

Abstract
Ichthyosis follicularis with atrichia and photophobia (IFAP syndrome) is a rare X-linked, oculocutaneous human disorder. Here, we assign the IFAP locus to the 5.4 Mb region between DXS989 and DXS8019 on Xp22.11-p22.13 and provide evidence that missense mutations exchanging highly conserved amino acids of membrane-bound transcription factor protease, site 2 (MBTPS2) are associated with this phenotype. MBTPS2, a membrane-embedded zinc metalloprotease, activates signaling proteins involved in sterol control of transcription and ER stress response. Wild-type MBTPS2 was able to complement the protease deficiency in Chinese hamster M19 cells as shown by induction of an SRE-regulated reporter gene in transient transfection experiments and by growth of stably transfected cells in media devoid of cholesterol and lipids. These functions were impaired in five mutations as detected in unrelated patients. The degree of diminished activity correlated with clinical severity as noted in male patients. Our findings indicate that the phenotypic expression of IFAP syndrome is quantitatively related to a reduced function of a key cellular regulatory system affecting cholesterol homeostasis and ability to cope with ER stress.
Introduction
IFAP syndrome (MIM 308205) is an X-linked oculocutaneous human disorder characterized by a peculiar triad of follicular ichthyosis, total or subtotal atrichia, and photophobia of varying degree (Figure 1).1 Histopathologically, the epidermal granular layer is generally well-preserved or thickened at the infundibulum. Hair follicles are poorly developed and tend to be surrounded by an inflammatory infiltrate. A subgroup of patients is described with lamellar rather than follicular ichthyosis. Well-demarcated psoriasiform plaques in addition to, or instead of, follicular ichthyosis are also described.2,3 Congenital atrichia is the most prominent feature of the syndrome. In most of the affected boys, total atrichia is noted at birth. Occasionally, some sparse and thin hair may be present.4,5 The third constant feature is photophobia possibly due to vascularizing keratitis or to anomalies in Bowman's membrane combined with intracellular epithelial adhesion defects.6 Nonconsistent findings include neurological abnormalities such as retarded psychomotor development, cerebral atrophy, temporal lobe malformation, and hypoplasia of corpus callosum,7 failure to thrive, nail dystrophy, atopic manifestations, inguinal hernia, and aganglionic megacolon, as well as renal, vertebral, and testicular anomalies.2,8,9 X linkage was predicted because the full phenotype is found in males only. Female carriers may present with a linear pattern of follicular ichthyosis, mild atrophoderma, hypotrichosis, and hypohidrosis. These lesions clearly follow the lines of Blaschko.10 In other obligate carrier females, extreme lyonization may give rise to nonpenetrance, thus blurring the difference between recessive and dominant X-linked inheritance. The rarity of IFAP syndrome has previously hampered genetic mapping. In this report, we show that IFAP syndrome is caused by functional deficiency of membrane-bound transcription factor protease, site 2 (MBTPS2 [MIM 300294]), a membrane-embedded zinc metalloprotease that activates signaling proteins involved in sterol control of transcription and ER stress response.
Figure 1.

Clinical Features of IFAP Syndrome
Male patient showing typical filiform follicular hyperkeratoses (“ichthyosis follicularis”) (A), atrichia of the scalp (B), half-closed eyelids reflecting severe photophobia, and absence of eyebrows and lashes (C).
Material and Methods
Study Subjects
This study included three extended multigeneration pedigrees as well as two unrelated male patients (4-I:1 and 5-I:1), all of European descent, and two affected brothers (6-II:1 and 6-II:4) from an Algerian family (Figure 2 and Table 1). Patient 4-I:1 had a mildly affected mother who was unavailable for molecular analysis. DNA from the clinically unaffected mother (6-I:2) of patients 6-II:1 and 6-II:4 was included in the mutation analysis. Patient 5-I:1 had been mentioned as individual IV:8 in a pedigree reported previously.2 Examination of medical records and mutation analyses were carried out under research protocols approved by the local ethics committee of the University of Marburg. Informed consent was obtained from patients, their parents, and controls. A total of 140 control DNA samples (225 X chromosomes) were taken from healthy individuals with self-reported European ancestry.
Figure 2.

Pedigrees of Three Families of European Descent with IFAP Syndrome
Partial pedigrees of families 1–3 show genotypes of X-chromosomal markers from Xpter to Xcen. Disease-associated haplotypes in families 1 and 2 are encased in a box. For clarity, not all analyzed markers listed in Table S1 are given. MBTPS2 mutations and the derived amino acid changes are indicated below the family identifier and are included as allele “mt” in the list of genotypes; “wt” represents wild-type MBTPS2. The status of females as unaffected carriers in families 2 and 3 is defined a posteriori according to the presence of a mutated MBTPS2 allele.
Table 1.
