

Abstract
The spondylometaphyseal dysplasias (SMDs) are a group of short-stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMD Kozlowski type (SMDK) is a well-defined autosomal-dominant SMD characterized by significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles similar to autosomal-dominant brachyolmia, which can result from heterozygosity for activating mutations in the gene encoding TRPV4, a calcium-permeable ion channel. Mutation analysis in six out of six patients with SMDK demonstrated heterozygosity for missense mutations in TRPV4, and one mutation, predicting a R594H substitution, was recurrent in four patients. Similar to autosomal-dominant brachyolmia, the mutations altered basal calcium channel activity in vitro. Metatropic dysplasia is another SMD that has been proposed to have both clinical and genetic heterogeneity. Patients with the nonlethal form of metatropic dysplasia present with a progressive scoliosis, widespread metaphyseal involvement of the appendicular skeleton, and carpal ossification delay. Because of some similar radiographic features between SMDK and metatropic dysplasia, TRPV4 was tested as a disease gene for nonlethal metatropic dysplasia. In two sporadic cases, heterozygosity for de novo missense mutations in TRPV4 was found. The findings demonstrate that mutations in TRPV4 produce a phenotypic spectrum of skeletal dysplasias from the mild autosomal-dominant brachyolmia to SMDK to autosomal-dominant metatropic dysplasia, suggesting that these disorders should be grouped into a new bone dysplasia family.
Introduction
Spondylometaphyseal dysplasias (SMDs) are characterized by abnormalities in the vertebrae and the metaphyses of the tubular bones. There are numerous distinct SMDs, and classification has been based on the severity of the clinical and radiographic findings1 and variations in vertebral and femoral neck involvement.2 Among these disorders, SMD Kozlowski type ([SMDK]; MIM 1842522) is the best defined SMD and is inherited in an autosomal-dominant manner.3,4 SMDK is primarily characterized by postnatal short stature and significant kyphoscoliosis leading to progressive deformity. Other clinical findings include normal facies, a short neck, pectus carinatum, and genu varus.5 Most cases are diagnosed after the age of two when the patients present with a waddling gait. The radiographic phenotype is distinct and includes odontoid hypoplasia, platyspondyly with vertebral bodies described as resembling an “open staircase,” overfaced vertebral pedicles, short square ilia, flat acetabular roofs, wide proximal femoral epiphyseal plates, metaphyseal irregularities, and markedly delayed carpal bone ossification.6
Metatropic dysplasia (MIM 156530) was first delineated by Maroteaux et al.7 in 1966, and the name is derived from the Greek term metatropos, meaning “changing patterns.” Genetic heterogeneity based on clinical and radiographic variability has been proposed,8 including a nonlethal dominant form and both lethal and nonlethal autosomal-recessive forms. Other investigators have suggested that all cases represent dominant forms with phenotypic variability.9,10 Patients often present with shortened limbs and a long, narrow trunk in the newborn period that evolves into a severe, progressive kyphoscoliosis, frequently requiring surgical correction.11 Affected individuals have a distinctive facies with a prominent forehead and squared-off jaw.9 Other significant clinical findings include a high incidence of odontoid hypoplasia, cervical myelopathy, and, in some cases, stenosis leading to significant neurologic sequelae,11,12 as well as respiratory compromise that can lead to death. Sensorineural deafness can also occur.9,10 Patients have been described as manifesting a coccygeal tail in the newborn period as a result of excessive ossification of the coccyx giving rise to a bump above the anus. The radiographic findings in metatropic dysplasia are characteristic and include wafer-like vertebral bodies in newborns, a halberd-shaped pelvis, irregular calcanei and tali, brachydactyly with delayed carpal ossification, and flared or “mushroomed” proximal and distal metaphyses of the femora leading to a “dumbbell-shaped bone.”6
Recently, a form of autosomal-dominant brachyolmia (MIM 113500) was shown to result from activating mutations in the gene that encodes TRPV413 (MIM 605427), a calcium-permeable ion channel of the vanilloid subfamily of TRP channels. The vertebral body abnormalities (severely flattened, irregular vertebrae and overfaced pedicles) in SMDK are similar to those in this form of brachyolmia, suggesting the possibility that the two disorders could be allelic. In addition, although the appendicular and vertebral abnormalities in metatropic dysplasia are much more severe and distinct from those in SMDK, there was some similarity in the generalized appearance of the vertebral bodies, the shape of the pelvis, and the delayed carpal ossification. Therefore, TRPV4 was analyzed for mutations in SMDK and metatropic dysplasia. All six of the SMDK cases examined were shown to be heterozygous for missense mutations in TRPV4, as were two cases of metatropic dysplasia. The SMDK mutations were further shown to result in altered calcium channel activity in vitro. These data revealed the molecular basis of SMDK and metatropic dysplasia and suggest that this group of disorders constitutes a new bone dysplasia family.
Material and Methods
Clinical Information
Patients and their families were assessed under an IRB-approved protocol. The diagnoses of SMDK and metatropic dysplasia were made based on clinical and radiographic information (Table 1).
Table 1.
