

Abstract
We have identified a consanguineous Pakistani family where oligodontia is inherited along with short stature in an autosomal-recessive fashion. Increased bone density was present in the spine and at the base of the skull. Using high-density single-nucleotide polymorphism microarrays for homozygosity mapping, we identified a 28 Mb homozygous stretch shared between affected individuals on chromosome 11q13. Screening selected candidate genes within this region, we identified a homozygous nonsense mutation, Y774X, within LTBP3, the gene for the latent TGF-β binding protein 3, an extracellular matrix protein believed to be required for osteoclast function.
Main Text
Congenital tooth agenesis of one or more teeth, which may be either hypodontia (agenesis of fewer than six teeth) or oligodontia (agenesis of six or more teeth), also known as selective tooth agenesis (STHAG), is the most common abnormality of human dentition. It affects permanent rather than deciduous dentition. Tooth agenesis may be caused by local trauma, chemotherapy, or radiotherapy, or it may occur as part of a systemic genetic syndrome or as an independent oral trait shown to be associated with mutations in several genes, including MSX1 (Selective Tooth Agenesis 1 [SHTAG1]; MIM 106600)1 and PAX9 (STHAG3; MIM 604625).2 Additional loci are believed to exist on chromosome 16q12.1 (STHAG2; MIM 602639)3 and elsewhere (STHAG5; MIM 610926,4 10q11.2; STHAG4; MIM 150400, locus not yet determined).5
In the present study, a single consanguineous family from the Kasur district of Punjab province, Pakistan, was ascertained. Approval from the institutional ethical review board was obtained for the study, and informed written consent was obtained for each family member. In this family, the dual traits of oligodontia and short stature appear to be inherited as an autosomal-recessive trait, and the unaffected parents are second cousins with ten offspring, three of whom are now deceased (Figure 1). Four of the living affected members, two male (II:6; II:7) and two female (II:2; II:3), presented with absence of many of the permanent teeth (Figure 2A). Dental history was obtained and revealed the presence of deciduous dentition, and no history of extensive extractions was reported; however, past dental records were not available for verification. In the studied cases (two males), apparent congenital oligodontia (absence of six or more teeth, including third molars) was demonstrated by radiological examination; twenty-three permanent teeth were missing in the most affected individual (II:7), and eighteen permanent teeth were absent in the second case (II:6), where fully formed impacted upper third molars were identified bilaterally and fully formed unerupted unilateral lower second and third molars were seen. Neither completely nor incompletely formed impacted teeth were noted. Alveolar bone was absent where the teeth were missing (i.e., because most teeth are missing in the affected individuals, this encompasses the majority of the mandible). The bone was otherwise healthy and robust looking. One non-affected individual studied (II:8) had complete, healthy dentition and an impacted fully formed lower third molar (Figure 2A).
Figure 1.
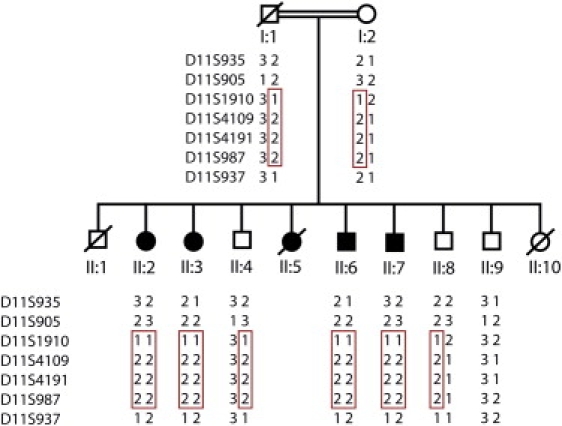
Pedigree of the Pakistani Oligodontia Family
Affected family members are indicated by shaded symbols, and deceased members are indicated by symbols with a line through them. Microsatellite markers D11S935, D11S905, D11S1910, D11S4109, D11S4191, D11S987, and D11S937 were genotyped in all available family members. Haplotypes are shown, and the disease haplotype is indicated with a red box. The haplotype for the deceased father was reconstructed from genotype information from the mother and offspring.
Figure 2.
