

Abstract
Many patients suffering from developmental disorders harbor submicroscopic deletions or duplications that, by affecting the copy number of dosage-sensitive genes or disrupting normal gene expression, lead to disease. However, many aberrations are novel or extremely rare, making clinical interpretation problematic and genotype-phenotype correlations uncertain. Identification of patients sharing a genomic rearrangement and having phenotypic features in common leads to greater certainty in the pathogenic nature of the rearrangement and enables new syndromes to be defined. To facilitate the analysis of these rare events, we have developed an interactive web-based database called DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources) which incorporates a suite of tools designed to aid the interpretation of submicroscopic chromosomal imbalance, inversions, and translocations. DECIPHER catalogs common copy-number changes in normal populations and thus, by exclusion, enables changes that are novel and potentially pathogenic to be identified. DECIPHER enhances genetic counseling by retrieving relevant information from a variety of bioinformatics resources. Known and predicted genes within an aberration are listed in the DECIPHER patient report, and genes of recognized clinical importance are highlighted and prioritized. DECIPHER enables clinical scientists worldwide to maintain records of phenotype and chromosome rearrangement for their patients and, with informed consent, share this information with the wider clinical research community through display in the genome browser Ensembl. By sharing cases worldwide, clusters of rare cases having phenotype and structural rearrangement in common can be identified, leading to the delineation of new syndromes and furthering understanding of gene function.
Main Text
Advances in molecular cytogenetic techniques and sequencing of the human genome now enable chromosome rearrangements to be analyzed at an unprecedented level of accuracy. The resolution of conventional Giemsa-banded chromosome analysis is approximately 5–10 Mb, whereas that of a typical high-density genomic array is approximately 100 kb or less, resulting in an at least 50-fold improvement in sensitivity. With high-resolution genomic-array analysis, disorders of chromosome imbalance (copy number change) can now be identified with such precision that mapping the rearrangements onto the reference sequence of the human genome becomes a realistic proposition.
Array comparative genomic hybridization (array-CGH) and genomic copy-number analysis with SNP genotyping arrays are proving particularly effective for the investigation of patients with developmental delay, learning disability, dysmorphic features, and/or congenital anomalies and are identifying the probable underlying cause of the disease phenotype in approximately 15% of previously undiagnosed cases.1–5 Moreover, a genomic basis to several later-onset disorders, e.g., early-onset Alzheimer disease with amyloid angiopathy (EOAD)6 and adult-onset autosomal-dominant leukodystrophy7, has now been defined. However, copy-number changes in many patients are novel or extremely rare, such that uncertainty remains as to whether the aberration is pathogenic or simply a benign variant. Identification of additional patients who share a region of genomic deletion or duplication and have phenotypic features in common allows greater certainty to be given to the pathogenic nature of the rearrangement and delineation of new syndromes.
Several studies have highlighted the presence of large numbers of deletions, insertions, and duplications, ranging from a few kilobases to several megabases in size, in the normal population.8,9 For example, using the complementary technologies of single-nucleotide polymorphism (SNP) genotyping arrays and clone-based comparative genomic hybridization, Redon et al10 generated a global map of copy-number variable regions (CNVRs) in the human genome and found 1447 CNVRs covering approximately 12% of the human genome. As more studies are completed and as the resolution of genomic array analysis increases, more CNVRs are being discovered. There are currently more than 6000 copy-number-variant loci listed in the Database of Genomic Variants (DGV). These regions contain several hundreds of genes, disease loci, and segmental duplication regions. Because patients also have these normal copy-number variants it has become a challenge to identify which changes are normal variants and which are likely to be associated with a phenotype.
Although many clinical centers are now applying genomic microarray technology to investigate patients with developmental delay, learning disability, and congenital malformation,11 the sporadic nature and rarity of the majority of these cases limits the ability of the individual clinician to interpret the molecular findings from genome-wide array analysis. Thus, there is a great need for international collaboration in the reporting and cataloguing of genotype-phenotype correlations such that clusters of individuals sharing similar genomic rearrangements and phenotypes can be identified. This will not only facilitate diagnosis and genetic counseling but also improve our understanding of gene function and disease.
The DECIPHER database (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources) and project (Figure 1) was initiated in 2004 with the general aim of providing a clinical and research tool to (1) aid in the interpretation of data from genomic microarray analysis, e.g., the differentiation between pathogenic and polymorphic copy-number changes; (2) utilise the human genome map via the Ensembl genome browser to define genes involved in a specific microdeletion, microduplication, translocation, or inversion; and (3) facilitate collaboration between clinical geneticists and molecular cytogeneticists to accelerate progress in the delineation of new syndromes and of gene function.
Figure 1.