Clinical and Radiographic in Findings in Subjects with SMD Kozlowski and Metatropic Dysplasia
| Patient |
R03-404 |
R97-261 |
R72-010 |
R07-490 |
R07-474 |
R08-216 |
R03-386 |
R00-067 |
|---|---|---|---|---|---|---|---|---|
| Diagnosis | SMDK | SMDK | SMDK | SMDK | SMDK | SMDK | MD | MD |
| Clinical Findings | ||||||||
| Age of ascertainment | 9 years | 8 years | 3–6 years | 5 years | 2.5 years | 8 months | newborn | newborn |
| High forehead/flat nasal bridge | - | - | - | - | + | + | + | + |
| Hearing loss | - | - | - | - | - | - | - | NA |
| Congenital scoliosis | NA | - | + | - | + | + | + | + |
| Respiratory compromise | - | - | - | - | - | +/mild | + | - |
| Waddling gait | - | - | + | + | - | NA | - | NA |
| Contracture | - | Elbow | - | - | - | - | + | + |
| Prominent joints | - | - | - | - | - | - | + | + |
| Coccygeal tail∗ | - | - | - | - | - | ∗ | - | - |
| Radiographic Findings | ||||||||
| Odontoid hypoplasia | - | NA | NA | - | - | - | + | NA |
| Clavicular pseudoarthrosis | + | - | - | - | - | - | + | - |
| Anterior rib splaying | - | NA | - | - | - | - | + | + |
| Platyspondyly | mild | + | + | + | + | + | + | + |
| Dense wafer vertebrae | - | - | - | - | - | - | + | + |
| Overfaced vertebral pedicles | + | + | + | + | + | +/− | - | - |
| Flared iliac wings | - | mild | mild | + | + | + | - | + |
| Halberd pelvis | - | - | mild | mild | + | + | + | + |
| Flat acetabular roof | - | mild | slanted | mild | - | + | + | - |
| Irregular proximal femoral growth plate | + | + | + | + | - | + | NA | NA |
| Narrow sacroiliac notches | - | + | + | + | + | - | mild | + |
| Dumbbell shaped femora/humeri | - | - | - | - | - | - | + | + |
| Phalangeal cone epiphyses | - | - | - | - | - | - | + | + |
| Brachydactyly | - | NA | - | - | - | mild | + | NA |
| Carpal ossification delay | + | NA | + | + | + | NA | NA | NA |
| Irregular calcanei/tali | - | NA | NA | - | - | - | + | + |
The following abbreviations are used: SMDK, spondylometaphyseal dysplasia, Kozlowski type; MD, metatropic dysplasia; ∗, sacral dimple; NA∗, not applicable because of age or not available; and NA, not applicable or not available.
Mutation Analysis
Genomic DNA was extracted by standard protocols, and the coding exons of TRPV4 were amplified by PCR and their sequences determined as previously described.13 Sequences were compared to the reference sequence for TRPV4, with nucleotide numbering starting from the A of the ATG initiation codon.
Expression of Normal and Mutant TRPV4 in HEK293 Cells
Human embryonic kidney cells (HEK293T) were grown in Dulbecco's modified Eagle's medium containing 10% (v/v) human serum, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin at 37°C in a humidity-controlled incubator with 10% CO2. As previously described,13 the cells were transiently transfected with a human TRPV4 (accession number NP_671737) vector, cloned as a BamHI fragment into the BcII-site of the pCAGGS IRES-GFP vector. Mutagenesis and transfection were performed as previously described.13
Electrophysiology
Whole-cell membrane currents were measured with an EPC-10 (HEKA Elektronik, Lambrecht, Germany) with ruptured patches sampled at 20 kHz and filtered at 2.9 kHz. Patch electrodes had a direct current (DC) resistance between 2 and 4 MΩ when filled with intracellular solution. An Ag-AgCl wire was used as a reference electrode. Series resistance was compensated (50% and 70%). A ramp protocol was applied consisting of a voltage step from the holding potential of 0 mV to −150 mV followed by a 400 ms linear ramp to +150 mV applied every 2 s. The voltage-step protocol consisted of 40 ms steps from −80 mV to + 200 mV. Mean cell-membrane capacitance was 7.43 ± 0.1 picofarads ([pF], n = 58) and very similar for all HEK cells; therefore, current densities were not calculated. For electrophysiological measurements, the standard extracellular solution contained 150 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose, 10 mM HEPES, buffered at pH 7.4 with NaOH. The osmolality of this solution, measured with a vapor-pressure osmometer (Wescor 5500, Schlag, Gladbach, Germany), was 320 ± 5 mOsm. The pipette solution was composed of 20 mM CsCl, 100 mM Asp, 1 mM MgCl2, 10 mM HEPES, 4 mM Na2ATP, 10 mM BAPTA, 2.93 mM CaCl2, adjusted to pH 7.2 with CsOH. The free Ca2+ concentration of this solution was 50 nM. The non-PKC-activating phorbol ester, 4α-phorbol 12,13-didecanoate (4α-PDD, Alexis Biochemicals Lausen/Switzerland), was applied at a 2 μM concentration from a 10 mM stock solution in ethanol, and arachidonic acid (Sigma) was applied to a final concentration of 10 μM. For hypotonic cell swelling, cells were superfused with a solution containing (in mM) 95 NaCl, 6 CsCl, 1 MgCl2, 1.5 CaCl2, 10 glucose, and 10 HEPES, buffered to pH 7.4 with NaOH. Iso-osmolarity was achieved by the addition of mannitol (125 mM) to this solution to reach 320 mOsm. Hypotonic cell swelling was induced by the removal of mannitol to lower the extracellular osmolarity to 200 mOsm. All measurements were carried out at room temperature, 22°C −25°C (for additional details, see 13,14).