Clinical Features and MBTPS2 Genotypes of IFAP Patients and Family Members
| Proband | Origin | Sex | Clinical Features | Genotype |
|---|---|---|---|---|
| 1-I:1 | G | m | n.a. | WT |
| 1-I:2 | G | f | dry skin, congenital bald patches of the scalp | c.680T/WT |
| 1-I:3 | G | m | n.a. | WT |
| 1-II:1 | G | m | n.a. | WT |
| 1-II:2 | G | f | dry skin, congenital bald patches of the scalp, asymmetrical distribution of body hair, linear lesions of scaling, atrophy and hypohidrosis on arms and back | c.680T/WT |
| 1-II:3 | G | m | n.a. | WT |
| 1-II:4 | G | m | n.a. | WT |
| 1-II:5 | G | m | IFAP triad with pronounced photophobia; death at 1 year of age. | n.t. |
| 1-III:1 | G | m | IFAP triad, eczematous changes of shoulders and limbs, frequent skin infections, nail dystrophy, ptosis of left upper eyelid | c.680T |
| 1-III:2 | G | f | dry skin, linear lesions of scaling and atrophy, predominantly involving the left arm, congenital bald area of the scalp | c.680T/WT |
| 2-I:2 | A | f | sparse hair | c.261A/WT |
| 2-II:2 | A | f | unaffected carrier | c.261A/WT |
| 2-II:3 | A | f | n.a. | WT/WT |
| 2-II:5 | A | m | n.a. | WT |
| 2-II:7 | A | m | n.a. | WT |
| 2-II:8 | A | m | alopecia, dystrophic finger nails | c.261A |
| 2-III:1 | A | m | IFAP triad, psoriasiform plaques on elbows, knees and heels, absence of eyebrows, dystrophic nails, recurrent skin infections, no neurological anomalies | c.261A |
| 2-III:2 | A | f | sparse scalp hair, dystrophic fingernails | c.261A/WT |
| 2-III:4 | A | f | n.a. | WT/WT |
| 2-III:5 | A | f | n.a. | WT/WT |
| 2-III:6 | A | f | n.a. | WT/WT |
| 2-III:7 | A | f | n.a. | WT/WT |
| 2-III:8 | A | m | n.a. | WT |
| 2-III:9 | A | f | patchy scalp hair, dystrophic fingernails | c.261A/WT |
| 2-III:10 | A | f | unaffected carrier | c.261A/WT |
| 2-III:11 | A | m | n.a. | WT |
| 2-III:12 | A | m | n.a. | WT |
| 2-IV:1 | A | m | n.a. | WT |
| 2-IV:2 | A | m | IFAP triad, dystrophic nails, perlèche | c.261A |
| 3-I:2 | G | f | dry skin | c.1286A/WT |
| 3-II:2 | G | f | linear lesions of atrophoderma, linear hairless scalp lesion, hyperkeratotic plaque on right knee, bilateral plantar keratoderma | c.1286A/WT |
| 3-II:4 | G | f | linear lesions of atrophoderma, partial absence of retinal rods (R eye) | c.1286A/WT |
| 3-II:5 | G | f | unaffected carrier | c.1286A/WT |
| 3-II:6 | G | m | atrichia, cleft hand, death in neonatal period | n.t. |
| 3-II:7 | G | m | atrichia, cleft hand, death in neonatal period | n.t. |
| 3-III:1 | G | f | unaffected carrier | c.1286A/WT |
| 3-III:2 | G | f | n.a. | WT/WT |
| 3-III:3 | G | m | IFAP triad, initially in the form of collodium baby; dermatitis on arms and legs, sensitiveness to cow's milk, motor retardation, general dystrophy, cleft palate, unilateral cleft hand, two butterfly vertebrae, absence of a kidney, bilateral inguinal hernia, omphalocele, stenosis of small intestine, Hirschsprung disease, death at 9 months of age | n.t. |
| 3-III:4 | G | m | IFAP triad, microcephaly, arachnoidal cyst, Arnold-Chiari malformation type I, thoracolumbar hydromyelia, seizures, psychomotor retardation, retrognathia, deficient growth, cleft hands, butterfly vertebra, wedge-shaped vertebra, atrial septum defect, arterial hypertension, recurrent infections of upper airways, absence of a kidney, hypospadia, choanal stenosis, inguinal hernia, Hirschsprung disease; death at 14 months of age | n.t. |
| 4-I:1 | S | m | IFAP triad with pronounced photophobia, dermatitis on arms and legs, corneal pannus (mother has dry skin and bald patches on her scalp) | c.677T |
| 5-I:1 | Ar | m | IFAP triad, hyperkeratosis around joints, perlèche, perianal erythema, plantar keratoderma, subungual hyperkeratoses, periungual erythema and hyperkeratosis, inguinal hernia, cryptorchidism | c.1424C |
| 6-I:2 | Al | f | n.a. | c.225-6A/WT |
| 6-II:1 | Al | m | IFAP triad | c.225-6A |
| 6-II:4 | Al | m | IFAP triad | c.225-6A |
IFAP triad: generalized scaling, complete atrichia, and photophobia. Al, Algeria; Ar, Argentina; A, Australia; G, Germany; S, Spain; n.a., not affected; n.t., not tested; WT, wild-type.
Cell Culture
CHO-M19 cells, a mutant line of CHO-K1 cells, auxotrophic for cholesterol and unsaturated fatty acids owing to a deletion in the S2P-gene,11,12 a generous gift of T.Y. Chang (Dartmouth Medical School, Hanover, NH, USA) were maintained under standard conditions in F12 media supplemented with 10% FCS, nonessential amino acids, and antibiotics.