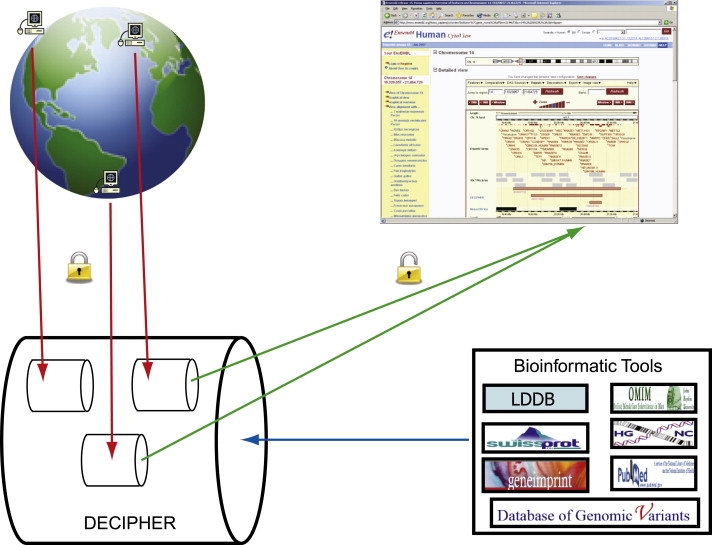
Overview of DECIPHER
The DECIPHER database is accessed via the World Wide Web so that patient phenotypic and genomic information can be deposited in a secure and private project areas by consortium members. Interaction with bioinformatic tools allows clinicians and scientists to interpret the genomic information. With informed consent, data become viewable within the Ensembl genome browser so that patients with overlapping copy-number changes can be readily identified.
The acronym DECIPHER was chosen for the database because the word means “To give the key to, to discover the meaning of [something obscure and perplexing].”12 DECIPHER was granted Multi-Centre Research Ethics Committee (MREC) approval (04/MRE05/50) in the UK. The project has been made possible by several independent but interlinked developments, including the Human Genome Mapping Project, bioinformatic integration of genomic information in genome browsers, and advances in molecular cytogenetics. The Human Genome Mapping Project provided a finished human genome sequence in October 200413,14, and this acts as a reference sequence, or assembly map, onto which chromosome rearrangements and the order and position of genes and predicted genes can be placed. Crucial to the operation of DECIPHER was the development of the Ensembl genome browser15 which allows users to select and view an annotated segment of the human genome map and connect to other relevant resources via hyperlinks. The clone resources developed in the Human Genome Mapping Project, the SNP Consortium Project, and indeed, the sequence itself have provided cytogeneticists with a new suite of molecular tools with which to analyze chromosome rearrangements. The use of fluorescence in situ hybridization (FISH) and, in particular, the introduction of genomic array analysis (“molecular karyotyping”) using array-CGH or high-density SNP genotyping has revolutionized the identification of subtle (and hitherto unidentifiable) cytogenetic imbalances.
Contributing to the DECIPHER database is a Consortium, comprising an international network of academic departments of clinical genetics now numbering more than 100 centers and having uploaded more than 2000 cases (current statistics can be found on the DECIPHER homepage). Each contributing center has a nominated clinical geneticist (with expertise in dysmorphology) and a nominated molecular cytogeneticist who are jointly responsible for data entry for their center. Each center maintains control of its own patient data (which are password protected within the center's own DECIPHER project) until patient consent is given to allow anonymous genomic and phenotypic data to become viewable within Ensembl. Once data are shared, consortium members are able to gain access to the patient report and contact each other to discuss patients of mutual interest, thus facilitating the delineation of new microdeletion and microduplication syndromes. With patient consent, positional genomic information together with a brief description of the associated phenotype becomes viewable without password protection via the DECIPHER track in Ensembl. This is of benefit not only to clinicians advising patients with similar findings but also to researchers working on the specific disorders (e.g., congenital heart disease or cleft lip) seen in that patient or working on the role of genes contained within the aberration.
Key Features of DECIPHER
(1) DECIPHER integrates seamlessly with the Ensembl genome browser and interrogates the current version of the human genome assembly displayed in the Ensembl genome browser. Because DECIPHER is a dynamic system, each time it is interrogated the most recent data with regard to gene content are returned into the patient report. Ensembl itself builds all its annotated genes and associated features onto the most recent version of the human genome assembly created by the NCBI (current assembly NCBI-36). The annotation of genes and their features are continually being reviewed, refined, and updated, culminating bimonthly in a new version of the annotation released onto the Ensembl website. New features and types of data are continually and routinely added via the distributed annotation system (DAS16), which allows a set of data to be dropped into place and viewed across the genome as a new feature (e.g., copy-number variation (CNV) data). This approach makes the whole process configurable and allows rapid updates to data to be made available.
