | 4 |
| Vanderbilt University | 2 | 16 | 18 |
Parametric and nonparametric linkage analyses were performed on autosomal and X chromosomes, via MERLIN and MINX (MERLIN in X) software.29 Parametric linkage analyses can be powerful methods for detecting linkage when the true mode of inheritance is unknown as long as both a dominant and a recessive model are used and locus heterogeneity across families is considered.30–34 Therefore, we used both dominant and recessive “affecteds-only” models for our parametric linkage analyses with model parameters consistent with our previous report.17 Affecteds-only models utilize genotypes from unaffected and unclear individuals only to establish linkage phase—only individuals considered affected contribute to the LOD score. Disease allele frequencies were set at 0.001 and 0.20 for the dominant and recessive model, respectively. Marker allele frequencies were estimated from all founders. We generated two-point and multipoint parametric maximum LOD (MLOD) scores allowing for heterogeneity. Moreover, we also calculated both two-point and multipoint nonparametric LOD (LOD∗) scores because nonparametric linkage (NPL) analysis is robust to model assumptions of inheritance.30 In order to avoid the potential drawback of missing important linkage regions, we classified regions with LOD scores greater than 1.5 as interesting, though this may increase the risk of following up false-positive results.17
Many interesting two-point MLOD scores (MLOD ≥ 2) were detected (data not shown). Following our approach in the original genomic screen,17 to choose the most promising intervals for follow-up, we also examined the data with multipoint linkage analysis methods and selected regions of interest that generated LOD scores >1.5 on both the two-point and multipoint analyses. A plot of multipoint MLOD scores for the genome-wide linkage screen is shown in Figure 1. Multipoint dominant MLOD (black), recessive MLOD (blue), and LOD∗ scores (red) are plotted. The parametric MLOD scores are generally higher than LOD∗ scores. The highest multipoint MLOD scores were obtained under a dominant model of inheritance in an 11 cM interval on chromosome 3q25, at RS755763 (MLOD = 2.0; 1 LOD-down support interval: 153 to 164 cM) and in a 9 cM interval on chromosome 18q11 at RS948384 (MLOD = 1.8; 1 LOD-down support interval: 40 to 49 cM). The proportions of linked families for these regions were 0.19 (3q25) and 0.18 (18q11). These were also the only two regions with both two-point and multipoint LOD scores >1.5. Table 2 shows the SNPs that have two-point MLOD scores greater than 1.5 within the two candidate regions. RS902432 on chromosome 3 gives two-point MLOD scores of 1.57 and 2.54 for the dominant and recessive models, respectively. RS1972602 on chromosome 18 gives two-point MLOD score of 1.52 for the recessive model. The corresponding multipoint MLOD scores at RS902432 and RS1972602 are 1.9 and 1.7, respectively, under the dominant model.
Figure 1.
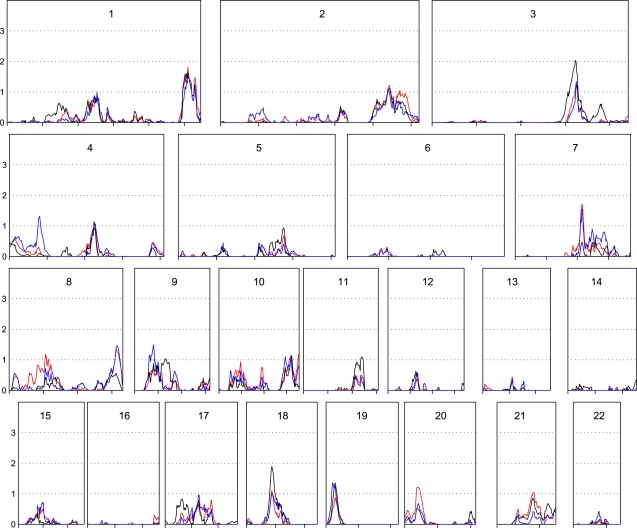
Multipoint Parametric and Nonparametric LOD Scores for the Overall Data Set
Multipoint maximized LOD scores for the dominant model (black) and the recessive model (blue) allowing for heterogeneity and nonparametric LOD∗ scores (red) are plotted as a function of marker location (scaled to the size of the chromosome). Chromosome number is at the top of each plot. Overall data set size, 278 families.