Intracellular Ca2+ Measurements
Intracellular [Ca2+]i was measured in Fura-2-loaded cells with a monochromator-based imaging system.14,15 Ca2+ influxes were monitored by CellM imaging software (Olympus, UK) with OLYMPUS IX81 (Olympus) with standard extracellular solution similar to that described for electrophysiology.
Results
Clinical
The subjects presented at ages ranging from the newborn period to age seven. The clinical and radiographic findings are summarized in Table 1. Four of the patients diagnosed with SMDK had typical clinical and radiographic findings that included scoliosis and a waddling gait, and radiographic abnormalities including platyspondyly, overfaced vertebral pedicles, flared iliac wings, a mildly flattened acetabular roof, irregular proximal capital femoral metaphyses, a relatively normal appendicular skeleton, and delayed carpal ossification (Figure 1). The mother of case R03-404 was also affected. Two of the SMDK cases (R07-474 and R08-216) were more severely affected and had some crossover findings with metatropic dysplasia, including a congenital scoliosis, mild facial dysmorphism, and a halberd-shaped pelvis. These two cases suggested a possible genetic relationship between the SMDK and metatropic dysplasia and showed the phenotypic continuum between the disorders. The two metatropic dysplasia cases studied both had typical clinical and radiographic findings that included a high forehead, scoliosis, joint contractures, odontoid hypoplasia, anterior rib splaying, severe platyspondyly, dense vertebral pedicles, a halberd-shaped pelvis, dumbbell-shaped femora and humeri, cone epiphyses, and irregularly shaped calcanei and tali (Figure 2).
Figure 1.
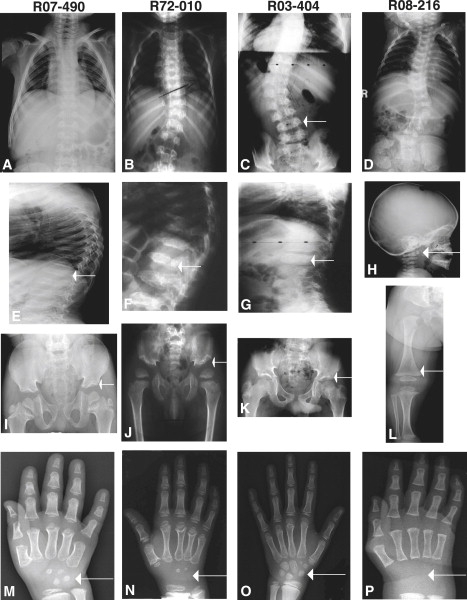
Radiographs of Individuals with SMDK with TRPV4 Mutations
Reference numbers are indicated above each column of radiographs. The ages of the subjects were 5 years (R07-490), 3 years (R72-010), 9 years (R03-404), and 8 months (R08-216).
(A–D) Anteroposterior (A/P) radiographs of the chest in four subjects, demonstrating the variability of the scoliosis from mild to more severe. The arrow in (C) shows the overfaced pedicles (lamina are seen medial to the vertebral edge).
(E–G) Lateral radiographs of the vertebrae showing flattened vertebrae (platyspondyly) and irregular margins.
(H) Lateral radiograph of the cervical vertebrae showing a small, irregular C1 vertebra (odontoid hypoplasia, arrow), a finding similar to metatropic dysplasia.
(I–K) A/P radiographs of the pelvis showing slightly flared ilia, wide epiphyseal plates, and mildly irregular acetabulae (arrows).
(L) A/P radiograph of lower extremity showing slightly widened metaphyses with normal epiphyses, somewhat reminiscent of metatropic dysplasia.
(M–P) A/P hand radiographs demonstrating the marked carpal ossification delay (arrows) for each subject's respective age.
Figure 2.
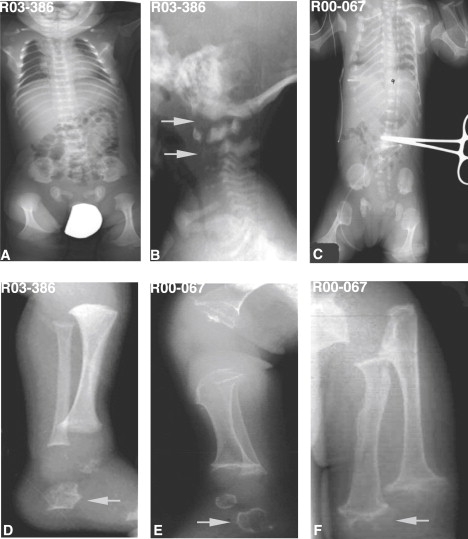
Radiographs of Metatropic Dysplasia in the Newborn Period
Reference numbers for the cases are indicated on each image.