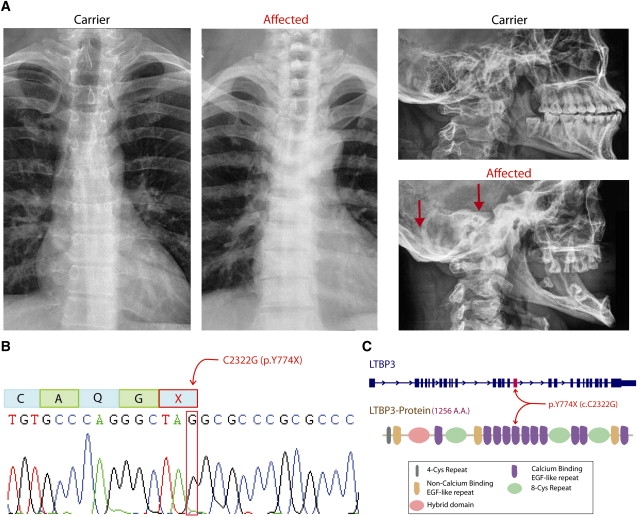
Radiological Findings and Details of LTBP3 Mutation
(A) Chest and skull X-rays of an unaffected (carrier) individual (individual II:8, Figure 1; age 25 years) and an affected individual (individual II:6, Figure 1; age 30 years) show increased bone density in the spine and the bottom of the skull (indicated by red arrows) in the affected individual.
(B) An electropherogram from affected individual II:6, with the position of the C-to-G mutation in LTBP3 indicated by a red box. Above this is indicated the effect on amino acid sequence, where the TAC codon for tyrosine is mutated to a TAG stop codon.
(C) The position of the Y774X mutation is shown relative to the genomic organization of LTBP3 (above) and the domain organization of the LTBP3 protein.
The affected individuals also presented with short stature (the tallest [II:6] was 4 ft 11 in, or 150 cm), with normal limb/torso ratios and proximal/distal limb ratios. Head circumference and intelligence were all within the normal range; varying degrees of scoliosis were observed, but no other skeletal anomalies were observed. A fifth affected female (II:5) died of an unrelated problem at age 17.
After the exclusion of MSX1 and PAX9 by direct sequencing, DNA samples of the four living affected individuals and one unaffected individual were analyzed with the Affymetrix GeneChip Mapping 500K array, NspI chip, which allowed us to genotype ∼260,000 SNPs. Sample processing, labeling, and hybridization were performed in accordance with the manufacturer's instructions (Affymetrix Mapping 500K Assay Manual). The arrays were scanned with a GeneChip Scanner, and the data were processed with GeneChip® Operating Software (GCOS) and GeneChip® Genotyping Analysis (GTYPE) Software (version 3.0.2) for the generation of SNP allele calls. A single large homozygous stretch of between 41.381 and 69.263 Mb (UCSC May 2004) on chromosome 11 was identified as common between the four affected individuals but not the unaffected individual. Microsatellite markers were genotyped for all eight available family members (mother and seven of her offspring) across this region, confirming the shared autozygosity. Linkage analysis using the easyLINKAGE plus v5.02 platform6 gave a maximum lod score of 2.85 for markers D11S1910, D11S4109, D11S4191, and D11S987 (46.15 Mb to 67.65 Mb, UCSC Genome Browser, May 2004). The 28 Mb autozygous region spans the centromere, including bands 11p12–q13.3, and contains ∼440 RefSeq annotated genes (UCSC Genome Browser, March 2006). We selected the following genes from this region for screening for coding mutations by direct automated sequencing of genomic DNA on the basis of known function and expression profiles: FGF4 (MIM 164980), FGF19 (MIM 603891), FIBP (MIM 608296), LRP5 (MIM 603506), EHD1(MIM 605888), FOSL1 (MIM 136515), and LTBP3 (MIM 602090) (FGF3 [MIM 164950], which lies just distal to the autozygous region, was also sequenced). Within LTBP3, the gene encoding latent transforming growth factor-β (TGF-β) binding protein-3, we identified the nucleotide substitution c.2322C > G, resulting in the nonsense mutation Y744X (Figure 2B). This sequence change was confirmed in both forward and reverse directions and was genotyped by the sequencing of all available family members (Table 1). No other homozygous changes were identified in the other candidate genes analyzed. This base substitution results in the creation of a number of restriction endonuclease sites, including NarI, EheI, and BbeI. The base substitution is not present in any SNP databases. We screened 240 unrelated unaffected individuals of Pakistani origin by sequencing and did not detect this mutation. To our knowledge, this is the first report of a mutation in LTBP3 associated with a human disease, or indeed any Latent TGF-β Binding Protein genes.
Table 1.
