

Abstract
The short-rib polydactyly (SRP) syndromes are a heterogenous group of perinatal lethal skeletal disorders with polydactyly and multisystem organ abnormalities. Homozygosity by descent mapping in a consanguineous SRP family identified a genomic region that contained DYNC2H1, a cytoplasmic dynein involved in retrograde transport in the cilium. Affected individuals in the family were homozygous for an exon 12 missense mutation that predicted the amino acid substitution R587C. Compound heterozygosity for one missense and one null mutation was identified in two additional nonconsanguineous SRP families. Cultured chondrocytes from affected individuals showed morphologically abnormal, shortened cilia. In addition, the chondrocytes showed abnormal cytoskeletal microtubule architecture, implicating an altered microtubule network as part of the disease process. These findings establish SRP as a cilia disorder and demonstrate that DYNC2H1 is essential for skeletogenesis and growth.
Main Text
Cilia are highly conserved microtubule-based organelles that project from the cell surface into the extracellular environment and play diverse roles in cellular motility, sensory transduction, and signaling. Cilia consist of a microtubule-based axoneme made of nine peripheral microtubule doublets arranged around a central core; motile cilia contain two central microtubules (9 + 2), whereas nonmotile or primary cilia do not (9 + 0).1,2 During early stages of cilia formation, the centriolar vesicle docks at the plasma membrane to form the axonemal shaft and basal body. Further elongation and maintenance of the cilia is dependent on the dynamic process of intraflagellular transport (IFT). IFT bidirectionally transports ciliary cargo with the help of motors along the outer microtubules of the axoneme. Anterograde movement of axonemal precursors from the basal body to the distal tip is driven by the heterotrimeric kinesin-II motor. Retrograde transport from the distal tip to the basal body is accomplished by the cytoplasmic dynein 2 complex.
In humans and mice, ciliary dysfunction is associated with a large spectrum of disorders,3–5 including defects in a variety of proteins necessary for IFT as well as in components of the primary cilia, basal body, and centrosome. Many of these phenotypes include polydactyly, and some include abnormal skeletogenesis. In particular, some cases of asphyxiating thoracic dystrophy (ATD [MIM 208500]) result from mutations in the gene encoding the anterograde transport protein IFT806 (MIM 611177). About two-thirds of patients with Ellis-van Creveld (EVC [MIM 225500]) syndrome have mutations in either EVC1 (MIM 604831) or EVC27–9 (MIM 607261), and the Evc1 gene product has been localized to the ciliary basal body in mouse chondrocytes, where it participates in hedgehog signal transduction.10
The short-rib polydactyly syndromes are among the most frequent autosomal-recessive osteochondrodysplasias.11 Four distinct types (SRP I [Saldino-Noonan type (MIM 263530)]; SRP II [Majweski type (MIM 263520)]; SRP III [Verma-Neumoff type (MIM 263510)]; and SRP IV [Beemer type (MIM 269860)]) are recognized, and the current nosology of the osteochondrodysplasias classifies types I and III together.12 However, many cases defy classification and demonstrate transitional findings between the types, suggesting that SRP could be considered a single group of disorders with a spectrum of variable phenotypic expression.13–16 SRPs manifest extremely shortened long bones, a small, narrow thorax, and frequent pre- and postaxial polydactyly. Other multisystem anomalies, including brain and cardiac malformations (including situs inversus), polycystic kidneys, gastrointestinal and genitourinary atresia, and occasional sex reversal, are also observed. Although genetically distinct in some instances,17 the SRPs share phenotypic and radiographic findings with ATD and EVC, supporting the hypothesis that these diseases comprise a family of disorders that may be functionally related.
Under an IRB-approved protocol for human subjects, we ascertained a consanguineous family (International Skeletal Dysplasia Registry reference number R01-314) with first-cousin parents and four affected offspring. Although a diagnosis of SRP type III was assigned, phenotypic analysis showed intrafamilial variability. At term, the proband (R01-314A, II-2) had shortened horizontal ribs, a trident pelvis, and metaphyseal spikes at the lateral ends of the femora, with neither polydactyly nor other organ system abnormalities (Figures 1A–1C). The other affected fetuses, R01-314B (II-3) (Figure 1F), R01-314C (II-4), and R01-314F (II-5) (20 weeks, 16 weeks, and 11 weeks gestational age, respectively) showed similar radiographic findings but also had postaxial polydactyly of the hands and feet. For R01-304B, C, and F (II-3, 4, and 5), prenatal ultrasound did not identify abnormalities in the brain, heart, kidneys, or liver.