(A and C) Body A/P radiographs in two cases showing severe platyspondyly (wafer-like), anterior and posterior rib cupping, widened ileum, and markedly flared proximal and distal femoral metaphyses (dumbbell-shaped femora).
(B) Lateral cervical spine radiograph demonstrating odontoid hypoplasia and hypoplasia of the cervical vertebral bodies with cervical kyphosis (arrows).
(D and E) Lateral lower-extremity radiograph showing characteristic irregular tali and calcanei (arrows) frequently seen in metatropic dysplasia.
(F) A/P radiograph of the upper extremity showing highly irregular metaphyses with popcorn appearance (arrow).
Mutation Analysis
For testing the hypothesis that SMDK and metatropic dysplasia could result from TRPV4 mutations, the sequences of the coding exons were determined in each case. Heterozygosity for mutations in TRPV4 was found in all eight patients studied (Table 2). In four patients with SMDK, R97-261, R72-010, R07-490, and R07-474, a recurrent nucleotide substitution c.G1781→A in exon 11, predicted to lead to the amino acid substitution p.R594H, was found. In two of these families, R07-490 and R07-474, this nucleotide substitution was not found in DNA from the unaffected parents, establishing the change as de novo (see Figure S1 available online). In SMDK case R03-404, heterozygosity for a transition, c.A992→G, in exon 6 was identified, predicting the amino acid substitution p.D333G. This change was inherited from the subject's affected mother. For SMDK case R08-216, heterozygosity for a transversion, c.G2146→T, was identified, predicting the amino acid substitution pA716S. This substitution was not seen in the parental DNA.
Table 2.
Summary of Mutations and Their Effect on TRPV4
| Patient | Diagnosis | TRPV4 Exon | Nucleotide Substitution | Predicted Amino Acid Change | Protein Domain | Basal [Ca2+]i Intracellular Activity |
|---|---|---|---|---|---|---|
| R03-404 | SMDK | 6 | cA992→G | p.D333G | ANK5 | ↑ |
| R72-010 | SMDK | 11 | c.G1781→A | p.R594H | cytoplasmic/S4 | ↑ |
| R97-261 | SMDK | 11 | c.G1781→A | p.R594H | cytoplasmic/S4 | ↑ |
| R07-490 | SMDK | 11 | c.G1781→A | p.R594H | cytoplasmic/S4 | ↑ |
| R07-474 | SMDK | 11 | c.G1781→A | p.R594H | cytoplasmic/S4 | ↑ |
| R08-216 | SMDK | 13 | c.G2146→T | p.A716S | cytoplasmic/S6 | Same as WT |
| R00-067 | Metatropic | 6 | c.A1080→T | p.I331F | ANK5 | unknown |
| R03-386 | Metatropic | 15 | c.C2396→T | p.P799L | cytoplasmic | unknown |
The following abbreviations are used: SMDK, spondlyometaphsyeal dysplasia; c, nucleotide; p, protein; ANK, anykrin; S, transmembrane domain; ↑, increased; and WT, wild-type.
Heterozygosity for mutations in TRPV4 was also found in the two metatropic dysplasia cases studied, R03-386 and R00-067. These probands were heterozygous for the nucleotide substitutions c.C2396→T and c.A991→T, predicting the amino acid substitutions, p.P799L and p.I331F, respectively (see Figure S1). In both cases, these substitutions were not found in DNA from the unaffected parents. The predicted amino acid substitutions identified in all of the SMDK and metatropic dysplasia cases occurred in residues that have been highly conserved through evolution (Homo sapiens through C. elegans), further supporting their pathogenicity. Furthermore, none of the sequence changes were found among at least 214 control chromosomes, indicating that none are common polymorphisms in the population16 and consistent with each change being causative.
Ca2+ Measurements and Electrophysiology
For determination of the effect of the SMDK mutations on TRPV4 activity, normal and mutant human TRPV4 were expressed in HEK cells.13 In comparison to wild-type TRPV4, the D333G and R594H mutations yielded much larger constitutive currents before agonist application (Figure 3A–3C and 3E). This was confirmed in whole-cell current measurements showing robust basal currents for the D333G and R594H mutants (Figure 3E). The basal currents were blocked in a voltage-dependent manner by the TRPV4 blocker ruthenium red (data not shown). The shape of the IV curve and the reversal potentials were not changed for the D333G and R594H mutants (see Figure S2A). The increased nonstimulated channel activity was also observed in calcium-imaging experiments on intact cells (Figure 4). The basal Fura-2 fluorescence ratio, which reflects basal [Ca2+]i and depends on Ca2+ influx, was significantly higher in cells expressing the D333G and R594H mutations than in those expressing wild-type (WT) TRPV4 (Figure 4E).
Figure 3.