Height and Head Circumference Data for the Oligodontia Family
| Family Member | Age (Years) | Oligodontia (Y/N) | LTBP3 Genotype | Height inches (cm) | OFC [Inches (cm)] |
|---|---|---|---|---|---|
| I:1. Fathera | 72a | N | c/g | 61b (154.9) | nm |
| I:2. Mother | 63 | N | c/g | 57 (145.8) | 22 (55.9) |
| II:2. Daughter | 41 | Y | g/g | 51 (129.5) | 21 (53.3) |
| II:3. Daughter | 39 | Y | g/g | 56 (142.2) | 21.5 (54.6) |
| II:4. Son | 34 | N | c/g | 62 (157.5) | 22 (55.9) |
| II:6. Son | 30 | Y | g/g | 59 (149.9) | 22.4 (56.9) |
| II:7. Son | 28 | Y | g/g | 56 (142.2) | 22 (55.9) |
| II:8. Son | 25 | N | c/g | 64 (162.6) | 22.8 (57.9) |
| II:9. Son | 23 | N | c/c | 63.5 (161.3) | 21.5 (54.6) |
Height and occipito-frontal head circumference (OFC) for family members (see Figure 1), affected status, and carrier status according to sequence analysis of c.2322C > G (p.Y744X) at LTBP3. Mean height for Pakistanis is taken as 66.14 ± 3.1 for adult males (mean age 33 years ± 12) and 59.8 ± 2.8 for adult females (mean age 40 years ± 16) (from Khan et al., 200811). nm = not measured.
Deceased; age at death.
As reported by family.
Quantitative RT-PCR analysis of LTBP3 with lymphoblast-derived cDNA from three affected individuals and one control individual was performed as follows. Reverse transcription was performed from 2 μg of total RNA with random hexamer primers and SuperScript III Reverse Transcriptase (RT) (Invitrogen, Carlsbad, CA), in accordance with the manufacturer's instructions. Real-time PCR was performed with the SYBR Green Dye I chemistry (Applied Biosystems, Foster City, CA) on the Applied Biosystems 7300 RT-PCR System with primers spanning exon 6 to exon 7 of LTBP3 (forward primer: 5′-CTCAACAACCCTGGCTCCTA; reverse primer: 5′- CGGAAACACAGGCTCTTCTC) at 300 nM final concentration. The product size was 110 bp. As a reference gene for normalization of LTBP3 expression, we used the housekeeping gene HPRT (MIM 308000). The reaction mixture contained 1× Power SYBR Green PCR Master Mix and the first-strand RT product at a final dilution of 1:100, plus AmpliTaq Gold DNA polymerase. The relative standard curve method was used for evaluating amplification efficiencies of primers used in the study and calculating the quantity of LTBP3 mRNA relative to HPRT and calibrating it against the control sample. The experimental data were analyzed with the 7500 Software v2.0.1. All real-time RT-PCR reactions were performed in triplicate, and results obtained from three reactions (n = 3) were used for calculating mean values and standard deviations shown in the histogram (Figure 3). Amplification for family members II:2, II:3, and II:6 shows the same size PCR product but an approximately 5-fold reduction in mRNA levels associated with the mutated allele, suggesting that nonsense-mediated decay is occurring (Figure 3). The change would also result in premature truncation of any translated protein product within the fifth out of 11 calcium-binding epidermal growth factor (EGF)-like domains (cbEGF).
Figure 3.
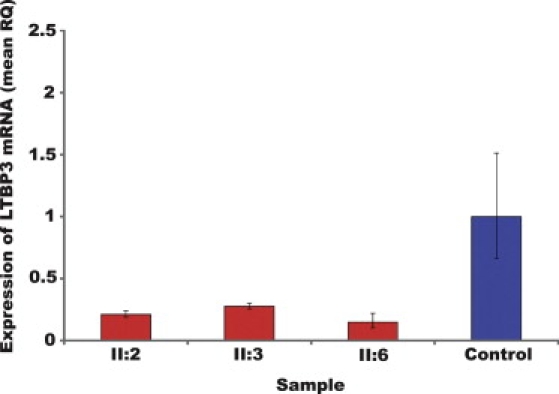
Summary of LTBP3 mRNA Expression
Summary of LTBP3 expression as quantified by qPCR with mRNA extracted from lymphoblastoid cell lines of three affected individuals (II:2, II:3, II:6) and a control. Data indicate a significantly lower quantity of LTBP3 mRNA in all three patients than in the control, as a result of nonsense-mediated decay. Standard error bars are shown.