(2) DECIPHER links to other genetic and medical databases, including HUGO Gene Nomenclature Committee (HGNC), On-line Mendelian Inheritance in Man (OMIM), PubMed, GeneReviews, Ensembl genes, Swiss-Prot, and a frequently updated list of emerging bioinformatics databases. DECIPHER not only displays detected aberrations in relation to the genome sequence but has also been programmed to allow the clinician or researcher to rapidly obtain clinically relevant information (publications) about the genes located in these regions.
(3) DECIPHER is designed to work with any number of technologies where position data can be mapped onto the reference sequence. For DECIPHER to interact directly with the Ensembl genome browser, positional data regarding the probes used for analysis must first be mapped onto the reference sequence. DECIPHER can be configured to utilize any molecular method (e.g., FISH, MAPH, MLPA, PCR, array-CGH, or SNP-genotyping) where the probes or SNPs are positioned on the reference sequence or simply base-pair position (sequence) itself. Any structural rearrangement defined in this way, including copy-number changes, translocations, and inversions, can be catalogued within DECIPHER.
(4) DECIPHER uses a restricted ontology of phenotype terms and speciically the well-known hierarchy of phenotype terms developed for the The Baraitser-Winter Neurogenetics Database. In this way, clinicians in different centers use a common set of terms, which facilitates consistent phenotype description and enables efficient data sharing and database searching.
(5) DECIPHER facilitates the characterization of copy-number changes. A major problem in the identification of potentially pathogenic changes is the large number of normal variants that are identified, particularly as array resolution increases. DECIPHER includes a feature graph tool that displays patient copy-number changes together with data from the DGV as well as variants identified in a selection of the major studies of copy-number variation in normal individuals. Together with the analysis of parental samples (see analysis of trios, number 8 below), each copy-number change in the patient can then be classified as de novo, CNV (normal variant), or familial variant. Only de novo changes and familial variants are then displayed, with consent, in Ensembl.
(6) DECIPHER provides detailed gene lists for rearrangements. The gene lists and gene displays generated by DECIPHER are filtered via HGNC to eliminate redundancy caused by synonyms and to ensure that DECIPHER is using currently approved gene names. Genes included in the OMIM Morbid database, i.e., those of known importance in human disease, are denoted with the suffix M so that the clinician is immediately alerted to the need to explore their potential clinical significance. Similarly, imprinted genes listed at the website GeneImprint are denoted by the suffix I because, in this instance, an inherited deletion or duplication, although present in a normal parent, can be potentially pathogenic.
(7) DECIPHER provides a comprehensive search facility. All of the consented data held within the DECIPHER database are searchable. Features that can be queried include chromosomal band, genomic position, chromosomes, and phenotypes or a combination of these. DECIPHER members can also use this search function to interrogate nonconsented data within their own project group. (Figure 2).
Figure 2.
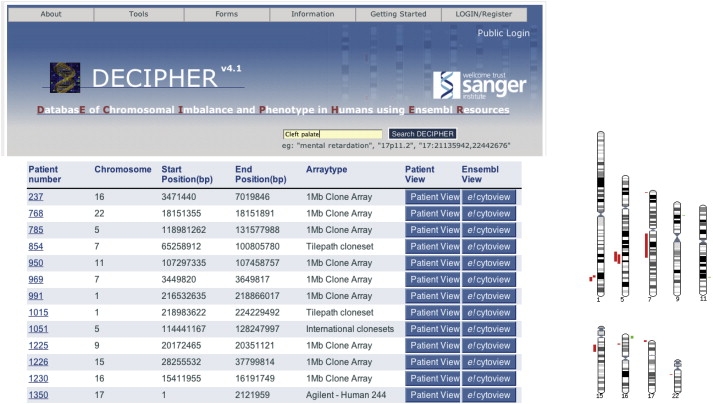
Searching the Genome by Phenotype
Using the phenotype search term “cleft palate” generates an ideogram (below) and list of all the consenting patients that DECIPHER knows to have cleft palate and displays their genomic aberrations. Two clusters, one on 1q and the other on 5q, are seen together.
(8) DECIPHER includes novel software to assist in the analysis of trios (DNA samples from a patient and both parents). DECIPHER collects information from array analysis of both parents and displays this in relation to copy-number changes found in the patient. This enables the user to identify whether a genomic aberration identified in a patient is of parental origin or has occurred as a de novo event.
(9) DECIPHER provides advanced text-mining tools for gene prioritization and for genotype-phenotype correlation. Identifying which gene or genes in a deleted or duplicated region may be responsible for features of the phenotype involves considerable literature and database searching. DECIPHER includes advanced text-mining tools that order genes in the region according to their likelihood of being associated in the literature with a phenotypic feature or group of features.17,18 In addition, because DECIPHER is directly linked to Ensembl, clinicians and researchers can identify and query genes that are located in affected regions of the genome by accessing gene-specific databases.