Figure 1.
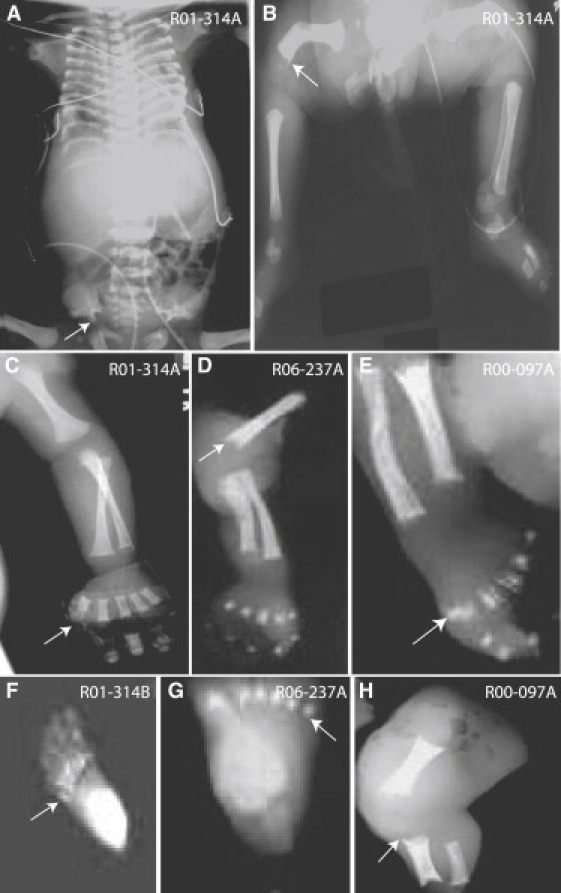
Radiographic Hallmarks of SRP
(A) R01-314A, newborn. A/P view showing the characteristic trident pelvis (arrow) and the long narrow chest with horizontal ribs.
(B) R01-314A, newborn. The arrow identifies the distal end of the femur, showing metaphyseal spikes.
(C) R01-314A, newborn. There is no polydactyly in the hand (arrow).
(D) R06-237A, 20 weeks gestation. The arrow identifies metaphyseal spikes of the distal humerus.
(E) R00-097A, 20 weeks gestation. The arrow identifies the preaxial polydactyly of the foot.
(F) R01-314B, 20 weeks gestation. The arrow identifies postaxial polydactyly of the foot.
(G) R06-237A, 20 weeks gestation. The arrow identifies postaxial polydactyly of the foot.
(H) R00-097A, 20 weeks gestation. The lower extremity shows a shortened femur, tibia, and fibula with metaphyseal spikes (arrow).
Two nonconsanguineous SRP type III cases were also studied. Case R00-097A (II-1) exhibited preaxial polydactyly and shortened long bones with some metaphyseal spikes (Figures 1E and 1H). Case R06-237A (II-1) had similar radiographic findings but exhibited postaxial rather than pre-axial polydactyly (Figures 1D and 1G). Abnormalities of the visceral organs were not noted in these cases.
To define the molecular basis of SRP, we used whole-genome SNP analysis to identify regions of homozygosity that were greater than 1 Mb and were shared among the affected individuals in the consanguineous family but not shared with the unaffected sibling. SNP data were generated with the NSPI array from the GeneChip Human Mapping 500K Array Set (Affymetrix) in the UCLA Microarray Core facility according to manufacturer-recommended protocols. Three intervals, totaling approximately 60.7 Mb, were identified on chromosomes 1, 11, and 16 (Figure 2). Under the hypothesis that SRP would result from a defect in cilia function, genes with a possible role in cilia were identified within these intervals via the cilia proteome database (see URLs below). One gene, DYN2CH1 (MIM 603297) at chromosome 11q21-22 was a particularly attractive candidate because homozygosity for either point mutations or null mutations in the mouse ortholog resulted in polydactyly.18–20 DYNC2H1 encodes a 4,307 amino acid ciliary protein that has a predicted molecular mass of approximately 492 kD and plays a role in retrograde transport in the cilium.21
Figure 2.