Recombinant Plasmids
Vectors expressing MBTPS2 mutants as fusion proteins with Yellow Fluorescent Protein (EYFP) were derived by mutagenesis via directional PCR product cloning from the full ORF expression clone IOH46558-pdEYFP-C1amp13 (imaGenes GmbH). For stable transfection, the EYFP-MBTPS2 wild-type or mutant fusion genes were subcloned into the pcDNA3.1-vector expressing a neomycin (G418) resistance gene (Invitrogen). A reporter plasmid pSRE-GL4.23 was generated in the expression vector pGL4.23[luc2/minP] (Promega) by inserting in tandem three copies of repeat 2+3 of the human LDL receptor promoter (Sterol Regulatory Element, SRE)14 upstream of the minimal promoter driving the Firefly-luciferase gene. The control luciferase plasmid pRL-SV40 (Renilla-luciferase driven constitutively by an SV40 promoter and enhancer) was obtained from Promega.
Genetic Mapping
Genomic DNA was extracted from peripheral blood lymphocytes in accordance with standard procedures and taken as a template for polymerase chain reaction (PCR).
For linkage analysis, we PCR-amplified 11 X-chromosomal microsatellite markers from the CHLC screening set ver. 6 using, with minor modifications, the conditions described in the Genome Database (GDB: DXS6807, DXS8022, DXS989, DXS1068, DXS1003, DXS7132, DXS6800, DXS8020, DXS1001, DXS1047, and DXS1193). The mean heterozygosity was >0.8. Allele sizes were standardized by comparison with individual 1347MO2 (Centre d’Études du Polymorphisme Humain, CEPH). Locus order and sex-averaged intermarker distances (8.44 cM on average) were used as provided by the Center for Medical Genetics, Marshfield Medicine Research Foundation. Corresponding allele frequencies were adopted from GDB, where possible, or set to be equally distributed (DXS7132 and DXS6800). A dense grid of five highly polymorphic microsatellite markers was used for fine mapping on chromosome Xp21-p22.1 covering a distance of 12.5 Mb (DXS8019, DXS7593, DXS1226, DXS989, and DXS8039).
For genetic mapping, we assumed an X-linked recessive mode of inheritance with 90% penetrance in female carriers and nearly complete penetrance (0.999) in affected males. Phenocopies were virtually excluded (0.001). The disease allele frequency was adjusted to 1:100.000.
Software Applied in Linkage and Haplotype Analyses
Parametric two-point linkage analyses were undertaken as described previously.15,16 LOD-score calculations were performed for each marker with the MLINK program of LINKAGE package, ver. 5.10. Multipoint analyses were performed for all the microsatellite markers employing the MINX program of MERLIN, ver. 1.0.1.17 MINX was also used to generate haplotypes.
HR-CGH: Microarray Construction and Probe Selection
Arrays for high resolution comparative genome hybridization (HR-CGH) covering the entire X chromosome were generated at NimbleGen Systems as described previously.18 The arrays were hybridized with Cy3-labeled genomic DNA samples from six unrelated IFAP patients (1-III:1, 2-III:1, 3-II:2, 4-I:1, 5-I:1, and 6-II:1; Table 1) in comparison with standard samples. Copy number variants were then searched by automated segmentation analysis of the signal data sets and visualized with the SignalMap software tool provided by NimbleGen.18
Mutation Analysis of Candidate Genes
Candidate genes MBTPS2, PTCHD1 (accession number NM_173495), SCML1 (MIM 300227), RAI2 (MIM 300217), and YY2 (MIM 300570), selected because of their inferred biological relevance for IFAP syndrome, were sequenced in the same six unrelated patients. Primer pairs used for amplification of the coding exons of PTCHD1, SCML1, RAI2, and YY2 are available on request. Primer pairs and PCR conditions for the amplification of the coding exons of MBTPS2 including the intron-exon boundaries are listed in Table S2 available online. We used the same oligonucleotides to sequence the PCR products on both strands. Mutations in the coding exons of MBTPS2 were confirmed by independent methods such as an Amplification Refractory Mutation System (ARMS test, family 1 and individual 6-II:1), single-strand conformation analysis (SSCA, family 2), and mutation-specific restriction enzyme digestion (family 3 with restriction enzyme TaiI, individual 4-I:1 with BstXI, and individual 5-I:1 with Eco130I. To exclude mutations that represented single-nucleotide polymorphisms (SNPs), we scrutinized DNA samples representing 225 X chromosomes from unrelated control individuals by the same procedures and searched the sequence of the mutated allele in NCBI dbSNP.
Histochemical Analysis of Cells Transiently Transfected with EYFP-MBTPS2 Expression Plasmids
Chinese hamster K1 cells and transformed African Green Monkey kidney fibroblast cells (COS-7 line) were cultured in Ham's F-12 or DMEM medium, respectively, supplemented with nonessential amino acids and glutamine, 100 units/ml of penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (FCS). A total of 2 × 105 cells each were seeded in 1 ml per well of two-well Lab-Tek chamber slides. Transfection complexes composed of 4 μl TurboFect (MBI Fermentas), and 500 ng of wild-type or mutant YFP-MBTPS2 recombinant expression construct DNA were mixed in 500 μl culture medium without serum, incubated for 10–15 min at room temperature to allow formation of transfection complexes, and subsequently added drop wise to the cells in each of the wells. The cells with the transfection complexes were incubated under normal growth conditions. After 12 hr, the medium was replaced with fresh growth media containing 10% FCS, and incubation was continued for 12 hr. Subsequent fluorescence microscopy detected in parallel the intracellular localization of YFP or YFP-tagged MBTPS2, DAPI-stained nuclei, and Golgi vesicles stained with monoclonal mouse golgin-97 antibody (Molecular Probes/Invitrogen: A21270, 1:200) and Alexa Fluor 594 goat anti-mouse immunoglobulin (Molecular Probes/Invitrogen: A11005) as secondary antibody (diluted 1:500). Immunostained cells were again washed thoroughly with PBS/0.5% Tween 20 and mounted in an antifade reagent with DAPI for staining of nuclei (Molecular Probes/Invitrogen: S30939). Images were taken on a Zeiss Axioplan microscope (Carl Zeiss MicroImaging GmbH) with a Leica DFC 300 FX camera (Leica Microsystems), and processed with the Leica imaging system 50 (IM50) software.