Table 2.
List of Markers with Two-Point LOD Scores > 1.5 within the Candidate Regions on Chromosomes 3 and 18 for the Overall Data Set
| SNP | Location | cM |
Two-Point MLOD |
Multipoint MLOD |
||
|---|---|---|---|---|---|---|
| Dominant | Recessive | LOD∗ | Dominant | |||
| rs902432 | 3q25 | 155 | 1.6 | 2.5 | 1.5 | 1.9 |
| rs1972602 | 18q11 | 46 | 1.0 | 1.5 | 1.5 | 1.7 |
The families in the final data set comprised those included in the original genomic screen (N = 158) and families recruited since that screen was completed (N = 120), and the current screen did not detect linkage overall in regions previously implicated (on chromosomes 5, 8, 9, 17, and X) in 2001, so we suspected that clinical and locus heterogeneity might exist. Therefore, we stratified the data set into families included in the original genome screen and new families added to the current screen. In the subset of 158 families included in the original screen, the linkage peaks were in general agreement with our previous genomic screen report (data not shown). However, in the 120 families not previously screened, the parametric linkage peaks under the dominant model on chromosome 3 and 18 were much stronger than the overall data set (Figure 2). Significant evidence for linkage to a 5 cM interval on chromosome 18 was obtained, with an MLOD of 4.1 at RS948384 (1 LOD-unit down support interval of 42 to 47 cM and proportion of linked families = 0.39). On chromosome 3, an MLOD of 2.5 was obtained in an 11 cM interval centered on RS755763 (1 LOD-unit down support interval from 153 to 164 cM and proportion of linked families of 0.29). There was little evidence for linkage to these regions overall in the original 158 families (MLOD = 0 in both regions). Generally, one expects linkage to grow stronger with additional families if the result is true; however, locus heterogeneity between the subsets might explain the unexpectedly stronger results in the newly screened families. A natural question is whether the strengthening of the linkage peaks on chromosomes 3 and 18 in the newly screened family data set is due to chance (i.e., a same size random sample can produce similar linkage peaks). We randomly selected 120 families (the number of newly screened families) without replacement from the overall data set and reran the linkage analysis. We replicated the procedure 100 times. Only one of the random selection of families gave linkage peaks as high as or higher than that observed in the newly screened family data set (p = 0.01). We conclude from this simulation that the improvement in the linkage analysis result by stratification into original and newly screened subsets was not due to chance, but rather an indicator of locus heterogeneity among the families. There is no difference in the mean number of individuals with PD sampled and the mean age at onset between the original families and the newly screened families, and all families were of European, non-Hispanic ancestry. The ascertainment and diagnostic criteria were also the same in the two sets of families. Although there is a difference in the proportion of families contributed by each ascertainment site to the two subsets and a greater percentage of families with affected relative pairs in the newly screened families (Table 1), there is no difference in evidence for linkage when stratifying families by site or type of relative pair (affected sibpair versus affected relative pair).
Figure 2.
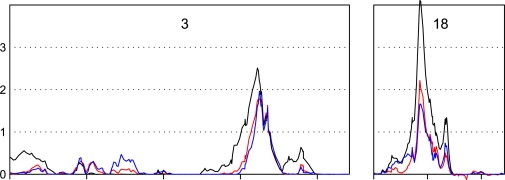
Multipoint Parametric and Nonparametric LOD Scores for the Newly Screened Family Set for Chromosomes 3 and 18
Multipoint maximized LOD scores for the dominant model (black) and the recessive model (blue) allowing for heterogeneity and nonparametric LOD∗ scores (red) are plotted as a function of marker location (scaled to the size of the chromosome). Chromosome number is at the top of each plot. Newly screened family set size, 120 families.