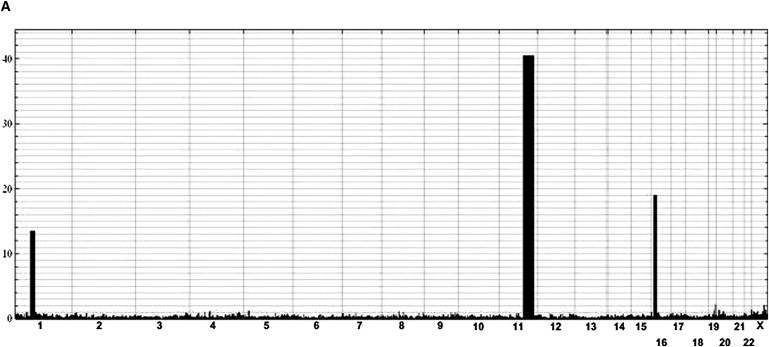
Homozygosity by Descent Mapping
(A) Homozygosity by descent mapping in family R01-314. All intervals of homozygosity shared among the four affected family members of family R01-314 are plotted across the genome. Chromosomal position is indicated on the x axis, and length of the homozygous interval is plotted on the y axis such that height of the filled-in graphic indicates the length of the measured homozygous intervals in centimorgans. Physical distances: chromosome 1 (rs11209979-rs4656100; 71,153,296-85,028,578bp), chromosome 11 (rs 10501715-rs1029278; 86,043.913-119,363,025bp), and chromosome 16 (rs12924823-rs12928078; 10,726,309-24,280,658bp).
The 90 coding exons of DYNC2H1 were amplified by PCR (oligonucleotide sequences; see Table S1 in the Supplemental Data available online), and the sequences of the PCR products were determined by bidirectional sequence analysis (MC Labs) and compared with the reference sequence for DYNC2H1 with Sequencher (Gene Codes). Nucleotides were numbered according to the mRNA sequence, starting from the A of the ATG initiation codon. The affected individuals in family R01-314 were all homozygous for an exon 12 missense mutation, c.1759C > T, predicted to lead to the amino acid substitution p.R587C (Figure 3A). This residue is conserved among DYNC2H1 orthologs from Homo sapiens through Caenorhabditis elegans. The unaffected parents were both heterozygous for the mutation, and the unaffected sibling carried only the wild-type sequence. The sequence change was not found among 214 control chromosomes, indicating that it is not a common polymorphism in the population.22
Figure 3.
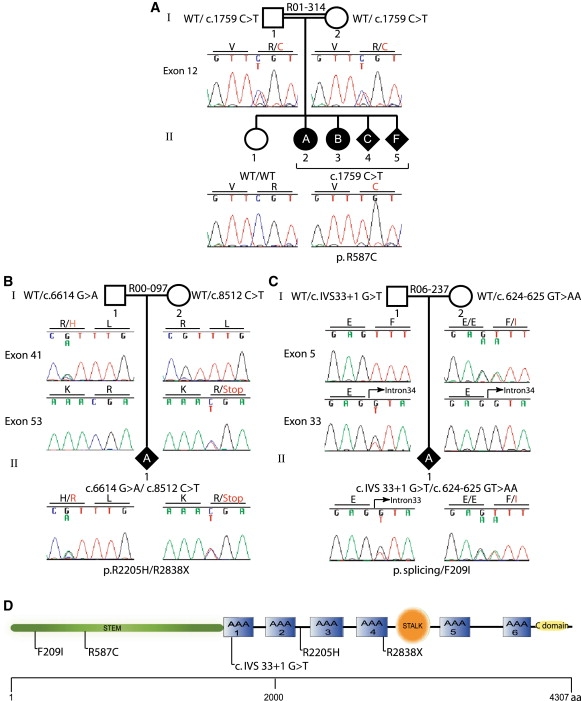
DYNC2H1 Mutation Analysis
Sequencing chromatograms demonstrating mutations in DYNC2H1 in the three families studied: (A) R01-314, (B) R00-097, and (C) R06-237. (D) Diagram of the DYNC2H1 molecule with the locations of the protein sequence changes predicted by the mutations. The scale below indicates the length of the protein. AAA, ATP binding and hydrolysis domains; C domain, carboxyl-terminal domain; aa, amino acid.