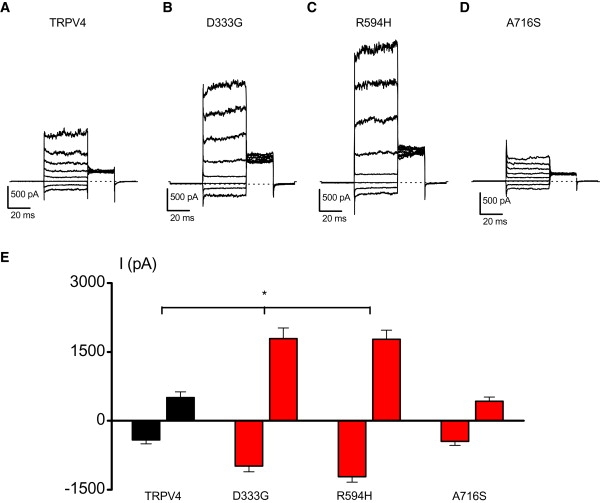
Basal Activity of TRPV4 Wild-Type and TRPV4 Mutants
(A–D) Basal currents through wild-type TRPV4 (A), D333G (B), R594H (C), and A716S (D) transfected HEK cells in response to a voltage step protocol (40 mV steps from −80 mV to +200 mV).
(E) Average basal inward and outward currents at −150 and +150 mV in wild-type TRPV4 (n = 8), D333G (n = 12), R594H (n = 16) and A716S (n = 9) expressing HEK cells. ∗ indicates significant differences when compared with cells expressing wild-type TRPV4 (one-sided Student's t test, p < 0.05). Error bars represent means ± SE (standard error).
Figure 4.
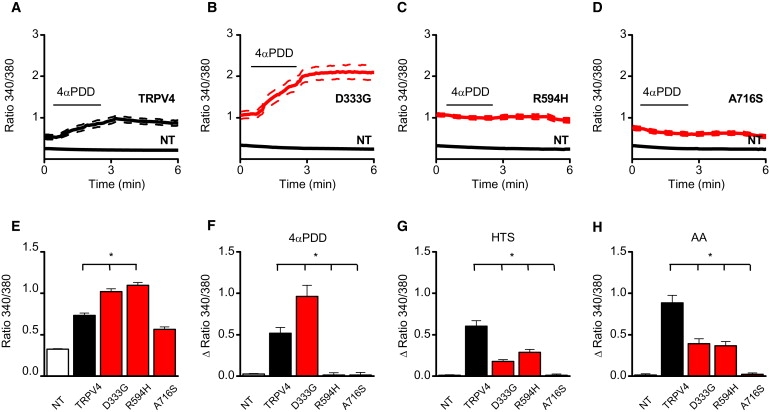
Effect of TRPV4 Mutagenesis on Activation of TRPV4 by Different Activation Stimuli
(A–D) Effect of stimulation with 2 μM 4αPDD on internal fluorescence ratio in wild-type TRPV4 (A), D333G (B), R594H (C), and A716S (D) transfected HEK cells.
(E) Basal internal fluorescence ratio, which is a reference for the basal [Ca2+]i, in nontransfected (NT) and the wild-type TRPV4 and TRPV4 mutant transfected cells, D333G, R594H, and A716S.
(F–H) Average increase in fluorescence ratio in response to 4αPDD (2 μM) (F), a hypotonic stimulus (HTS) (G), and 10 μM arachidonic acid (AA) (H). For every condition, n > 24 in at least three independent recordings. ∗ indicates significant differences when compared with cells expressing wild-type TRPV4 (one-sided Student's t test, p < 0.05). Error bars represent means ± SE.
The mutations also affected the responses to the TRPV4-specific agonist 4αPDD (Figures 4A–4C, and 4F), and Figure S2D). The R594H mutant was fully insensitive to 4αPDD stimulation, whereas the response of the D333G mutant was significantly increased when compared with WT TRPV4. However, reduced current and Ca2+ responses were measured upon stimulation with hypotonic solution (reduction of extracellular osmolarity from 320 to 200 mOsm) or by application of 10 μM arachidonic acid (AA) (Figure 4 and Figures S2 and S3). It is possible that the reduced response of these channels to agonist or mechanical stimulation reflects basal activities that approach maximal channel activation.
In contrast, the A716S mutation showed a similar basal activity as WT TRPV4 (Figures 3D and 3E). However, calcium-imaging experiments and electrophysiological recordings performed on A716S transfected HEK cells showed that this mutant was fully unresponsive to stimulation by 4αPDD (Figures 4D and 4F, and Figures S2A and S2D), cell swelling (Figure 4G and Figures S2B and S3D) and AA (Figure 4G and Figures S2C and S3H). Two lines of evidence indicate that the lack of response to TRPV4-activating stimuli in this mutant was not due to an effect on channel expression or protein targeting to the membrane. First, HEK cells expressing the A716 mutant exhibited increased basal currents and intracellular Ca2+ levels when compared to nontransfected HEK cells, indicating that the channel is functional (Figure 4E). Second, cell-surface biotinylation assays did not reveal significant differences in the level of TRPV4 at the plasma membrane between cells expressing mutant and WT TRPV4 (data not shown).
Discussion
In mammals, the transient receptor potential (TRP) superfamily consists of 28 cation channels that are permeable to both monovalent and divalent cations.17–19 These channels play a key role in ion homeostasis and are unique cellular sensors that function as important integrators of sensory information required for taste, vision, nociception, and the detection of temperature and mechanical forces.20,21 TRPV4 is involved in promoting regulatory-volume decrease by sensing a hypotonic stimulus and mediating the influx of extracellular calcium that then activates pathways leading to the secretion of K+ and Cl−, followed by the loss of intracellular water.22–24 Additionally, channel activity appears to be regulated by a calmodulin-dependent mechanism with negative feedback loops.