Luciferase Reporter Assays
This assay, an indirect measure of the ability of MBTPS2 cDNAs to restore sterol-regulated transcriptional activity in transfected M19 cells with a pSRE-Luciferase reporter plasmid, essentially followed the rationale outlined by Zelenski and coworkers.19 A total of 1.5 × 105 M19 cells per well were set up in triplicate in 12-well plates in a 1:1 mixture of Ham's F-12 medium and Dulbecco's Modified Eagle Medium (DMEM) containing GlutaMax (Invitrogen), nonessential amino acids, penicillin, streptomycin (Medium A), supplemented with 5% fetal calf serum, and maintained at 37°C, 5% CO2. After 16 hr, the cells were washed once with PBS, and incubation was continued with 2 ml of medium A with 5% lipoprotein-deficient fetal calf serum (medium B) supplemented with 20 μM sodium oleate. After 7 hr, we transfected the M19 cells by adding to each well a transfection mixture containing 200 μl serum-free F12/DMEM, 2 μl TurboFect in vitro Transfection Reagent (MBI Fermentas), 500 ng of an expression plasmid without MBTPS2 cDNA insert (pdEYFP-C1amp, kindly provided by Dr. S. Wiemann, Heidelberg) or a wild-type or mutant EYFP-tagged MBTPS2 cDNA together with 400 ng of pSRE-GL4.23 reporter plasmid, and 50 ng of pRL-SV40 as control for transfection efficiency. After incubation for 3 hr at 37°C, 5% CO2, the cells were switched to medium B containing 50 μM sodium mevalonate and 50 μM sodium compactin, either supplemented with 10 μg/ml water-soluble cholesterol, 1 μg/ml 25-hydroxycholesterol or without sterols. After 16 hr at 37°C, 5% CO2, cell lysates were prepared and analyzed for Firefly- and Renilla-luciferase activities in a luminometer Auto Lumat LB953 (Berthold Technologies) with a dual luciferase reporter assay system as suggested by the supplier (Promega). These experiments were repeated three times.
Complementation Analysis: Growth of Stably Transfected M19 Cells in Sterol-Deficient Medium
A total of 106 CHO-M19 cells, each, were set up in 75 ml cell-culture flasks in medium A, supplemented with 10% FCS, and transfected with 1.5 μg of a plasmid (pcDNA3.1, Invitrogen) expressing a neomycin (G418) resistance gene alone or together with EYFP-MBTPS2 wild-type or mutant fusion genes in accordance with the procedure described for luciferase reporter assays. Stable transfectants were selected by growth for 14 days at 37°C, 5% CO2 in medium C, a mixture of medium A, 5% fetal calf serum, water-soluble cholesterol (5 μg/ml), sodium mevalonate (1 mM), and sodium oleate (20 μM), supplemented with 750 μg/ml G418. This medium was replaced every 1–3 days. A total of 105 CHO-M19 cells stably transfected with tagged wild-type or one of five mutant MBTPS2 genes were seeded in 5.5 cm diameter Petri dishes and grown for 6 days at 37°C, 5% CO2 in medium B or C, respectively, each supplemented with 500 μg/ml G418. At day 6, the cells surviving in the Petri dishes were washed once with PBS, fixed for 15 min in 4% PFA in PBS, washed briefly in distilled water, stained for 20 min in 1% Crystal Violet, washed again three times in water, dried, and photographed for documentation. To quantify the number of cells able to grow in sterol-depleted media, these experiments were repeated three times in 75 ml cell-culture flasks. On day 6, the surviving cells were harvested and their number was determined in a hemocytometer.
Results
To map the IFAP locus by parametric linkage analysis, identify the gene responsible for the trait, and study functional deficiencies that might explain the phenotype, we analyzed three families of European descent (families 1, 2, and 3) in which IFAP segregated according to an X-linked pattern of transmission, as well as IFAP patients from three unrelated families (families 4, 5, and 6) (Figure 2 and Table 1). Only minor intrafamilial phenotypic variability between male patients was observed, whereas in different families, the clinical severity of the syndrome varied to a large degree. Affected males of family 2 mainly displayed the IFAP triad of symptoms comprising total alopecia, ichthyotic scaling (sometimes with psoriasiform plaques), and mild or severe photophobia. Individual 1-II:5 was completely hairless and avoided daylight because of severe photophobia.10 Patient 4-I:1 showed similar features. In contrast, male patients of families 3 and 5 suffered from a more severe form of IFAP syndrome. Individuals 3-II:6, 3-II:7, 3-III:3, and 3-III:4 had multiple additional malformations and died within 2 years after birth. Male patients from family 4 had seizures, mental retardation, and inguinal hernia in addition to the IFAP triad.2 Female carriers in all families were either phenotypically normal or showed rather mild symptoms such as asymmetric distribution of body hair, patchy alopecia, or linear lesions of follicular ichthyosis after the lines of Blaschko (Table 1).