Compound heterozygosity for DYNC2H1 mutations was identified in the two nonconsaguineous SRP type III cases. In case R00-097A (II-1), mutations in exons 41 (c.6614 G > A) and 53 (c.8512 C > T), predicted to lead to the amino acid substitution p.R2205H, and a nonsense mutation, p.R2838X, were found (Figure 3B). These sequence changes were not found among 210 control chromosomes. For case R06-237A (II-1), a change of two consecutive base pairs was identified in exon 5 (c.624-625 GT > AA); the first nucleotide change altered the last base of codon 208 without changing the encoded amino acid, and the second change predicted the amino acid substitution F209I (Figure 3C). This sequence change was not found among 210 control chromosomes. The second mutation, which was not found among 210 control chromosomes, altered the splice donor in intron 33 (c.IVS 33+1, G > T). To examine the consequences of the splice donor mutation on splicing, we carried out RT-PCR on RNA extracted from cultured chondrocytes. Primary chondrocytes were isolated from distal femurs of affected individuals or age-matched normal controls by incubation of cartilage with 0.03% bacterial collagenase II (Sigma-Aldrich) at 37°C for 8 hr. Liberated chondrocytes were propagated in DMEM with 10% FBS. RNA was isolated with Trizol (Invitrogen) according to standard protocols. The RT-PCR experiments demonstrated that the mutation did not result in the skipping of exon 33 (data not shown). However, when primers E4F: 5′ TGAGTTCCAGTTTTGGATAGAACA3′ and E6R: 5′ATTTTTCCATGGATGAGGAACATA3′ were used for amplifying a 512 bp pair product spanning exons 4-6, sequence analysis of the resulting RT-PCR products containing exon 5 showed that most of the transcripts were derived from the c.624-625 GT > AA missense allele (Figure 4A). These data were consistent with loss of the exon 5 wild-type sequence derived from the allele with the splice-donor mutation. The diminished abundance of the products of the splice-donor mutant allele was partially reversed when the cells were incubated with cyclohexamide (Sigma-Aldrich) at a concentration of 100 μg/ml at 37°C for 8 hr (Figure 4B), consistent with loss of the transcripts by nonsense-mediated decay.23 To corroborate these results, we performed quantitative PCR analysis for DYNC2H1 chondrocyte cDNA in triplicate (SABioscience) (Stragene, MX3005P). Results showed an almost 50% reduction in DYNC2H1 cDNA levels in R06-237A compared to controls (Figure 4C). We thus conclude that the intron 33 mutation in R06-237A (c.IVS 33+1, G > T) results in a null allele.
Figure 4.
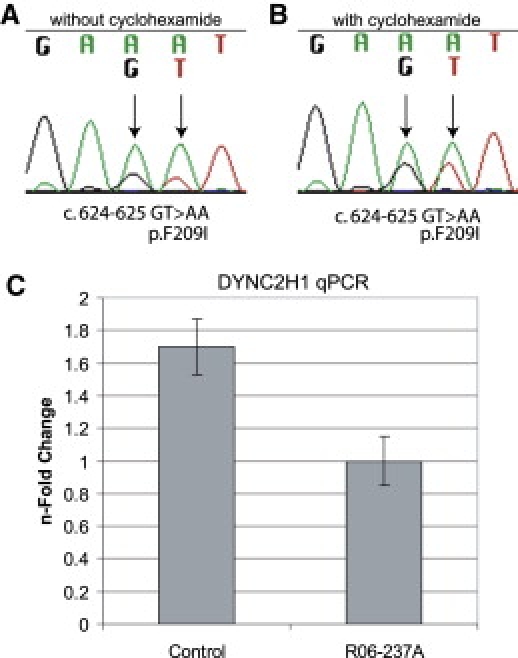
c. IVS33+1 G > T Allele Found in R06-237 Leads to Nonsense-Mediated Decay
Sequencing chromatograms of RT-PCR products from R06-237A chondrocytes generated in the absence (A) and presence (B) of cycloheximide. (C) Quantitative PCR analysis showed an almost 50% reduction in DYNC2H1 transcripts in R06-237A chondrocytes compared to stage-matched control chondrocytes. Vertical bars indicate the standard deviation.
On the basis of the function of DYNC2H1 and the abnormalities seen in the mouse mutants,18–20 the SRP mutations identified would be predicted to lead to morphologically abnormal cilia that could be visualized by scanning electron microscopy. To examine the cilia, we seeded primary chondrocytes on 13 mm round Thermanox coverslips. After a 24 hr incubation in DMEM with 10% FBS, confluent cells were serum starved in DMEM with 0.5% FBS for 48 hr. Cells were fixed in 2.5% glutaraldehyde/DMEM for 40 min, washed with 0.1 M sodium phosphate buffer, and fixed in Karnovsky's fixative (2% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 M sodium phosphate buffer) for 1.5 hr. After fixation, cells were washed with 0.1 M sodium phosphate buffer, dehydrated through a graded ethanol series, and processed for critical point drying and coating. Samples were imaged with a Cambridge 360 scanning electron microscope (Carl Zeiss, Thornwood, NY). The cilia in mutant cultured chondrocytes from case R01-314B were shortened and had bulbous distal ends (Figures 5B and 5D) in comparison to controls (Figures 5A and 5C). Although shortening of all mutant cilia was observed, some chondrocytes had no discernable cilia projecting from their cell surfaces.