These linkage regions (and their 1-LOD unit down support intervals) have not, to our knowledge, been previously reported by other genome-wide linkage screens of PD. Neither have they been implicated as associated with PD in two published genome-wide association screens (GWAS) for PD.24,26 Although the publicly available summary results for both data sets available in dbGaP show there is modest evidence (smallest p = 0.003) for association with individual SNPs in these two regions, the results do not implicate a single, common area of the interval and do not withstand correction for multiple comparisons. According to NCBI, there are 90 and 76 annotated genes in the chromosome 3 and 18 candidate regions, respectively. None of these genes has previously been associated with PD. However, a recently published study of a large Amish pedigree with parkinsonism and progressive supranuclear palsy found significant evidence for linkage to a microsatellite marker (D3S1764, 153cM, LOD = 3.6) near the peak of our chromosome 3 region.27 These results suggest that a gene influencing development of a broader parkinsonism phenotype might exist in this region. Therefore, an interval-wide, tag-SNP-based association screen is warranted to identify the genes underlying the evidence for linkage.
By using a dense (intermarker spacing < 1 cM) genome-wide linkage SNP panel, we identified two autosomal regions linked to PD. Linkage evidence to these two regions improved when stratifying the sample into previously screened and newly screened family subsets, indicating that locus heterogeneity is a prominent feature of this complex disease and that this heterogeneity must be carefully considered when selecting samples for gene identification efforts. Identification of chromosomal regions linked to PD is a step toward identifying the genes that, together and when interacting with the environment, influence development of this complex disorder. Fine mapping of the linkage region through traditional position cloning and candidate gene evaluation or high-density association mapping (such as can be achieved with GWAS genotyping arrays) will also be required to identify the particular variants responsible for influencing risk of PD in these regions.
Acknowledgments
We are grateful to the families who participated in this study. We thank the members of the PD Genetics Collaboration: Martha A. Nance, Ray L. Watts, Jean P. Hubble, William C. Koller, Kelly Lyons, Rajesh Pahwa, Matthew B. Stern, Amy Colcher, Bradley C. Hiner, Joseph Jankovic, William G. Ondo, Fred H. Allen, Jr., Christopher G. Goetz, Gary W. Small, Donna Masterman, Frank Mastaglia, and Jonathan L. Haines who contributed families to the study. Some of the samples used in this study were collected while the Udall PDRCE was based at Duke University. This work was supported by National Institutes of Health grant NS39764.
Web Resources
The URLs for data presented herein are as follows:
Genetic Data Analysis, http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php
NCBI website, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Note Added in Proof
In a recently published genome-wide association study of 857 PD cases and 867 controls (Pankratz, N., Wilk, J.B., Latourelle, J.C., DeStefano, A.L., Halter, C., Pugh, E.W., Doheny, K.F., Gusella, J.F., Nichols, W.C., Foroud, T., et al. (2009). Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124, 593–605), one intronic SNP (rs12638253), at the distal edge of our chromosome 3 linkage region, was among the 30 providing the strongest evidence for association with PD (p < 0.0001) under an additive model. These results support the existence of a PD locus in this region on chromosome 3.