(10) DECIPHER generates both detailed clinical reports and summary family reports. Once all the phenotype and genotype data of a patient have been entered into DECIPHER, two types of printable reports are directly available from the website. The first report comprises a summary of the full clinical phenotypic description and an overview of genes affected by the aberration. This report can be used as a clinical report and includes the karyotype and ideogram of the affected chromosome(s) and any uploaded patient images. Alternatively, a simplified version of the full clinical report can be printed out for counseling patients and families.
(11) In order to protect patient privacy, data in DECIPHER are served over an encrypted SSL (secure socket layer) connection, similar to a security level employed by banks and financial institutions. Clinical photographs are an optional element of DECIPHER. When consent is given for these to be held in DECIPHER, they are password protected so that they are only accessible to logged-in members of the consortium. Photographs are digitally watermarked for additional security. Only cases with documented full informed consent are visible within the Ensembl genome browser. Nonconsented data are viewable only by the submitting center. Public access to consented data is restricted to a basic report identifying copy-number changes and the phenotype but not the identity of the submitting center. Requests for further patient information are made to the DECIPHER administration, who pass on the request to the submitting center.
(12) DECIPHER maintains a series of syndrome reports. The syndromes pages within DECIPHER provide a single, curated resource of information and web links. As shown in Figure 3, for each syndrome entity DECIPHER provides the following: a brief clinical synopsis; information regarding the size and origin of a deletion or duplication; an ideogram of the location of the deletion or duplication on the relevant chromosome; a list of the genes contained within the aberrant interval; a direct clickable link to a visualization of the deletion or duplication in Ensembl; an up-to-date publication reference list and; a link to relevant support groups and further information (e.g., GeneReviews). DECIPHER Syndromes is supported by a panel of expert advisors (see Acknowledgments), each responsible for reviewing and updating the entry for a specific syndrome on an annual basis.
Figure 3.

DECIPHER Syndrome Report for 8p23.1 Deletion Syndrome
Detailed information about known syndromes is maintained by expert advisors within DECIPHER. The copy-number change is displayed on a ideogram (1), in a genome browser showing normal copy-number variants (2), and with links to OMIN (3) and Ensembl (4).
Case Studies
Cases are entered into DECIPHER from standard web browsers, and the data are mapped directly to the latest human genome assembly via the Ensembl genome browser. For consenting patients, the aberrant region is displayed within Ensembl with phenotype information and any other similar cases within the database, as well as with other genomic features such as gene content, segmental duplication, and regions of normal copy-number variation.
Case Study 1 (DECIPHER 00000797)—Importance of Knowing the Gene Content
A 28-year-old woman presented to the genetics clinic for diagnosis. She had mild to moderate learning difficulties and dysmorphic facial features (See Figure 4A). After an uneventful pregnancy, she was born with bilateral talipes, which required surgery in infancy. As a young child, she had been reviewed by pediatricians and geneticists and had been given a diagnosis of “peroneal muscular atrophy.”
Figure 4.
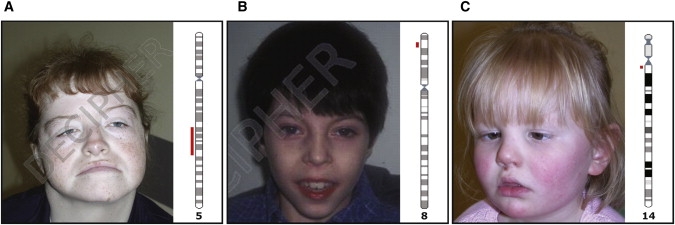
Patient Images
(A) Case 1.
(B) Case 2.
(C) Case 3.
At the clinic she reported experiencing fatigue and lassitude. She appeared anaemic, but there was no history of menorrhagia, blood loss, or dietary deficiency. Investigations revealed a severe iron-deficiency anaemia, Hb 7.1g/dl, and ferritin < 1, and chromosome analysis revealed a visible deletion of 5q21-q22. FISH analysis refined the breakpoints, and the deletion was then uploaded into DECIPHER. Genes located within the deleted region included the APC gene (MIM 611731). Germline mutations of the APC gene have been found to be responsible for Familial adenomatous polyposis (FAP [MIM 175100])19,20, a cancer-predisposition syndrome in which hundreds to thousands of precancerous colonic polyps develop during adolescence. Most mutations in the APC gene are protein truncating and are spread throughout the coding region of the gene. However, exonic deletions and whole-gene deletions are also described.20,21 In view of the high risk of colorectal malignancy in adults with FAP, young people with a diagnosis of FAP are usually enrolled in a colorectal surveillance program from the ages of 10–12 years and undergo prophylactic colectomy in early adult life.22
As a result of these findings, the patient was referred urgently to a colorectal surgeon for further investigation and management. Colonoscopy revealed relative sparing of the rectum, but numerous adenomas were seen throughout the colon, and there was a large sessile lesion at the splenic flexure. Furthermore, upper GI endoscopy demonstrated some gastric fundal and duodenal polyps. These findings confirmed the diagnosis of FAP, which had been predicted from the molecular cytogenetic results. On account of her high risk of colorectal malignancy, the patient was admitted for a colectomy with ileorectal anastamosis, and periodic surveillance of the upper GI tract and rectum was instituted. Sadly, despite this intervention, she died 18 months later from metastatic carcinoma.