Figure 5.
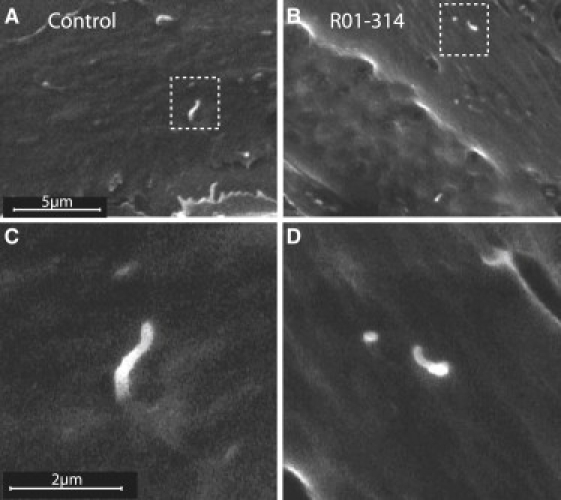
p.R587C Allele Found in R01-314 Produces Abnormal Primary Cilia in Chondrocytes
(A) Cilia visualized by scanning electron microscopy showed normally shaped cilia in control chondrocytes. The boxed area is shown at higher magnification in (C). (B) Cilia present on chondrocytes from case R01-314B were short and had an abnormal bulge at the tip of the axoneme. The boxed area is shown at higher magnification in (D).
Additional structural information for the chondrocyte cilia was provided by fluorescence microscopy. For these studies, primary chondrocytes were seeded on four-well chamber slides (Lab-Tek). After 24 hr of growth, confluent cells were serum starved in DMEM with 0.5% FBS for 48 hr. Cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, permeablized with PBS plus 0.1% Tween-20, blocked with 10% goat serum (Zymed) for 1 hr, and incubated with monoclonal acetylated α-tubulin antibody (1/1000, Clone 6-11B-1, Sigma-Aldrich) and γ-tubulin antibody (1/500, T3559, Sigma-Aldrich) diluted in 1% goat serum/PBST overnight at 4°C. Primary antibodies were detected with Alexa Fluor 568 goat anti-mouse antibody (Molecular Probes, Invitrogen) diluted 1/400 and Alexa Fluor 488 goat anti-rabbit antibody (Molecular Probes, Invitrogen) in 1% goat serum/PBST at room temperature for 1 hr. Slides were coverslipped with Vectashield and DAPI. Confocal images were taken sequentially on a Leica TCS-SP2 AOBS inverted confocal microscope (Mannheim, Germany) equipped with a 405 nm blue diode laser, argon laser (488 nm blue excitation: JDS Uniphase), and three helium neon lasers (543 nm, green excitation; 594 nm, near-red excitation; and 633 nm far-red excitation). As observed by SEM, immunofluorescence for acetylated α-tubulin, a known marker for cilia, in cultured chondrocytes from cases R01-314B and R06-237A demonstrated that, in comparison to control cells (Figures 6A and 6D), the cilia were shorter and abnormally shaped and had a bulbous tip (Figures 6, 6C, 6E, and 6F). Distinct from the SEM findings, the acetylated α-tubulin antibody identified abbreviated, cilia-like structures in all mutant chondrocytes (Figures 6B and 6C). Cilia length was an average of 3.2 ± 0.57 μm in control cell and 1.9 ± 0.44 μm in SRP chondrocytes (p < 0.01). Interrogation with γ-tubulin antibody (Figures 6G–6I) identified the basal body in SRP cilia and confirmed that the bulbus projection seen by SEM in the axoneme was at the distal tip (Figures 6H and 6I). Together, these data suggest that the early stages of cilia formation, which involve docking of the basal body to the plasma membrane, remained intact, whereas elongation and/or maintenance of the axoneme by IFT to form the mature primary cilium was abnormal.
Figure 6.