Employing genotyping results from families 1 and 2, we assigned the IFAP locus by linkage analysis to the 5.5 Mb region between DXS989 and DXS8019 in Xp22.11-p22.13, encompassing approximately 35 known and predicted genes (Figure 2, Figure 3, and Table S1).
Figure 3.
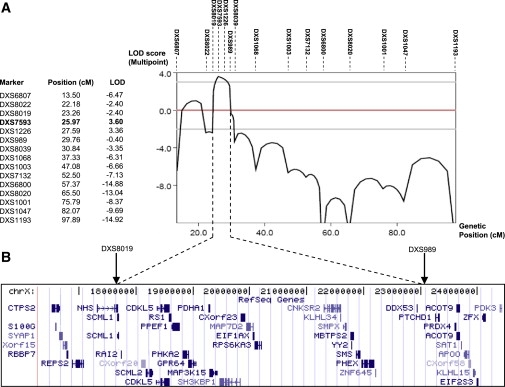
Multipoint Linkage Analysis of the IFAP Locus
Combined parametric multipoint linkage analysis with the MINX program of MERLIN confirms the two-point linkage results placing the IFAP locus between DXS8019 and DXS989 (Table S1).
(A) Table and graphical representation of multipoint LOD scores calculated with X-chromosomal marker genotypes of pedigrees 1 and 2 (Figure 2). The highest LOD score of 3.60 at DXS7593 is marked in bold.
(B) NCBI RefSeq genes (adopted from the UCSC genome browser) from the critical interval on the X chromosome between markers DXS8019 (telomeric) and DXS989 (centromeric).
Because array CGH did not detect any copy number variants in this region (data not shown), candidate genes were searched for mutations in genomic DNA of six unrelated IFAP patients. In five patients missense mutations exchanging highly conserved amino acids were identified in the gene MBTPS2 at Xp22.11 by sequencing the coding exons including intron-exon boundaries from genomic DNA amplified by PCR (family 1: c.680A→T; p.H227L; family 2: c.261G→A; p.M87I; family 3: c.1286G→A; p.R429H; family 4: c.667G→T; p.W226L; family 5: c.1424T→C; p.F475S; Table 1, Figure 4, and Figure S1A). In family 6, a c.225-6T→A transition introduced a putative splice acceptor site in intron 2. SpliceView, an online system for splice-site prediction, predicted this site is used with similar probability as the wild-type sequence 6 bp upstream, implicating that it may lead to a frame shift and consequently a truncated gene product consisting of 74 amino acid residues. Usage of this site might reduce the amount of functional MBTPS2. Cells of these patients were unavailable for confirmation of functional MBTPS2 deficiency. The mutations, each, were confirmed by independent methodology listed above (data not shown). They were not detected in 225 X chromosomes of unrelated individuals nor listed in NCBI dbSNP as known SNPs. In male individuals, the MBTPS2 mutations segregated with the IFAP phenotype. In addition to female carriers showing clinical features of the syndrome, also obligate carriers without IFAP symptoms and females for whom the carrier status was not predictable from the pedigree were heterozygous for the mutations (Figure 2 and Table 1).
Figure 4.
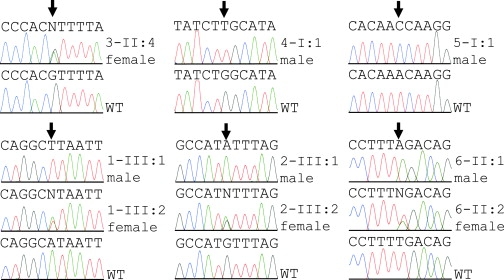
MBTPS2 Mutation Analysis
DNA sequence electropherograms of WT MBTPS2 and mutations observed in male or female IFAP patients from six unrelated families. The positions of the mutant bases are indicated by arrows. Phenotypes of the individuals analyzed are listed in Table 1.
The exchange of highly conserved amino acids in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response, and the segregation of the missense mutations with the phenotype suggest a causative role of enzyme dysfunction in IFAP syndrome (Figure 2 and Figure S1). We tested the effect of the five MBTPS2 missense mutations detected in IFAP patients on the cellular localization of the enzyme in cultured cells and on the potential to complement S2P deficiency in Chinese hamster CHO-K1-M19 cells. Transient transfection of molecular YFP-fusion constructs with WT or mutated MBTPS2 into cultured cells resulted in the synthesis of fluorescent proteins which localized preferentially to the endoplasmatic reticulum. None of the mutations interferes with synthesis or cellular localization of the proteins (Figure 5A). M19 cells in which the orthologous Mbtps2 is deleted are unable to grow in cholesterol and lipid-deficient culture media.11,12 Wild-type MBTPS2 stably transfected into M19 cells complemented this defect and restored wild-type growth characteristics (Figure 5B). None of five mutants detected in IFAP patients has retained this property comparably to the wild-type gene; however, a great variation in residual activity was observed. In mutants R429H and F475S, almost no survival was detected (Figure 5B). When correlated to the severity of the clinical phenotype it is evident that these mutants with little residual growth potential are observed in the most seriously affected male patients (Table 1). Because M19 cells are deficient in S2P as a result of a genomic rearrangement that disrupts Mbtps2, they fail to process SREBPs in response to cholesterol deprivation.11,12 A luciferase reporter gene under transcriptional control of sterol responsive elements (SRE) was active in cells grown in sterol-deficient media when wild-type MBTPS2 was cotransfected (Figure 5C). However, sterol responsiveness of the cells transfected with the mutants was restored to a lower extent than with wild-type and differed considerably among the mutants (Figure 5C). No significant difference was obtained between the wt and the H227L mutant. The lowest residual activity was retained in mutant R429H.