Case Study 2 (DECIPHER 00000128)—A Rare Microdeletion Syndrome
An 8-year-old boy with complex cyanotic congenital heart disease, including an atrioventricular septal defect, reflux nephropathy, behavioural problems (impulsivity and hyperkinetic conduct disorder), and mild learning disability was seen in the genetics clinic (see Figure 4B). His first cardiac surgery was undertaken at the age of 4 months, and he had a stormy post-operative course, during which he spent several weeks in intensive care. He was reviewed by a number of pediatricians and a pediatric psychiatrist, and it was uncertain to what extent his perioperative complications were the cause of his learning and behavior problems.
An array-CGH study revealed a small deletion of approximately 4 Mb in size on chromosome 8p23.1.2 From DECIPHER it was immediately apparent that this deletion is a rare syndrome, the “8p23.1 deletion syndrome,” which includes the gene GATA4 (MIM 600576), which encodes a transcription factor involved in heart formation.23 The 8p23.1 deletion syndrome is characterized by “developmental delay and a characteristic behavior profile with hyperactivity and impulsiveness,” which explains many aspects of this child's phenotype, including his behavioral and developmental problems.
Case Study 3 (DECIPHER 00000126)—Importance of Collaboration to Further Clinical and Scientific Understanding
This case has been previously reported.24 A 4-year-old girl with developmental delay and poor eye contact was seen in the genetics clinic for diagnosis (Figure 4C). She was hypotonic in infancy, and the first year of her life was characterized by a lack of social interaction: she did not develop eye contact or a social smile. She had a normal G-banded karyotype but harbored a de novo submicroscopic deletion of approximately 1.1 Mb defined by clones RP11-203M5 (19.853 Mb to 20.059 Mb) and RP11-524O1 (20.736 Mb to 20.932 Mb) mapping to chromosome 14q11.2. The case was entered into DECIPHER, and initially no overlapping patients were seen. The family was counseled that the de novo deletion did not appear to overlap any known copy-number variable regions and was likely to be the cause of her neurobehavioural problems. However, because this deletion had not been reported previously, we could not be certain whether or to what extent it explained her phenotype.
A year later, two other cases with submicroscopic deletions of 14q11 (DECIPHER 00000976 and 00000977) were added by colleagues in Vancouver. Evaluation of the three cases together by Zahir et al24 revealed that all shared a characteristic pattern of learning disability and a similar facial appearance (widely spaced eyes, prominent epicanthic folds, a very short nose with flat nasal bridge, long philtrum, prominent Cupid's bow of the upper lip, full lower lip, and similar anomalies of the auricles). Comparison of the three cases revealed a minimal critical region of 35 kb on chromosome 14q11.2, containing only two HGNC genes: SUPT16H (MIM 605012) and CHD8 (MIM 610528), which are both plausible candidates for genes involved in neurodevelopment.
Genomic microarray analysis brings the opportunity for much higher diagnostic yield than conventional chromosome analysis for patients with developmental delay, learning disability, dysmorphic features and/or congenital anomalies. However, this high-resolution information also brings challenges for the clinical team in the interpretation of unfamiliar findings.
DECIPHER in the Clinical Evaluation of Unfamiliar Aberrations
(1) DECIPHER facilitates the identification of whether a rearrangement has been seen before in the normal population by displaying copy-number-variation tracks from external studies and databases for each rearrangement identified in the patient.
(2) DECIPHER identifies when a finding overlies a known syndrome. Whereas clinicians are familiar with the small number of well-recognized microdeletion disorders that predate genomic microarray analysis, the rapid proliferation of new syndromes over the past few years (e.g., 17q21.325–27 [MIM 610443], 15q2428, TAR syndrome susceptiblity locus29 [MIM 274000], 14q11.224, and 1q2130,31 [MIM 612474]) provides a challenge to clinicians in staying abreast of new findings.
(3) DECIPHER displays whether an aberration has been seen before by another member of the DECIPHER consortium and if so how the phenotypes of the patients compare.
(4) DECIPHER reports the gene content of the aberration. For submicroscopic imbalances, the size of the deletion or duplication is a poor guide to its gene content because the density of genes in the human genome is very uneven. Some regions are particularly gene rich, whereas other regions are virtual gene deserts. Gene content plays an important role in assessment of the possible pathogenicity of a novel aberration.