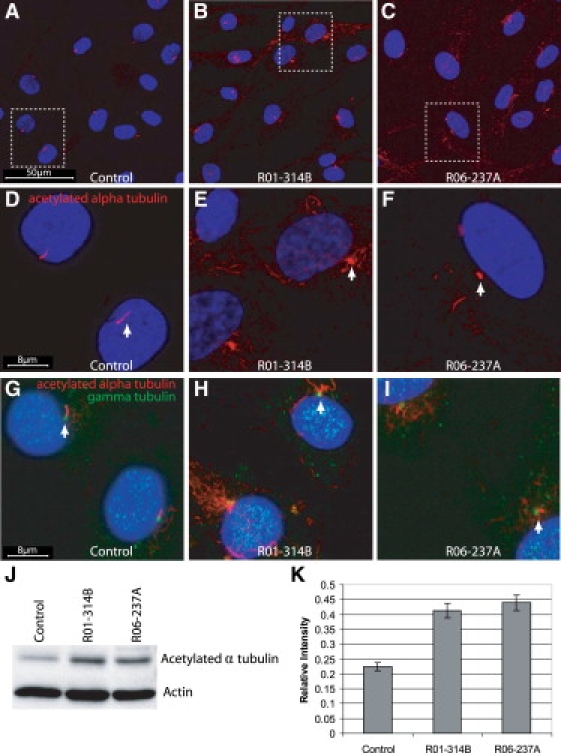
SRP Chondrocytes Have Abnormal Primary Cilia and Increased Levels of Acetylated α-tubulin
(A and D) Immunofluorescence for acetylated α-tubulin (red) and DAPI (blue) in primary chondrocytes shows normal presentation of cilia in control cells. Chondrocytes from R01-314B (B and E) and R06-237A (C and F) show abnormal cilia and increased amounts of cytoplasmic acetylated α-tubulin. (D–F) Higher-magnification views of the boxed regions in (A)–(C). Cilia length measured an average of 3.2 ± 0.57 μm in control cells and 1.9 ± 0.44 μm in SRP chondrocytes (p, 0.01, Student's t test). (G–I) Immunofluorescence for acetylated α-tubulin (red), γ tubulin (green), and DAPI (blue) shows the presence of a basal body in both control and SRP chondrocytes. (J) Immunoblot analysis of acetylated α-tubulin expression in control, R01-314A, and R06-237B chondrocytes showed an increase of acetylated α-tubulin expression in mutant chondrocytes compared to the control. β-actin was used as a protein loading control. (K) Quantitation of the ratio of acetylated α-tubulin relative to β-actin from the immunoblots (n = 4) demonstratied about a 2-fold increase in the relative level of acetylated α-tubulin in mutant chondrocytes compared to the normal control. Vertical bars indicate the standard deviation.
In addition to the structural defect in the cilia, the immunofluorescence studies of SRP chondrocytes showed both abnormally fragmented and increased amounts of cytoplasmic acetylated α-tubulin (Figures 6B, 6C, 6E, 6F, 6H, and 6I) in comparison to controls (Figures 6A, 6D, and 6G), consistent with a defect in cytoskeletal microtubule architecture. To examine these findings quantitatively, we carried out immunoblot analysis for acetylated α-tubulin (Figure 6J). Proteins from primary chondrocytes were extracted in RIPA buffer and resolved under reducing conditions by electrophoresis on a 10% SDS PAGE gel. Proteins were transferred to a nitrocellulose membrane (BioRad) and blocked for 1 hr in 5% milk/TBS and 0.1% Tween-20 (TBST). Membranes were probed with monoclonal acetylated tubulin antibody (Clone 6-11B-1, Sigma-Aldrich) diluted 1/2000 or β-actin antibody (Cell Signaling) diluted 1/1000 in 5% BSA/TBST overnight at 4°C. Membranes were washed in TBST and incubated with HRP-conjugated goat anti-mouse secondary antibody (Immuno Pure, Thermo Science) diluted 1/5000 in 1% milk/TBST for 1 hr at room temperature. Immunoreactivity was detected with a chemiluminescence detection system (Cell Signaling Technology). Immunoblots were performed in quadruplicate, and quantitation of band intensity demonstrated an approximately 2-fold increase in acetylated α-tubulin in the SRP samples relative to the control (Figure 6K).
Microtubules exhibit dynamic instability in that they build and disassemble in response to the needs of the cell.24 Alterations in stabilization of the microtubules through acetylation of α-tubulin and destabilization by α-tubulin tyrosination are known to disrupt cellular functions.25 In Dictyostelium, the dynein complex stabilizes interphase microtubule arrays, and disruptions result in a disorganized microtubule cytoskeleton.26,27 Because the cytoplasmic dyneins in the basal body of the cilium have a microtubule-binding domain and interact with microtubules via the microtubule-organizing center of the centrosome, the microtubule structural abnormalities identified in SRP raise the possibility that loss of DYNC2H1 function could affect movement of organelles and transport of critical cargo necessary for signal transduction and skeletal development.