References
- 1.McDonald W.M., Richard I.H., DeLong M.R. Prevalence, etiology, and treatment of depression in Parkinson's disease. Biol. Psychiatry. 2003;54:363–375. doi: 10.1016/s0006-3223(03)00530-4. [DOI] [PubMed] [Google Scholar]
- 2.Tanner C.M., Ottman R., Goldman S.M., Ellenberg J., Chan P., Mayeux R., Langston J.W. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341–346. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- 3.Klein C., Schlossmacher M.G. Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. 2007;69:2093–2104. doi: 10.1212/01.wnl.0000271880.27321.a7. [DOI] [PubMed] [Google Scholar]
- 4.Martin E.R., Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Koller W.C., Lyons K., Pahwa R., Stern M.B., Colcher A. Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA. 2001;286:2245–2250. doi: 10.1001/jama.286.18.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 2004;351:1972–1977. doi: 10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 6.van der Walt J.M., Noureddine M.A., Kittappa R., Hauser M.A., Scott W.K., McKay R., Zhang F., Stajich J.M., Fujiwara K., Scott B.L. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am. J. Hum. Genet. 2004;74:1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satake W., Mizuta I., Suzuki S., Nakabayashi Y., Ito C., Watanabe M., Takeda A., Hasegawa K., Sakoda S., Yamamoto M. Fibroblast growth factor 20 gene and Parkinson's disease in the Japanese population. Neuroreport. 2007;18:937–940. doi: 10.1097/WNR.0b013e328133265b. [DOI] [PubMed] [Google Scholar]
- 8.van der Walt J.M., Nicodemus K.K., Martin E.R., Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Haines J.L., Koller W.C., Lyons K. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa P., Checkoway H., Levy D., Smith-Weller T., Franklin G.M., Swanson P.D., Costa L.G. Association of a polymorphism in intron 13 of the monoamine oxidase B gene with Parkinson disease. Am. J. Med. Genet. 1997;74:154–156. doi: 10.1002/(sici)1096-8628(19970418)74:2<154::aid-ajmg7>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]
- 10.Mellick G.D., Buchananm D.D., McCann S.J., James K.M., Johnson A.G., Davis D.R., Liyou N., Chan D., Le Couteur D.G. Variations in the monoamine oxidase B (MAOB) gene are associated with Parkinson's disease. Mov. Disord. 1999;14:219–224. doi: 10.1002/1531-8257(199903)14:2<219::aid-mds1003>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Parsian A., Racette B., Zhang Z.H., Rundle M., Perlmutter J.S. Association of variations in monoamine oxidases A and B with Parkinson's disease subgroups. Genomics. 2004;83:454–460. doi: 10.1016/j.ygeno.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Kang S.J., Scott W.K., Li Y.J., Hauser M.A., van der Walt J.M., Fujiwara K., Mayhew G.M., West S.G., Vance J.M., Martin E.R. Family-based case-control study of MAOA and MAOB polymorphisms in Parkinson disease. Mov. Disord. 2006;21:2175–2180. doi: 10.1002/mds.21151. [DOI] [PubMed] [Google Scholar]
- 13.Wu R.M., Cheng C.W., Chen K.H., Lu S.L., Shan D.E., Ho Y.F., Chern H.D. The COMT L allele modifies the association between MAOB polymorphism and PD in Taiwanese. Neurology. 2001;56:375–382. doi: 10.1212/wnl.56.3.375. [DOI] [PubMed] [Google Scholar]
- 14.Gao X., Scott W.K., Wang G., Mayhew G., Li Y.J., Vance J.M., Martin E.R. Gene-gene interaction between FGF20 and MAOB in Parkinson disease. Ann. Hum. Genet. 2008;72:157–162. doi: 10.1111/j.1469-1809.2007.00418.x. [DOI] [PubMed] [Google Scholar]
- 15.Hancock D.B., Martin E.R., Fujiwara K., Stacy M.A., Scott B.L., Stajich J.M., Jewett R., Li Y.J., Hauser M.A., Vance J.M. NOS2A and the modulating effect of cigarette smoking in Parkinson's disease. Ann. Neurol. 2006;60:366–373. doi: 10.1002/ana.20915. [DOI] [PubMed] [Google Scholar]