(5) DECIPHER shows genes of known clinical importance within the aberration and provides direct links to supporting literature to allow the clinician to evaluate the extent to which these genes may explain the phenotype seen in the patient. As illustrated in case 1 (above), this enables clinicians to be alerted to any genes of potential clinical importance, e.g., a tumor suppressor gene that, when deleted, may have important implications for patient management.
Currently, our understanding of the clinical consequences of changes in copy number is incomplete. When evaluating a novel copy-number change, one must bear in mind a large number of different potential mechanisms of inheritance before dismissing a change as not linked to the phenotype. These include imprinting; unmasking of a recessive mutation on the other allele; deletion, duplication, or rearrangement of regulatory elements; complex inheritance in which the phenotype results from the interplay of more than one genomic alteration and/or SNP; the possibility that regions that are recognized copy-number variants in the normal population may have a pathogenic effect on a specific genetic background; and the possibility that there may be thresholding effects at some loci where, for example, duplication may be better tolerated than triplication or further amplification. These factors, together with awareness that the human genome map continues to evolve toward a more complete reference sequence with consequent repositioning of genes and other elements, need to be considered32,33 because they necessarily imply a degree of caution in interpreting and using data in DECIPHER.
Although DECIPHER is to our knowledge the only web-accessible database of microscopic imbalance containing consented cases with open access and automated instant display in a public genome browser, similar data from other patients are accessible elsewhere. Of note is the database ECARUCA, which catalogues large cytogenetic imbalances as well as data from microarray analyses, accessible to registered members. The combination of all data sources into a single location would be an ideal, but in practice this is difficult to achieve due to incompatibilities between datasets. However, display of data from different databases within a genome browser where other diverse genomic information as well as common disease associations can also be visualized provides a practical and efficient solution to this problem.
DECIPHER and the Importance of Rare Dominant Disease Phenotypes
Many genes in the human genome are still without known function in human development and disease. For other genes, the function may be inferred from cross-species comparison, and for dominantly inherited disorders (where the phenotype does not abolish reproductive fitness), genes can be mapped by linkage analysis. Similarly, homozygosity mapping can refine the position of genes responsible for recessively inherited disorders in consanguineous families. However, mapping genes for severe de novo dominant disorders that are reproductively lethal typically relies upon identifying chromosomal aberrations to obtain positional information about gene location. The importance of genomic rearrangement as a cause of sporadic traits has only recently been recognized.34 Indeed, the de novo rate for genomic rearrangement may be orders of magnitude greater than for point mutations.
Chromosome rearrangements have historically been important in identifying disease genes by a variety of approaches: (1) the coincidence of a sporadic occurrence of the disorder with an apparently balanced de novo autosomal reciprocal translocation, e.g., the identification of SATB2 (MIM 608148) as a cleft-palate gene35 and SOX2 (MIM 184429) as an anophthalmia gene36, (2) the segregation of a phenotype with a translocation, e.g., the identification of EOMES (MIM 604615) as a cause of severe neurological malformation (microcephaly with polymicrogyria and agenesis of the corpus callosum)37, and (3) the fine mapping of visible deletions, e.g., the identification of the TSC2 gene38 (MIM 191092). Because high-resolution genomic microarray analysis has the potential to identify very small aberrations that may contain only a few genes, it offers an enhanced opportunity for the discovery of gene function, as was the case for the identification of CHD7 (MIM 608892) as the CHARGE syndrome gene.39
DECIPHER is designed specifically to bring together genomic and phenotypic data about submicroscopic chromosome aberrations and other rearrangements and display them openly in the Ensembl genome browser. In this way, DECIPHER enables clinical scientists worldwide not only to maintain records of phenotype and chromosome rearrangement for their patients but also, with informed consent, to share this information with the wider research community. By sharing cases worldwide, critical numbers of rare cases with phenotypes and structural rearrangements in common can be identified. In classical genetics, individuals with common phenotypes are identified, and a clinical syndrome is deliniated, e.g., supravalvular aortic stenosis and characteristic facies and neurodevelopmental profile in Williams-Beuren syndrome (WBS [MIM 194050]).40 In due course, a common genetic basis to the clinical syndrome is recognized (e.g., a recurrent microdeletion on 7q11.23 was recognized in the case of WBS41). DECIPHER provides a mechanism for such clinical advances to be made not only for as-yet-undiscovered recurrent rearrangements but also for the many other patients for whom rearrangements are sporadic but overlap a common genomic region. In this way genomic microarray analysis is enabling the delineation of new syndromes that have less distinctive clinical features by a process that can be termed “reverse dysmorphology.” Here, the classical practice of identification of the phenotype first and analysis of the genotype second is reversed. In this way, clusters of cases are first recognised by their overlapping copy number changes and subsequently the phenotypes of the individuals in the cluster are compared. This new approach permits the recognition of common clinical features that were initially too subtle or too variable (when seen in patients of different age, sex, and ethnicity) to enable a new syndrome to be identified solely on clinical grounds. This approach may be particularly valuable for the investigation of disorders with high locus heterogeneity, e.g., learning disability.