The TRPV4 molecule is composed of three ankyrin repeats located at the amino terminus and six putative transmembrane domains with a cation-permeable pore region located between transmembrane domains 5 and 625–27 (TM5 and TM6; Figure 5). Heterozygosity for mutations in TRPV4 produces autosomal-dominant brachyolmia, a relatively mild skeletal dysplasia, primarily characterized by modest short-trunk short stature, scoliosis, and platyspondyly with overfaced vertebral pedicles apparent on radiographs.13,28 To date, two missense mutations that reside within the TM5 domain, R616Q and V620I (Figure 5), have been found to produce the disorder. Patch-clamp experiments using transfected HEK cells showed that both mutations resulted in a significant gain of function characterized by increased constitutive activity and elevated channel activation by a variety of mechanisms including mechanostimulation, and agonist stimulation by arachidonic acid and the TRPV4-specific agonist 4α-phorbol 12,13-didecanoate (4αPDD).13 The mutations are localized at the cytoplasmic end of TM5, a region that has been recently discussed as part of a “hand-handle” domain transferring a gating signal from the TM1-TM4 unit to the TM5-TM6 pore unit.29 It thus appears that, in brachyolmia, the primary consequence of these mutations is to increase intracellular calcium via increased channel activity.
Figure 5.
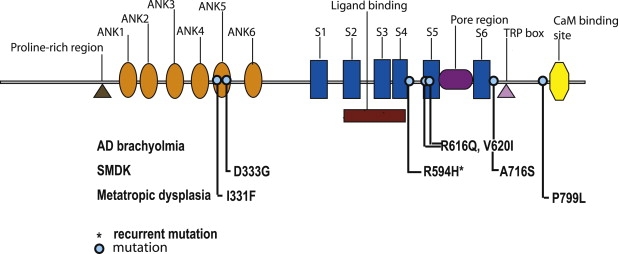
Diagram of the TRPV4 Molecule
The locations of the mutations relative to the domains of the molecule for autosomal-dominant brachyolmia, spondylometaphyseal dysplasia, Kozlowski type, and metatropic dysplasia.
SMDK shares some features with autosomal-dominant brachyolmia, including scoliosis, platyspondyly, and the radiographic appearance of the vertebral bodies. However, the skeletal phenotype of SMDK is more severe than that of brachyolmia, especially the spinal changes. Relative to brachyolmia, SMDK patients exhibit more significant short stature over time, have odontoid hypoplasia, more severe metaphyseal changes in the appendicular skeleton, and carpal ossification delay. One of the unexpected clinical findings in the cohort of patients described in this study was that three out of six patients presented with congenital scoliosis, questioning the original literature suggesting that SMDK patients typically present with a waddling gait and scoliosis around age two.3 In addition, there was significant phenotypic variability within the SMDK cohort. Among the four patients harboring the R594H mutation, there was a spectrum in the degree of spinal abnormalities and one patient, R07-474, had radiographic abnormalities that had some overlapping findings with metatropic dysplasia. Thus it is difficult to establish a direct relationship between mutation and phenotype.
In contrast to the brachyolmia cases, the TRPV4 mutations found in SMDK did not alter TM5. Two mutations, D333G and A716S, were found to alter cytoplasmic portions of the molecule (Figure 5), the functions of which have not been determined to date. D333 is located in the ankyrin repeat domains of TRPV4, a region considered to act as a putative binding region for other regulatory proteins.25 The cytoskeletal organizer Pacsin 3 binds to the N terminus of TRPV4, thereby modulating its activation modes.30 Therefore, it is possible that mutations in this region could render the channel nonresponsive to important protein-protein interactions.
There was one recurrent mutation, R594H, that occurred independently in four cases, revealing a mutational hotspot within the gene. This specific amino acid, arginine 594, is highly conserved among the members of the TRPV subfamily of TRP channels and has been found to be critical for the detection and transduction of chemical stimuli.14 Cells expressing the R594A substitution were unresponsive to chemical stimulation but remained responsive to heat stimulation. Other amino acid substitutions, R594Q and R594L, showed varying responses to stimuli and it appears that a charged residue is needed to detect and transduce stimuli.14 Patch-clamp experiments using HEK cells expressing the R594H SMDK mutation showed increased constitutive and agonist-responsive TRPV4 activity similar to what was found for the brachyolmia mutations. The D333G substitution also increased the level of constitutive channel activity and responded to additional stimulation. Thus, similar to mutations found in autosomal-dominant brachyolmia, these two SMDK mutations probably produce disease by resulting in a net increase in intracellular calcium in chondrocytes. However, the relevant TRPV4-activating stumuli in chondrocytes is unknown.