LTBP3 is an extracellular matrix (ECM) protein, first identified through its similarity to human fibrillin-1 epidermal growth factor-like repeats. It is coexpressed with TGF-β and also immunoprecipitates with the TGF-β1 precursor.7 LTBPs are required for targeting and activation of TGF-β, and LTBP3 in particular regulates the bioavailability of TGF-β in chondrocytes.8 In studies of ltbp3 null mice, the adult knockout mice were more than 50% smaller than sex-matched wild-type littermates.8 Thoracic kyphosis and distorted ribcage were observed, along with changes in skull shape as a result of ossification of cranial base synchondroses. The animals also developed osteosclerosis and osteoarthritis. No dental abnormalities comparable to the human phenotype were reported, although the upper jaw appeared shortened, and thus the lower incisors extended outside the upper incisors. In addition, an osteopetrosis-like phenotype, possibly due to a bone-resorption defect resulting from decreased TGF-β levels in cartilage and bone, was reported.9
Studies of two members of the Pakistani family (individuals II:7 and II:8) were carried out with dual-energy X-ray absorptiometry (DEXA), as well as standard anterior and posterior X-rays of skull, chest and spine, pelvis, hands, and knees. High bone mineral density (BMD) was observed in the spine of a homozygous affected (II:7: L2-L4 BMD = 1.353 g/cm2; z-score = 1.14; age 25 years [z score indicates BMD relative to reference controls]) versus a heterozygous unaffected family member (II:8: L2-L4 BMD = 0.9503 g/cm2; z-score = −1.05; age 28 years). Similar measures for the femur showed no BMD differences between one affected and one unaffected family member. In addition to the DEXA measurements, radiographs suggested a higher spinal bone density for affected individuals than for unaffected individuals as well as an increase in bone density at the base of the skull (Figure 2A). The vertebrae in the affected individual shown (Figure 2A) appear wider, but this is not indicated in X-ray images from a second affected male. These bone-density and radiographic findings are reminiscent of, although not identical to, the bone phenotype observed in ltbp3 null mice. Similar to the situation in the mice, there were no apparent differences in bone density in the long bones or ribs. We also examined radiographic images for evidence of osteoarthritis (as reported in ltbp3 null mice aged 6–9 months8) by looking for signs of osteophytes, subchondral cysts or sclerosis or for evidence of joint space narrowing indicated by asymmetry at the knee joint between the medial and lateral sides10; however, we found no such evidence. The findings in mice and our patients suggest an important role for LTBP3-mediated transcription in development of the axial skeleton.
Height and occipito-frontal head circumference measurements for living family members suggests that short stature is also inherited with the same mutation (see Table 1); however, it is not clearly recessive because in several cases the height of the affected individuals is not significantly lower than that of some of the carriers or unaffected family members, or the Pakistani national average.11 The possibility that genetic variation at the LTBP3 locus may exert an incompletely dominant or codominant effect on height cannot be excluded, although the significance of the height differences cannot be shown with such a small sample size. Although recently published genome-wide linkage or association studies show little support for the idea that a common locus determines adult height around 11q13,12–16 rare, single-gene defects causing short stature are numerous in the literature.17
A number of genes in the TGF-β signaling pathway have been implicated in skeletal developmental disorders, emphasizing the important role for this pathway in bone growth. For instance, mutations in the TGF-β gene, TGFB1 (MIM 190180), cause the autsosomal-dominant Camurati-Engelmann disease (CED; MIM 131300)—a bone disease that presents as increased bone density of the long bones of the arms and legs and occasionally in the skull and hip bones and disproportionately long limbs and often leads to scoliosis and joint contractures.18 Mutations in the TGF-β receptor genes, TGFBR1 (MIM 190181) and TGFBR2 (MIM 190182), cause Loeys-Dietz syndrome (MIM 609192)—an autosomal-dominant aortic aneurysm syndrome with skull and skeletal developmental abnormalities, frequently including marfanoid habitus, kyphoscoliosis, craniosynostosis, and micrognathia or microretrognathia.19
In summary, to our knowledge, LTBP3 is the third gene identified as causing oligodontia or selective tooth agenesis (STHAG), but the first autosomal-recessive STHAG gene, and the first such gene also to show a possible link to deficit in stature. LTBP3 is now added to the growing list of members of the TGF-β signaling pathway that have been linked with disorders of human bone and skeletal development.
Acknowledgments
We wish to indicate our deep gratitude to the Pakistani family members for their participation. In addition, we wish to acknowledge the assistance of Dr. Abbas Cheema and Mr. H.M. Ashfaq Bhatti. A.N. is supported by a Canadian Institutes of Health Research (CIHR) Collaborative Graduate Training Program in Molecular Medicine. C.W. is supported by a Centre for Addiction and Mental Health Postdoctoral Fellowship. J.B.V. is supported by a National Alliance for Research on Schizophrenia and Depression (NARSAD) Independent Investigator Award.