The application of chromosome microarray analysis to patients with developmental delay, learning disability, dysmorphic features, and/or congenital anomalies is revolutionizing clinical genetic practice. DECIPHER has the aim of being a key tool for the clinician in harnessing this new high-resolution cytogenetic data for the benefit of patients and their families. DECIPHER has already been instrumental in defining new syndromes (e.g., 17q21.327, 14q11.224, and 19q13.1142; Figure 5) and will become more powerful not only for clinical diagnosis but also for genomic research as the number of records in the database grows.
Figure 5.
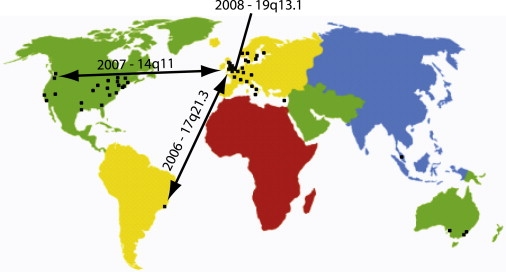
International Collaboration through DECIPHER
New syndromes defined with the help of DECIPHER: 17q21.327 in 2006, 14q11.224 in 2007, and 19q13.1144 in 2009.
Web Code
The web code supporting the website is a Perl CGI or mod_perl web interface with SSL encrypted transport (HTTPS), Apache web server, Perl Object-Oriented API, MySQL relational database back-end, and DAS data integration with Ensembl.
Acknowledgments
This work was supported by the Wellcome Trust (grant number WT077008). We thank the patients and their families for permission to include their details in this paper. We thank Jim Lupski and Martin Bobrow for their helpful comments on the draft manuscript, Anthony V. Cox for his bioinformatics expertise, and Lionel Willatt for conducting the FISH studies. We wish to thank all of the DECIPHER Syndrome Expert advisors together with the members of the DECIPHER Advisory Board for giving so generously of their expertise. Finally, we would like to thank all of the DECIPHER Consortium members, a list of whom can be found on the DECIPHER website.
Contributor Information
Helen V. Firth, Email: hvf21@cam.ac.uk.
Nigel P. Carter, Email: npc@sanger.ac.uk.
Web Resources
The URLs for data presented herein are as follows:
DECIPHER, https://decipher.sanger.ac.uk/
Ensembl, http://www.ensembl.org/
Ensembl Human, http://www.ensembl.org/Homo_sapiens/
Database of Genomic Variants, http://projects.tcag.ca/variation/
HUGO Gene Nomenclature Committee, http://www.gene.ucl.ac.uk/nomenclature/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM
GeneReviews, http://www.geneclinics.org/
PubMed, http://www.pubmed.gov/
GeneImprint, http://www.geneimprint.com/
The Baraitser-Winter Neurogenetics Database, http://lmdatabases.com/about_lmd.html
References
- 1.Lu X., Shaw C.A., Patel A., Li J., Cooper M.L., Wells W.R., Sullivan C.M., Sahoo T., Yatsenko S.A., Bacino C.A. Clinical implementation of chromosomal microarray analysis: Summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw-Smith C., Redon R., Rickman L., Rio M., Willatt L., Fiegler H., Firth H., Sanlaville D., Winter R., Colleaux L. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stankiewicz P., Beaudet A.L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr. Opin. Genet. Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Veltman J.A. Genomic microarrays in clinical diagnosis. Curr. Opin. Pediatr. 2006;18:598–603. doi: 10.1097/MOP.0b013e3280105417. [DOI] [PubMed] [Google Scholar]
- 5.Vissers L.E., de Vries B.B., Osoegawa K., Janssen I.M., Feuth T., Choy C.O., Straatman H., van der Vliet W., Huys E.H., van Rijk A. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 7.Padiath Q.S., Saigoh K., Schiffmann R., Asahara H., Yamada T., Koeppen A., Hogan K., Ptacek L.J., Fu Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 8.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 9.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Maner S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 10.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermeesch J.R., Fiegler H., de Leeuw N., Szuhai K., Schoumans J., Ciccone R., Speleman F., Rauch A., Clayton-Smith J., Van Ravenswaaij C. Guidelines for molecular karyotyping in constitutional genetic diagnosis. Eur. J. Hum. Genet. 2007;15:1105–1114. doi: 10.1038/sj.ejhg.5201896. [DOI] [PubMed] [Google Scholar]
- 12.Brown L., editor. The New Shorter Oxford English Dictionary. Oxford University Press; New York: 1993. [Google Scholar]