Figure 5.

Cellular Localization and Complementation Analysis of MBTPS2 Mutant Proteins
(A) In cultured cells transiently transfected with clones expressing YFP alone (vector), with clone IOH46558-pdEYFP-C1amp expressing a YFP-MBTPS2-wild-type protein (WT), or with five clones expressing mutated YFP-MBTPS2 proteins as indicated at the bottom of (C), the YFP-MBTPS2 fusion proteins localize preferentially in the cytoplasm but not in the nucleus, whereas YFP is found both in nucleus and cytoplasm (green fluorescence, upper panel). For clarity, nuclei are stained in blue with DAPI; the Golgi shows red color and is orange in superposition with green fluorescence (lower panel).
(B) Stable transfection of CHO-K1-M19 cells lacking hamster Mbtps2 with WT human MBTPS2 complements the enzyme defect, thereby allowing growth in cholesterol-deficient media. Complementation analysis with either of five mutants exchanging highly conserved amino acids indicated at the bottom of (C) supports growth in cholesterol-deficient media to various degrees correlated with the severity of the phenotype (Table 1). The proportion of cells capable of growth in spite of cholesterol deprivation is documented as photographs of the cultures in absence or presence of sterols and, graphically, by counts of growing stably transfected cells under the same conditions. Data are represented as mean ± SEM. Absence of sterols in the culture media is indicated by a red frame encasing the photographs of culture dishes and red columns in the diagrams; growth in media with sterols is indicated by blue frame or columns.
(C) Transient cotransfection of a construct expressing wild-type MBTPS2 (WT) with a luciferase reporter gene under transcriptional control of sterol responsive elements (SRE) into M19 cells restores sterol responsiveness of the cells in sterol-deficient media (red columns) but not in sterol-containing media (blue columns). Replacement of the wild-type with mutant MBTPS2 expression constructs results in reduced induction by sterols to different extent. Transfection efficiency is normalized to cotransfected Renilla luciferase. The results shown are representative of three independent transfection experiments. Data are represented as mean ± SEM.
In (A)–(C), ”Vector” represents different empty vectors used to clone WT and mutant MBTPS2 as described in Material and Methods.
Discussion
MBTPS2 codes in 11 exons for a membrane-embedded zinc metalloprotease, generally named site-2-protease (S2P), which cleaves within Golgi membranes after a first luminal proteolytic cut by site-1-protease (MBTPS1), cytosolic fragments from prospective signaling proteins such as the transcription factors SREBF1 and SREBF2 (MIM 184756 and MIM 600481), a prerequisite for their transport to the nucleus and for sterol control of target gene transcription.20 The same pair of proteases appears to control unfolded protein response (UPR) within the endoplasmatic reticulum by cleavage of membrane-bound transcription factors, serving as putative ER stress sensors, such as ATF6 (MIM 605537), CREB3L1 (GeneID 90993), or CREB3L4 (MIM 607138).21–23
Deficiency in either function, sterol or ER homeostasis, could lead to disturbed differentiation of epidermal structures evoking the triad of ichthyosis follicularis, atrichia, and photophobia in the IFAP phenotype: SREBF2 signaling is required for cholesterol biosynthesis. Many of the ichthyoses are associated with inherited disorders of lipid metabolism, mostly as a consequence of abnormal skin permeability barrier function causing pathophysiology through stimulation of epidermal hypoplasia (reviewed by Elias et al.24). For example, in recessive X-linked ichthyosis (XLI [MIM 308100]), because of mutation of the gene for steroid sulfatase (STS [MIM 300747]), accumulation of the amphipathic lipid cholesterol sulfate, the substrate of this enzyme, provokes both a typical scaling phenotype and permeability barrier dysfunction.25 Likewise, genetic defects in enzymes of the Kandutsch pathway result in disorders associated with ichthyosiform lesions and atrichia such as deficiency of NSDHL (NADPH steroid dehydrogenase-like protein [MIM 300275]) in CHILD syndrome (MIM 308050)26 and of emopamil-binding protein (EBP [MIM 300205]) in Conradi-Hünermann-Happle syndrome (CDPX2 [MIM 302960]).27
Photophobia in IFAP patients, similarly, is caused by an epithelial disturbance. Ulceration and vascularization lead to progressive corneal scarring and subsequent photophobia. Ultrastructural findings suggest a primary defect of epithelial adhesion at the corneal epithelial-stromal junction with anomalies of desmosomes, tonofilaments, and, subsequently, basement membrane and anchoring fibrils causing secondary changes in the corneal stroma. Such changes include deposition of diffusely arranged electron-dense amyloid material, degenerated keratocytes (mesenchymal cells) with cytoplasmic vesicles, and thinned collagen fibrils.6,28 These features may reflect a deficiency of unfolded protein response in the ER.