Cytoplasmic dyneins are composed of heavy-chain homodimers with differing accessory subunits. All dyneins share a common structure consisting of an amino-terminal stem region, a linker region, and six identifiable ATP-binding and -hydrolysis domains (AAA), and a stalk between AAA domains 4 and 5 is implicated in microtubule binding (Figure 3D).28,29 The amino-terminal stem of the molecule is involved in homodimerization and binding of accessory units to form the complex that binds cargo. The F209I and R587C substitutions found in the SRP cases alter the amino-terminal stem and thus may interfere with these functions. The R2205H substitution is localized in an AAA unit and could hinder ATP hydrolysis and generation of adequate force for microtubule movement, although the exact mechanism by which dyneins convert the energy released by ATP hydrolysis into force is not fully understood.29 The nonconsanguineous cases were both compound heterozygotes for one missense and one null mutation. It is therefore apparent that synthesis of inadequate amounts of the gene product along with expression of a mutant protein produces SRP in these cases. We speculate that homozygosity for two null alleles would result in early embryonic lethality but that a range of phenotypes could result from a combination of a null mutation and a variety of missense mutations in this large and complex molecule.
The abnormalities in SRP are primarily related to the effect on the skeleton, reflecting an essential role for DYNC2H1 in cilia function in cartilage. Chondrocyte cilia are embedded in a rich extracellular matrix that includes growth factors and signaling molecules that signal through cell surface receptors30 to organize the development of a properly patterned skeleton capable of promoting linear growth. The ciliary abnormalities in SRP suggest that cilia are critical to chondrocytes' ability to detect and transmit these signals, as well as mechanosensory signals,31 in that they provide a link through which the cell can monitor the extracellular environment and translate cues that facilitate cellular processes important for chondrocyte proliferation.32–34 Identification of the specific signaling pathways or cargos altered in SRP will aid in determining how defects in DYNC2H1 produce their profound effect on the skeleton. Our findings firmly establish SRP as a member of the increasingly large group of cilia disorders and reveal an essential function for DYNC2H1 in skeletogenesis and growth.
Acknowledgments
We thank the families for their active and longstanding participation in this project and particularly acknowledge the [srps] family network for their advocacy on behalf of the study. This project was supported in part by a grant from the NIH (HD22657) and the Joseph Drown Foundation. Fluorescence microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Facility at UCLA.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.cov/Omim
Cilia proteome website, http://www.ciliaproteome.org/
Mouse Genome Informatics database, http://www.informatics.jax.org/
[srps] family network, www.srps.net
Accession Numbers
The NCBI accession number for DYNC2H1 is NM_001080463.
References
- 1.Scholey J.M. Intraflagellar transport. Annu. Rev. Cell Dev. Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- 2.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 3.Eggenschwiler J.T., Anderson K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bisgrove B.W., Yost H.J. The roles of cilia in developmental disorders and disease. Development. 2006;133:4131–4143. doi: 10.1242/dev.02595. [DOI] [PubMed] [Google Scholar]
- 5.Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz-Perez V.L. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000;25:125. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- 8.Galdzicka M., Patnala S., Hirshman M.G., Cai J.F., Nitowsky H., Egeland J.A., Ginns E.I. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol. Genet. Metab. 2002;77:291–295. doi: 10.1016/s1096-7192(02)00178-6. [DOI] [PubMed] [Google Scholar]