Although expression of the carboxyl-terminal A716S mutation did produce a slightly increased basal intracellular Ca2+ concentration, this mutant had diminished channel activity relative to the other mutations under both basal and stimulated conditions. The properties of this channel suggest that the increased basal Ca2+ level conferred by the mutation, rather than an increased channel response upon stimulation, may be the key factor in producing disease. However it is also possible that this mutation causes disease by a completely different mechanism.
Metatropic dysplasia has been viewed as genetically complex because of the broad range of clinical severity and the possibility of different patterns of inheritance, with both autosomal-dominant and -recessive forms proposed. The hypothesis that metatropic dysplasia could be allelic to SMDK was suggested by phenotypic findings of severe platyspondyly, an often relentless scoliosis, a flared ileum, and carpal ossification delay. In two cases with characteristic findings of the nonlethal dominant form of metatropic dysplasia that included congenital scoliosis, a long narrow thorax, dense wafer-like vertebral bodies, and delayed carpal ossification, mutations in TRPV4 were identified. The two missense mutations, predicting I331F and P779L substitutions, altered cytoplasmic residues at the amino and carboxyl termini of the molecule, respectively (Figure 5). The more amino-terminal substitution, I331F, lies between the fourth and fifth ankryin repeats, and the more carboxyl-terminal mutation, P799L, is near the putative calmodulin-binding domain at residues 812–831. However, despite the proximity of these amino acids to established domains in TRPV4, the biological function of these residues is largely unknown. Because only two metatropic dysplasia cases were studied, it remains possible that mutations in other genes could also produce the phenotype.
Mutations in TRPV4 have now been identified in three dominantly inherited skeletal phenotypes, autosomal-dominant brachyolmia, SMDK and metatropic dysplasia, revealing a critical role for the molecule in the growth plate. The abnormalities in brachyolmia and SMDK imply abnormalities in growth-plate chondrocyte differentiation in the vertebrae and long bones. The more severe metatropic dysplasia phenotype, which by contrast with SMDK includes increased mineralization of the vertebrae, suggests possible involvement of both cartilage and bone. Recently, a role for TRPV4 in bone has been elucidated on the basis of the observation that inactivation of Trpv4 in mice leads to a defect in osteoclast differentiation and overmineralized bone.31 The relationship between these findings and the phenotypes and mechanisms resulting from altered TRPV4 remains to be defined.
The findings in this study demonstrate that mutations in TRPV4 produce a phenotypic spectrum of autosomal-dominant skeletal dysplasias from mild brachyolmia to SMDK to metatropic dysplasia. The overlapping phenotypic findings among the patients and the allelic series of mutations in TRPV4 suggest that these disorders comprise a new bone dysplasia family.
Supplemental Data
Supplemental Data include three figures and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.cov/Omim/
Acknowledgments
The authors thank the patients and their families for their participation, and thank Maryann Priore for her efforts on behalf of this study. This project was supported by National Institutes of Health grant HD22657 to D.K., D.C., R.S.L., and D.L.R. and by the Belgian Federal Government, the Flemish Government, and the Onderzoeksraad KU Leuven (Interuniversity Poles of Attraction Program, Prime Minister's Office IUAP Nr.3P4/23 and Excellentiefinanciering EF/95/010) to B.N. J.V. is a postdoctoral fellow of the Flemish Research Foundation (FWO).
References
- 1.Kozlowski K., Beemer F.A., Bens G., Dijkstra P.F., Iannaccone G., Emons D., Lopez-Ruiz P., Masel J., van Nieuwenhuizen O., Rodriguez-Barrionuevo C. Spondylo-metaphyseal dysplasia. (Report of 7 cases and essay of classification) Prog. Clin. Biol. Res. 1982;104:89–101. [PubMed] [Google Scholar]
- 2.Maroteaux P., Spranger J. The spondylometaphyseal dysplasias. A tentative classification. Pediatr. Radiol. 1991;21:293–297. doi: 10.1007/BF02018629. [DOI] [PubMed] [Google Scholar]
- 3.Kozlowski K., Maroteaux P., Spranger J.W. La dysostose spondylometaphisaire. Presse Med. 1967;75:2769–2774. [Google Scholar]
- 4.Kozlowski K. Spondylometaphyseal dysplasia. In: Kaufmann H.J., editor. Progress in Pediatric Radiology, Volume 4: Intrinsic Diseases of Bones. S. Karger; Basel: 1973. pp. 229–308. [Google Scholar]
- 5.Nural M.S., Diren H.B., Sakarya O., Yalin T., Dagdemir A. Kozlowski type spondylometaphyseal dysplasia: A case report with literature review. Diagn. Interv. Radiol. 2006;12:70–73. [PubMed] [Google Scholar]
- 6.Lachman R.S. Fifth Edition. Mosby-Year Book; St. Louis: 2007. Taybi and Lachman's Radiology of Syndromes, Metabolic Disorders and Skeletal Dysplasias. 1080–1081. [Google Scholar]