References
- 1.Vastardis H., Karimbux N., Guthua S.W., Seidman J.G., Seidman C.E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 1996;13:417–421. doi: 10.1038/ng0896-417. [DOI] [PubMed] [Google Scholar]
- 2.Stockton D.W., Das P., Goldenberg M., D'Souza R.N., Patel P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000;24:18–19. doi: 10.1038/71634. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad W., Brancolini V., Faiyaz ul Haque M., Lam H., ul Haque S., Haider M., Maimon A., Aita V.M., Owen J., Brown D. A locus for autosomal recessive hypodontia with associated dental anomalies maps to chromosome 16q12.1. Am. J. Hum. Genet. 1998;62:987–991. doi: 10.1086/301799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu W., Wang H., Zhao S., Zhao W., Bai S., Zhao Y., Xu S., Wu C., Huang W., Chen Z. The novel gene locus for agenesis of permanent teeth (He-Zhao deficiency) maps to chromosome 10q11.2. J. Dent. Res. 2001;80:1716–1720. doi: 10.1177/00220345010800080701. [DOI] [PubMed] [Google Scholar]
- 5.Woolf C.M. Missing maxillary lateral incisors: A genetic study. Am. J. Hum. Genet. 1971;23:289–296. [PMC free article] [PubMed] [Google Scholar]
- 6.Lindner T.H., Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 7.Yin W., Smiley E., Germiller J., Mecham R.P., Florer J.B., Wenstrup R.J., Bonadio J. Isolation of a novel latent transforming growth factor-beta binding protein gene (LTBP-3) J. Biol. Chem. 1995;270:10147–10160. doi: 10.1074/jbc.270.17.10147. [DOI] [PubMed] [Google Scholar]
- 8.Dabovic B., Chen Y., Colarossi C., Zambuto L., Obata H., Rifkin D.B. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice: A role for Ltbp in TGF-beta presentation. J. Endocrinol. 2002;175:129–141. doi: 10.1677/joe.0.1750129. [DOI] [PubMed] [Google Scholar]
- 9.Dabovic B., Levasseur R., Zambuto L., Chen Y., Karsenty G., Rifkin D.B. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone. 2005;37:25–31. doi: 10.1016/j.bone.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 10.Vignon E., Conrozier T., Piperno M., Richard S., Carrillon Y., Fantino O. Radiographic assessment of hip and knee osteoarthritis. Recommendations: Recommended guidelines. Osteoarthritis Cartilage. 1999;7:434–436. doi: 10.1053/joca.1999.0235. [DOI] [PubMed] [Google Scholar]
- 11.Khan A., Haq F.U., Pervez M.B., Saleheen D., Frossard P.M., Ishaq M., Hakeem A., Sheikh H.T., Ahmad U. Anthropometric correlates of blood pressure in normotensive Pakistani subjects. Int. J. Cardiol. 2008;124:259–262. doi: 10.1016/j.ijcard.2006.12.040. [DOI] [PubMed] [Google Scholar]
- 12.Perola M., Sammalisto S., Hiekkalinna T., Martin N.G., Visscher P.M., Montgomery G.W., Benyamin B., Harris J.R., Boomsma D., Willemsen G. Combined genome scans for body stature in 6,602 European twins: Evidence for common Caucasian loci. PLoS Genet. 2007;3:e97. doi: 10.1371/journal.pgen.0030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gudbjartsson D.F., Walters G.B., Thorleifsson G., Stefansson H., Halldorsson B.V., Zusmanovich P., Sulem P., Thorlacius S., Gylfason A., Steinberg S. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008;40:609–615. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- 14.Lettre G., Jackson A.U., Gieger C., Schumacher F.R., Berndt S.I., Sanna S., Eyheramendy S., Voight B.F., Butler J.L., Guiducci C. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sammalisto S., Hiekkalinna T., Schwander K., Kardia S., Weder A.B., Rodriguez B.L., Doria A., Kelly J.A., Bruner G.R., Harley J.B. Genome-wide linkage screen for stature and body mass index in 3.032 families: Evidence for sex- and population-specific genetic effects. Eur. J. Hum. Genet. 2008;17:258–266. doi: 10.1038/ejhg.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmert M.R., Hirschhorn J.N. Genetic approaches to stature, pubertal timing, and other complex traits. Mol. Genet. Metab. 2003;80:1–10. doi: 10.1016/s1096-7192(03)00107-0. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita A., Saito T., Tomita H., Makita Y., Yoshida K., Ghadami M., Yamada K., Kondo S., Ikegawa S., Nishimura G. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat. Genet. 2000;26:19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 19.Loeys B.L., Chen J., Neptune E.R., Judge D.P., Podowski M., Holm T., Meyers J., Leitch C.C., Katsanis N., Sharifi N. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]