- 16.Hancock D.B., Martin E.R., Vance J.M., Scott W.K. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson's disease. Neurogenetics. 2008;9:249–262. doi: 10.1007/s10048-008-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Koller W.C., Lyons K., Pahwa R., Stern M.B., Colcher A., Hiner B.C. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;286:2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- 18.Destefano A.L., Golbe L.I., Mark M.H., Lazzarini A.M., Maher N.E., Saint-Hilaire M., Feldman R.G., Guttman M., Watts R.L., Suchowersky O. Genome-wide scan for Parkinson's disease: the GenePD Study. Neurology. 2001;57:1124–1126. doi: 10.1212/wnl.57.6.1124. [DOI] [PubMed] [Google Scholar]
- 19.Pankratz N., Nichols W.C., Uniacke S.K., Halter C., Rudolph A., Shults C., Conneally P.M., Foroud T. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am. J. Hum. Genet. 2002;71:124–135. doi: 10.1086/341282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hicks A.A., Petursson H., Jonsson T., Stefansson H., Johannsdottir H.S., Sainz J., Frigge M.L., Kong A., Gulcher J.R., Stefansson K. A susceptibility gene for late-onset idiopathic Parkinson's disease. Ann. Neurol. 2002;52:549–555. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- 21.Bertoli-Avella A.M., Giroud-Benitez J.L., Bonifati V., Alvarez-Gonzalez E., Heredero-Baute L., van Duijn C.M., Heutink P. Suggestive linkage to chromosome 19 in a large Cuban family with late-onset Parkinson's disease. Mov. Disord. 2003;18:1240–1249. doi: 10.1002/mds.10534. [DOI] [PubMed] [Google Scholar]
- 22.Martinez M., Brice A., Vaughan J.R., Zimprich A., Breteler M.M., Meco G., Filla A., Farrer M.J., Betard C., Hardy J. Genome-wide scan linkage analysis for Parkinson's disease: the European genetic study of Parkinson's disease. J. Med. Genet. 2004;41:900–907. doi: 10.1136/jmg.2004.022632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foltynie T., Hicks A., Sawcer S., Jonasdottir A., Setakis E., Maranian M., Yeo T., Lewis S., Brayne C., Stefansson K. A genome wide linkage disequilibrium screen in Parkinson's disease. J. Neurol. 2005;252:597–602. doi: 10.1007/s00415-005-0686-2. [DOI] [PubMed] [Google Scholar]
- 24.Maraganore D.M., De Andrade M., Lesnick T.G., Strain K.J., Farrer M.J., Rocca W.A., Pant P.V., Frazer K.A., Cox D.R., Ballinger D.G. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertoli-Avella A.M., Dekker M.C., Aulchenko Y.S., Houwing-Duistermaat J.J., Simons E., Testers L., Pardo L.M., Rademaker T.A., Snijders P.J., van Swieten J.C. Evidence for novel loci for late-onset Parkinson's disease in a genetic isolate from the Netherlands. Hum. Genet. 2006;119:51–60. doi: 10.1007/s00439-005-0108-7. [DOI] [PubMed] [Google Scholar]
- 26.Fung H.C., Scholz S., Matarin M., Simon-Sanchez J., Hernandez D., Britton A., Gibbs J.R., Langefeld C., Stiegert M.L., Schymick J. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 27.Lee S.L., Murdock D.G., McCauley J.L., Bradford Y., Crunk A., McFarland L., Jiang L., Wang T., Schnetz-Boutaud N., Haines J.L. A genome-wide scan in an Amish pedigree with parkinsonism. Ann. Hum. Genet. 2008;72:621–629. doi: 10.1111/j.1469-1809.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Epstein M.P., Duren W.L., Boehnke M. Improved inference of relationship for pairs of individuals. Am. J. Hum. Genet. 2000;67:1219–1231. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 30.Kong A., Cox N.J. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg D.A., Abreu P., Hodge S.E. The power to detect linkage in complex disease by means of simple LOD score analysis. Am. J. Hum. Genet. 1998;63:870–879. doi: 10.1086/301997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abreu P.C., Greenberg D.A., Hodge S.E. Direct power comparisons between simple LOD scores and NPL scores for linkage analysis in complex diseases. Am. J. Hum. Genet. 1999;65:847–857. doi: 10.1086/302536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durner M., Vieland V.J., Greenberg D.A. Further evidence for the increased power of LOD scores compared with nonparametric methods. Am. J. Hum. Genet. 1999;64:281–289. doi: 10.1086/302181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenberg D.A., Abreu P.C. Determining trait locus position from multipoint analysis: accuracy and power of three different statistics. Genet. Epidemiol. 2001;21:299–314. doi: 10.1002/gepi.1036. [DOI] [PubMed] [Google Scholar]