- 7.Maroteaux P., Spranger J., Wiedemann H.R. Arch. Kinderheilkd. 1966;173:211–226. [PubMed] [Google Scholar]
- 8.Beck M., Roubicek M., Rogers J.G., Naumoff P., Spranger J. Heterogeneity of metatropic dysplasia. Eur. J. Pediatr. 1983;140:231–237. doi: 10.1007/BF00443368. [DOI] [PubMed] [Google Scholar]
- 9.Kannu P., Aftimos S., Mayne V., Donnan L., Savarirayan R. Metatropic dysplasia: Clinical and radiographic findings in 11 patients demonstrating long-term natural history. Am. J. Med. Genet. A. 2007;143A:2512–2522. doi: 10.1002/ajmg.a.31941. [DOI] [PubMed] [Google Scholar]
- 10.Genevieve D., Le Merrer M., Feingold J., Munnich A., Maroteaux P., Cormier-Daire V. Revisiting metatropic dysplasia: Presentation of a series of 19 novel patients and review of the literature. Am. J. Med. Genet. A. 2008;146A:992–996. doi: 10.1002/ajmg.a.32191. [DOI] [PubMed] [Google Scholar]
- 11.Leet A.I., Sampath J.S., Scott C.I., MacKenzie W.G. Cervical spinal stenosis in metatropic dysplasia. J. Pediatr. Orthop. 2006;26:347–352. doi: 10.1097/01.bpo.0000214923.95314.ad. [DOI] [PubMed] [Google Scholar]
- 12.Shohat M., Lachman R., Rimoin D.L. Odontoid hypoplasia with vertebral cervical subluxation and ventriculomegaly in metatropic dysplasia. J. Pediatr. 1989;114:239–243. doi: 10.1016/s0022-3476(89)80789-9. [DOI] [PubMed] [Google Scholar]
- 13.Rock M.J., Prenen J., Funari V.A., Funari T.L., Merriman B., Nelson S.F., Lachman R.S., Wilcox W.R., Reyno S., Quadrelli R. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 2008;40:999–1003. doi: 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vriens J., Owsianik G., Janssens A., Voets T., Nilius B. Determinants of 4 alpha-phorbol sensitivity in transmembrane domains 3 and 4 of the cation channel TRPV4. J. Biol. Chem. 2007;282:12796–12803. doi: 10.1074/jbc.M610485200. [DOI] [PubMed] [Google Scholar]
- 15.Vriens J., Owsianik G., Fisslthaler B., Suzuki M., Janssens A., Voets T., Morisseau C., Hammock B.D., Fleming I., Busse R. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- 16.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedersen S.F., Owsianik G., Nilius B. TRP channels: An overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 18.Nilius B. Transient receptor potential (TRP) cation channels: Rewarding unique proteins. Bull. Mem. Acad. R. Med. Belg. 2007;162:244–253. [PubMed] [Google Scholar]
- 19.Nilius B., Owsianik G., Voets T., Peters J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 20.Clark K., Middelbeek J., van Leeuwen F.N. Interplay between TRP channels and the cytoskeleton in health and disease. Eur. J. Cell Biol. 2008;87:631–640. doi: 10.1016/j.ejcb.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Damann N., Voets T., Nilius B. TRPs in our senses. Curr. Biol. 2008;18:R880–R889. doi: 10.1016/j.cub.2008.07.063. [DOI] [PubMed] [Google Scholar]
- 22.Becker D., Blase C., Bereiter-Hahn J., Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J. Cell Sci. 2005;118:2435–2440. doi: 10.1242/jcs.02372. [DOI] [PubMed] [Google Scholar]
- 23.Pan Z., Yang H., Mergler S., Liu H., Tachado S.D., Zhang F., Kao W.W., Koziel H., Pleyer U., Reinach P.S. Dependence of regulatory volume decrease on transient receptor potential vanilloid 4 (TRPV4) expression in human corneal epithelial cells. Cell Calcium. 2008;44:374–385. doi: 10.1016/j.ceca.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedersen S.F., Nilius B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol. 2007;428:183–207. doi: 10.1016/S0076-6879(07)28010-3. [DOI] [PubMed] [Google Scholar]
- 25.Gaudet R. TRP channels entering the structural era. J. Physiol. 2008;586:3565–3575. doi: 10.1113/jphysiol.2008.155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 2008;4:372–379. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phelps C.B., Huang R.J., Lishko P.V., Wang R.R., Gaudet R. Structural analyses of the ankyrin repeat domain of TRPV6 and related TRPV ion channels. Biochemistry. 2008;47:2476–2484. doi: 10.1021/bi702109w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner J., Beighton P. Brachyolmia: An autosomal dominant form. Am. J. Med. Genet. 1994;49:308–312. doi: 10.1002/ajmg.1320490313. [DOI] [PubMed] [Google Scholar]
- 29.Myers B.R., Saimi Y., Julius D., Kung C. Multiple unbiased prospective screens identify TRP channels and their conserved gating elements. J. Gen. Physiol. 2008;132:481–486. doi: 10.1085/jgp.200810104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D'Hoedt D., Owsianik G., Prenen J., Cuajungco M.P., Grimm C., Heller S., Voets T., Nilius B. Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J. Biol. Chem. 2008;283:6272–6280. doi: 10.1074/jbc.M706386200. [DOI] [PubMed] [Google Scholar]
- 31.Masuyama R., Vriens J., Voets T., Karashima Y., Owsianik G., Vennekens R., Lieben L., Torrekens S., Moermans K., Vanden Bosch A. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.