- 13.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 14.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Flicek P., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T. Ensembl 2008. Nucleic Acids Res. 2008;36:D707–D714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dowell R.D., Jokerst R.M., Day A., Eddy S.R., Stein L. The distributed annotation system. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebholz-Schuhmann D., Kirsch H., Arregui M., Gaudan S., Riethoven M., Stoehr P. EBIMed–text crunching to gather facts for proteins from Medline. Bioinformatics. 2007;23:e237–e244. doi: 10.1093/bioinformatics/btl302. [DOI] [PubMed] [Google Scholar]
- 18.Van Vooren S., Thienpont B., Menten B., Speleman F., De Moor B., Vermeesch J., Moreau Y. Mapping biomedical concepts onto the human genome by mining literature on chromosomal aberrations. Nucleic Acids Res. 2007;35:2533–2543. doi: 10.1093/nar/gkm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishisho I., Nakamura Y., Miyoshi Y., Miki Y., Ando H., Horii A., Koyama K., Utsunomiya J., Baba S., Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 20.Bodmer W.F., Bailey C.J., Bodmer J., Bussey H.J., Ellis A., Gorman P., Lucibello F.C., Murday V.A., Rider S.H., Scambler P. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614–616. doi: 10.1038/328614a0. [DOI] [PubMed] [Google Scholar]
- 21.Michils G., Tejpar S., Thoelen R., van Cutsem E., Vermeesch J.R., Fryns J.P., Legius E., Matthijs G. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): A Belgian study. Hum. Mutat. 2005;25:125–134. doi: 10.1002/humu.20122. [DOI] [PubMed] [Google Scholar]
- 22.Galiatsatos P., Foulkes W.D. Familial adenomatous polyposis. Am. J. Gastroenterol. 2006;101:385–398. doi: 10.1111/j.1572-0241.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 23.Garg V., Kathiriya I.S., Barnes R., Schluterman M.K., King I.N., Butler C.A., Rothrock C.R., Eapen R.S., Hirayama-Yamada K., Joo K. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 24.Zahir F., Firth H.V., Baross A., Delaney A.D., Eydoux P., Gibson W.T., Langlois S., Martin H., Willatt L., Marra M.A. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J. Med. Genet. 2007;44:556–561. doi: 10.1136/jmg.2007.050823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 26.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 27.Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 28.Sharp A.J., Selzer R.R., Veltman J.A., Gimelli S., Gimelli G., Striano P., Coppola A., Regan R., Price S.M., Knoers N.V. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet. 2007;16:567–572. doi: 10.1093/hmg/ddm016. [DOI] [PubMed] [Google Scholar]
- 29.Klopocki E., Schulze H., Strauss G., Ott C.E., Hall J., Trotier F., Fleischhauer S., Greenhalgh L., Newbury-Ecob R.A., Neumann L.M. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupski J.R. Structural variation in the human genome. N. Engl. J. Med. 2007;356:1169–1171. doi: 10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
- 33.Lupski J.R., Stankiewicz P. Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lupski J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 35.FitzPatrick D.R., Carr I.M., McLaren L., Leek J.P., Wightman P., Williamson K., Gautier P., McGill N., Hayward C., Firth H. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum. Mol. Genet. 2003;12:2491–2501. doi: 10.1093/hmg/ddg248. [DOI] [PubMed] [Google Scholar]
- 36.Fantes J., Ragge N.K., Lynch S.A., McGill N.I., Collin J.R., Howard-Peebles P.N., Hayward C., Vivian A.J., Williamson K., van Heyningen V. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 37.Baala L., Briault S., Etchevers H.C., Laumonnier F., Natiq A., Amiel J., Boddaert N., Picard C., Sbiti A., Asermouh A. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007;39:454–456. doi: 10.1038/ng1993. [DOI] [PubMed] [Google Scholar]
- 38.European Chromosome 16 Tuberous Sclerosis Consortium Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 39.Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 40.Williams J.C., Barratt-Boyes B.G., Lowe J.B. Supravalvular aortic stenosis. Circulation. 1961;24:1311–1318. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]
- 41.Ewart A.K., Morris C.A., Atkinson D., Jin W., Sternes K., Spallone P., Stock A.D., Leppert M., Keating M.T. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- 42.Malan V., Raoul O., Firth H.V., Rover G., Turleau C., Bernheim A., Willatt L., Munnich A., Vekemens M., Lyonnet S. 19q13.11 deletion syndrome: anovel clinicaly recognizable genetic condition identified by array-CGH. J. Med. Genet. 2009 doi: 10.1136/jmg.2008.062034. in press. [DOI] [PubMed] [Google Scholar]