The measurements of sterol responsiveness indicate that the sterol regulatory element-binding protein site 2 protease activity of the MBTPS2 enzyme is diminished as a consequence of the IFAP mutations. MBTPS2 belongs to a family of HEXXH-containing zinc endopeptidases. In these enzymes, the catalytic zinc atom, typically, is located in the interphase between two alpha helices. One helix contains the HEXXH sequence, and the other contains a glutamic acid that also coordinates with the zinc atom. In MBTPS2 both motifs are located in hydrophobic segments of the protein predicted to be adjacent in the Golgi membrane.19 The mutations detected so far do not affect the HEIGH motif or the LD467G sequence coordinating the zinc at the active site of the enzyme (Figure S1). However, when projected upon a hypothetic model of MBTPS2,19 the interfamilial phenotypic differences between male IFAP patients and the properties of the mutants in functional assays invite predictions about a genotype-phenotype correlation: clinical severity in male IFAP patients may range from mild forms of the triad of ichthyosis follicularis, atrichia, and photophobia, which can be categorized as a deficiency in epithelial development compatible with survival and fertility of male patients as evident in family 2, to severe manifestations which, in addition to these features, include mental retardation and various developmental defects that may even cause early death (families 3 and 5). It appears that mutations close to the hydrophobic, presumably intramembranous domain encompassing the LDG motif at the active site are more detrimental to development than an amino acid exchange in the amino-terminal part of the protein (Figure S1).
The spectrum of potential substrates for MBTPS2 has not fully been elucidated. Therefore, it is premature to speculate whether the demonstrated effect upon sterol-regulated expression is the predominant deficiency in all patients or whether activation of different transcription factors by binding and cleavage via the S1P/S2P mechanism is affected by structural changes within critical motifs. Detection of further mutations in MBTPS2 associated with meticulous clinical description of male IFAP patients, unveiling of the crystal structure of the protease, and progress in understanding of the biochemical mechanism of its enzymatic action must be awaited to substantiate genotype-phenotype predictions.
Functional MBTPS2 appears to be essential for development. Consequently, deletions of this gene in all cells of a male embryo might not be tolerated. As recently shown for the X-linked dominant, male-lethal trait focal dermal hypoplasia (FDH [MIM 305600]), complete loss of an essential function in male patients is only compatible with survival when present as a postzygotic mosaic or in males with more than one X chromosome.18 Accordingly, a mosaic situation should be considered in sporadic male IFAP patients in which mutations in MBTPS2 are not detected in DNA from blood.
The notion that, at least, a residual protease activity is needed for survival is compatible with the lack of knockout mouse models for this enzyme. Our description of a human-inherited disease associated with functional deficiency of MBTPS2 opens a new avenue for studying the role of this important intramembrane protease.
Supplemental Data
Supplemental data include two tables and one figure and can be found with this article at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Center for Medical Genetics, Marshfield Medicine Research Foundation, http://www.marshmed.org/genetics/
Genome Database (GDB), http://www.gdb.org
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/SNP/snp_blastByOrg.cgi
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim
SpliceView online splice site prediction, http://bioinfo.itb.cnr.it/oriel/splice-view.html
UCSC Genome Browser, http://genome.cse.ucsc.edu/
Acknowledgments
We are grateful to the patients and their families for their cooperation, and we thank Howard Cann, CEPH, Paris, for a control DNA sample, M. Soufi, Department of Internal Medicine, Philipp University, Marburg, for a supply of delipidated FCS, Dr. J.L. Goldstein, UT Southwestern Medical School, Dallas, Texas, for advice on where to trace research materials, T.Y. Chang, Dartmouth Medical School, Hanover, N.H., for providing CHO-K1-M19 cells, and S. Wiemann, DKFZ, Heidelberg, for the plasmid pdEYFP-C1amp. This research was supported by BMBF (Netzwerk für Ichthyosen und verwandte Verhornungsstörungen, NIRK) and EU (Coordination Action Geneskin). R.C.B. is a recipient of an Emmy Noether grant from the German Research Foundation (DFG).