- 9.Tompson S.W., Ruiz-Perez V.L., Blair H.J., Barton S., Navarro V., Robson J.L., Wright M.J., Goodship J.A. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet. 2007;120:663–670. doi: 10.1007/s00439-006-0237-7. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Perez V.L., Blair H.J., Rodriguez-Andres M.E., Blanco M.J., Wilson A., Liu Y.N., Miles C., Peters H., Goodship J.A. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007;134:2903–2912. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- 11.Krakow D., Alanay Y., Rimoin L.P., Lin V., Wilcox W.R., Lachman R.S., Rimoin D.L. Evaluation of prenatal-onset osteochondrodysplasias by ultrasonography: A retrospective and prospective analysis. Am. J. Med. Genet. A. 2008;146A:1917–1924. doi: 10.1002/ajmg.a.32269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 13.Franceschini P., Guala A., Vardeu M.P., Signorile F., Franceschini D., Bolgiani M.P. Short rib-dysplasia group (with/without polydactyly): Rreport of a patient suggesting the existence of a continuous spectrum. Am. J. Med. Genet. 1995;59:359–364. doi: 10.1002/ajmg.1320590316. [DOI] [PubMed] [Google Scholar]
- 14.Elcioglu N.H., Hall C.M. Diagnostic dilemmas in the short rib-polydactyly syndrome group. Am. J. Med. Genet. 2002;111:392–400. doi: 10.1002/ajmg.10562. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein R., Isdale J., Pinto M., Du Toit Zaaijman J., Jenkins T. Short rib-polydactyly syndrome: A single or heterogeneous entity? A re-evaluation prompted by four new cases. J. Med. Genet. 1985;22:46–53. doi: 10.1136/jmg.22.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Frias M.L., Bermejo E., Urioste M., Huertas H., Arroyo I. Lethal short rib-polydactyly syndromes: Further evidence for their overlapping in a continuous spectrum. J. Med. Genet. 1993;30:937–941. doi: 10.1136/jmg.30.11.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krakow D., Salazar D., Wilcox W.R., Rimoin D.L., Cohn D.H. Exclusion of the Ellis-van Creveld region on chromosome 4p16 in some families with asphyxiating thoracic dystrophy and short-rib polydactyly syndromes. Eur. J. Hum. Genet. 2000;8:645–648. doi: 10.1038/sj.ejhg.5200507. [DOI] [PubMed] [Google Scholar]
- 18.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 20.Ocbina P.J., Anderson K.V. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev. Dyn. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pazour G.J., Dickert B.L., Witman G.B. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J. Cell Biol. 1999;144:473–481. doi: 10.1083/jcb.144.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 24.Desai A., Mitchison T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 25.Yoon S.Y., Choi J.E., Choi J.M., Kim D.H. Dynein cleavage and microtubule accumulation in okadaic acid-treated neurons. Neurosci. Lett. 2008;437:111–115. doi: 10.1016/j.neulet.2008.03.083. [DOI] [PubMed] [Google Scholar]
- 26.Koonce M.P., Samso M. Overexpression of cytoplasmic dynein's globular head causes a collapse of the interphase microtubule network in Dictyostelium. Mol. Biol. Cell. 1996;7:935–948. doi: 10.1091/mbc.7.6.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dujardin D.L., Vallee R.B. Dynein at the cortex. Curr. Opin. Cell Biol. 2002;14:44–49. doi: 10.1016/s0955-0674(01)00292-7. [DOI] [PubMed] [Google Scholar]
- 28.Sakato M., King S.M. Design and regulation of the AAA+ microtubule motor dynein. J. Struct. Biol. 2004;146:58–71. doi: 10.1016/j.jsb.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Vallee R.B., Hook P. Autoinhibitory and other autoregulatory elements within the dynein motor domain. J. Struct. Biol. 2006;156:175–181. doi: 10.1016/j.jsb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 30.McGlashan S.R., Jensen C.G., Poole C.A. Localization of extracellular matrix receptors on the chondrocyte primary cilium. J. Histochem. Cytochem. 2006;54:1005–1014. doi: 10.1369/jhc.5A6866.2006. [DOI] [PubMed] [Google Scholar]
- 31.Jensen C.G., Poole C.A., McGlashan S.R., Marko M., Issa Z.I., Vujcich K.V., Bowser S.S. Ultrastructural, tomographic and confocal imaging of the chondrocyte primary cilium in situ. Cell Biol. Int. 2004;28:101–110. doi: 10.1016/j.cellbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Yang S.S., Langer L.O., Jr., Cacciarelli A., Dahms B.B., Unger E.R., Roskamp J., Dinno N.D., Chen H. Three conditions in neonatal asphyxiating thoracic dysplasia (Jeune) and short rib-polydactyly syndrome spectrum: A clinicopathologic study. Am. J. Med. Genet. Suppl. 1987;3:191–207. doi: 10.1002/ajmg.1320280523. [DOI] [PubMed] [Google Scholar]
- 33.Erzen M., Stanescu R., Stanescu V., Maroteaux P. Comparative histopathology of the growth cartilage in short-rib polydactyly syndromes type I and type III and in chondroectodermal dysplasia. Ann. Genet. 1988;31:144–150. [PubMed] [Google Scholar]
- 34.Corsi A., Riminucci M., Roggini M., Bianco P. Short rib polydactyly syndrome type III: Histopathogenesis of the skeletal phenotype. Pediatr. Dev. Pathol. 2002;5:91–96. doi: 10.1007/s10024-001-0122-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.