References
- 1.Macleod J.M.H. Three cases of “ichthyosis follicularis” associated with baldness. Br. J. Dermatol. 1909;21:165–189. [Google Scholar]
- 2.Boente M.C., Bibas-Bonet H., Coronel A.M., Asial R.A. Atrichia, ichthyosis, follicular hyperkeratosis, chronic candidiasis, keratitis, seizures, mental retardation and inguinal hernia: A severe manifestation of IFAP syndrome? Eur. J. Dermatol. 2000;10:98–102. [PubMed] [Google Scholar]
- 3.Sato-Matsumura K.C., Matsumura T., Kumakiri M., Hosokawa K., Nakamura H., Kobayashi H., Ohkawara A. Ichthyosis follicularis with alopecia and photophobia in a mother and daughter. Br. J. Dermatol. 2000;142:157–162. doi: 10.1046/j.1365-2133.2000.03260.x. [DOI] [PubMed] [Google Scholar]
- 4.Cambiaghi S., Barbareschi M., Tadini G. Ichthyosis follicularis with atrichia and photophobia (IFAP) syndrome in two unrelated female patients. J. Am. Acad. Dermatol. 2002;46:S156–S158. doi: 10.1067/mjd.2002.112930. [DOI] [PubMed] [Google Scholar]
- 5.Khandpur S., Bhat R., Ramam M. Ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome treated with acitretin. J. Eur. Acad. Dermatol. Venereol. 2005;19:759–762. doi: 10.1111/j.1468-3083.2005.01318.x. [DOI] [PubMed] [Google Scholar]
- 6.Traboulsi E., Waked N., Mégarbané H., Mégarbané A. Ocular findings in ichthyosis follicularis-alopecia-photophobia (IFAP) syndrome. Ophthalmic Genet. 2004;25:153–156. doi: 10.1080/13816810490514405. [DOI] [PubMed] [Google Scholar]
- 7.Keyvani K., Paulus W., Traupe H., Kiesewetter F., Cursiefen C., Huk W., Raab K., Orth U., Rauch A., Pfeiffer R.A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: Clinical and neuropathological observations in a 33-year-old man. Am. J. Med. Genet. 1998;78:371–377. doi: 10.1002/(sici)1096-8628(19980724)78:4<371::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Mégarbané H., Zablit C., Waked N., Lefranc G., Tomb R., Mégarbané A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: Report of a new family with additional features and review. Am. J. Med. Genet. A. 2004;124A:323–327. doi: 10.1002/ajmg.a.20352. [DOI] [PubMed] [Google Scholar]
- 9.Happle R. What is IFAP syndrome? Am. J. Med. Genet. A. 2004;124A:328. doi: 10.1002/ajmg.a.20353. [DOI] [PubMed] [Google Scholar]
- 10.König A., Happle R. Linear lesions reflecting lyonization in women heterozygous for IFAP syndrome (ichthyosis follicularis with atrichia and photophobia) Am. J. Med. Genet. 1999;85:365–368. doi: 10.1002/(sici)1096-8628(19990806)85:4<365::aid-ajmg12>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Hasan M.T., Chang C.C., Chang T.Y. Somatic cell genetic and biochemical characterization of cell lines resulting from human genomic DNA transfections of Chinese hamster ovary cell mutants defective in sterol-dependent activation of sterol synthesis and LDL receptor expression. Somat. Cell Mol. Genet. 1994;20:183–194. doi: 10.1007/BF02254759. [DOI] [PubMed] [Google Scholar]
- 12.Rawson R.B., Zelenski N.G., Nijhawan D., Ye J., Sakai J., Hasan M.T., Chang T.Y., Brown M.S., Goldstein J.L. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell. 1997;1:47–57. doi: 10.1016/s1097-2765(00)80006-4. [DOI] [PubMed] [Google Scholar]
- 13.Simpson J.C., Wellenreuther R., Poustka A., Pepperkok R., Wiemann S. Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 2000;1:287–292. doi: 10.1093/embo-reports/kvd058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briggs M.R., Yokoyama C., Wang X., Brown M.S., Goldstein J.L. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J. Biol. Chem. 1993;268:14490–14496. [PubMed] [Google Scholar]
- 15.Lisch W., Büttner A., Oeffner F., Boddeker I., Engel H., Lisch C., Ziegler A., Grzeschik K.-H. Lisch corneal dystrophy is genetically distinct from Meesmann corneal dystrophy and maps to xp22.3. Am. J. Ophthalmol. 2000;130:461–468. doi: 10.1016/s0002-9394(00)00494-3. [DOI] [PubMed] [Google Scholar]
- 16.O'Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 18.Grzeschik K.-H., Bornholdt D., Oeffner F., König A., del Carmen Boente M., Enders H., Fritz B., Hertl M., Grasshoff U., Höfling K. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat. Genet. 2007;39:833–835. doi: 10.1038/ng2052. [DOI] [PubMed] [Google Scholar]
- 19.Zelenski N.G., Rawson R.B., Brown M.S., Goldstein J.L. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999;274:21973–21980. doi: 10.1074/jbc.274.31.21973. [DOI] [PubMed] [Google Scholar]
- 20.Sakai J., Duncan E.A., Rawson R.B., Hua X., Brown M.S., Goldstein J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 21.Lai E., Teodoro T., Volchuk A. Endoplasmic reticulum stress: Signaling the unfolded protein response. Physiology (Bethesda) 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 22.Stirling J., O'Hare P. CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol. Biol. Cell. 2006;17:413–426. doi: 10.1091/mbc.E05-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye J., Rawson R.B., Komuro R., Chen X., Dave U.P., Prywes R., Brown M.S., Goldstein J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 24.Elias P.M., Williams M.L., Holleran W.M., Jiang Y.J., Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: Inherited disorders of lipid metabolism. J. Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elias P.M., Crumrine D., Rassner U., Hachem J.P., Menon G.K., Man W., Choy M.H., Leypoldt L., Feingold K.R., Williams M.L. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J. Invest. Dermatol. 2004;122:314–319. doi: 10.1046/j.1523-1747.2003.22258.x. [DOI] [PubMed] [Google Scholar]
- 26.König A., Happle R., Bornholdt D., Engel H., Grzeschik K.-H. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am. J. Med. Genet. 2000;90:339–346. [PubMed] [Google Scholar]
- 27.Braverman N., Lin P., Moebius F.F., Obie C., Moser A., Glossmann H., Wilcox W.R., Rimoin D.L., Smith M., Kratz L. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat. Genet. 1999;22:291–294. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 28.Cursiefen C., Schlötzer-Schrehardt U., Holbach L.M., Pfeiffer R.A., Naumann G.O.H. Ocular findings in ichthyosis follicularis, atrichia, and photophobia syndrome. Arch. Ophthalmol. 1999;117:681–684. doi: 10.1001/archopht.117.5.681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.