

Abstract
The Epstein–Barr virus (EBV) nuclear protein 2 (EBNA2) and herpes simplex virion protein 16 (VP16) acidic domains that mediate transcriptional activation now are found to have affinity for p300, CBP, and PCAF histone acetyltransferases (HATs). Transcriptionally inactive point mutations in these domains lack affinity for p300, CBP, or PCAF. P300 and CBP copurify with the principal HAT activities that bind to EBNA2 or VP16 acidic domains through velocity sedimentation and anion-exchange chromatography. EBNA2 binds to both the N- and C-terminal domains of p300 and coimmune-precipitates from transfected 293T cells with p300. In EBV-infected Akata Burkitt's tumor cells that do not express the EBV encoded oncoproteins EBNA2 or LMP1, p300 expression enhances the ability of EBNA2 to up-regulate LMP1 expression. Through its intrinsic HAT activity, PCAF can further potentiate the p300 effect. In 293 T cells, P300 and CBP (but not PCAF) can also coactivate transcription mediated by the EBNA2 or VP16 acidic domains and HAT-negative mutants of p300 have partial activity. Thus, the EBNA2 and VP16 acidic domains can utilize the intrinsic HAT or scaffolding properties of p300 to activate transcription.
Epstein–Barr virus (EBV) nuclear protein 2 (EBNA2) is an activator of viral and cellular gene transactivation (1) and is essential for EBV-mediated primary B lymphocyte growth transformation (2, 3). EBNA2 binds to DNA sequence-specific cell proteins RBP-Jκ (4, 5) and PU.1/Spi-1(6) and activates transcription through its C-terminal acidic activation domain (7). By reverse genetic analyses, the acidic and RBPJκ/PU.1 interacting domains are the principal EBNA2 domains essential for lymphocyte growth transformation (8–11).
The acidic domain of EBNA2 is similar to herpes simplex VP16 acidic domain (12). Both acidic domains can bind transcription factors including RPA70, TAF40, TFIIB (13–15), and TFIIH (16). Point mutations in a key hydrophobic residue in these domains that abolish transcriptional activation also abolish biochemical interaction with these transcription factors, providing a genetic linkage between the biochemical interaction and transcriptional activity (8, 17). Moreover, the core VP16 domain can substitute for the core EBNA2 domain in transactivation and in chimeric EBV recombinant-mediated transformation of primary B lymphocytes (12).
More than 20 transcriptional activators have been shown to interact with coactivators such as p300, CBP, PCAF, GCN5, and TAF250 that are histone acetyltransferases (HATs) (18, 19). The HAT activities of CBP, PCAF, and GCN5 are important for their coactivating effects (20–23). In the experiments reported here, we set out to determine whether the EBNA2 and VP16 transcriptional activators also interact with coactivators that have intrinsic HAT activity.
Materials and Methods
Cell Culture.
The EBV-negative human Burkitt's lymphoma B cell line BJAB and the EBV-positive Burkitt's lymphoma B cell line Akata were grown in RPMI 1640 medium supplemented with 10% FCS (R10). 293 T cells were grown in DMEM supplemented with 10% FCS (D10). All cultures were maintained in a 5% CO2 atmosphere at 37°C.
Plasmids.
The glutathione S-transferase (GST) fusion to acidic domains of EBNA2 (amino acids 427–483) or VP16 (amino acids 413–490) and their transcriptionally null mutants EBNA2W454T (8) or VP16F442P (17) have been described. The GST-p300 fusion plasmids were described before (24). For transfection assays, GAL4-EBNA2 or GAL4-VP16 acidic domain fusion has been described (7). The reporter plasmid pFR-Luc is from Stratagene. The pCI-p300, p300 HAT mutants, pCI-PCAF, and PCAF HAT mutants expression vectors are a gift from Yoshihiro Nakatani at the National Institutes of Health. The pSG5-p300 used in Akata cell experiments was subcloned from CMVβ-p300 (25) as a HindIII-NotI fragment into pSG5.
Purification of His-Tagged Protein and GST Binding.
His-tagged p100 and EBNA2 were purified with Ni-nitrilotriacetic acid (NTA) beads (Qiagen). The wild-type and mutant forms of GST-EBNA2 or GST-VP16 fusion proteins were adsorbed to glutathione beads and used for pull-down assays from BJAB cell extracts (15).
HAT Assays and Purification of HAT Activity.
The HAT activity that bound to GST fusion-coated beads was determined by using 5 μl of 50% bead slurry in a liquid HAT assay (26). To purify EBNA2 acidic domain-associated HAT activity, lysates from 2 liters of BJAB cell culture were precleared with GST beads for 1 hr at 4°C in lysis buffer that consists of 10 mM Hepes (pH 8.0), 1% Nonidet P-40, 1 mM DTT, and protease inhibitors, with 100 mM NaCl. The precleared extracts were incubated with 2 ml of 50% GST-EBNA2 slurry in lysis buffer with 100 mM NaCl at 4°C for 4 hr. The beads then were washed in the same buffer five times. HAT activity was eluted from the GST-EBNA2 beads by five 10-min incubations in 1.2 ml of lysis buffer containing 200 mM NaCl. A final elution with 1.2 ml of lysis buffer containing 500 mM NaCl resulted in a 20% increase in HAT activity, and this usually was combined with the previous elutes. Aliquots of 2 ml of the eluates were loaded on one 35-ml, 10–30% sucrose gradient and centrifuged at 25,000 × g for 20 hr at 4°C in a SW27 rotor. Fractions of 1.5 ml were collected from the bottom, and a 5-μl sample from each fraction was assayed for HAT activity. Fractions containing HAT activity were pooled and applied to an anion-exchange column, UNO Q (Bio-Rad). Proteins were eluted with a linear 50–300 mM NaCl gradient. Fractions of 0.5 ml were collected, and a 2-μl fraction was used to assay HAT activity.
Transfections.
Ten million Akata cells were electroporated with a Bio-Rad Gene Pulser at 200 V and 960 μF. Cells were harvested 40 hr later, lysed directly in 250 μl SDS of sample buffer, and sonicated briefly. Whole-cell lysate (10 μl) was separated by SDS/PAGE and blotted with S12 to detect LMP1 expression. For 293 T cell transfection, 5 × 105 cells were transfected with 15 μl of Superfect solution (Qiagen). Cells were harvested 20 hr later for assaying luciferase activity with the luciferase assay kit from Promega.
Coimmunoprecipitation.
Transfected 293 T cells were resuspended in lysis buffer containing 170 mM NaCl. After the cell debris was spun down, supernatant was incubated with 15 μl of M2 beads at 4°C overnight. Beads were washed extensively in the same lysis buffer and then subjected to SDS/PAGE. The coimmunoprecipitation of EBNA2 was detected by PE2.
Results
HAT Binding to the EBNA2 and VP16 Acidic Domains.
To ascertain whether HATs specifically associate with the EBNA2 or VP16 acidic domains, GST fusion proteins with the wild-type or transcriptional null point mutant EBNA2 (8) or VP16 (17) acidic domains were used as affinity matrices for HATs in extracts from BJAB B cell lymphoma cells. As shown in Fig. 1, HATs did not bind to GST or to GST fusions to the transcriptional null point mutant EBNA2 or VP16 acidic domains. In contrast, considerably more HAT activity bound to GST fusions to wild-type EBNA2 or VP16 acidic domains than could be detected in the whole-cell extracts. This indicates that the EBNA2 and VP16 acidic domains have significant specific affinity for HATs. The HATs that bound to EBNA2 and VP16 acidic domains acetylated histones H3 and H4 (data not shown). When these HATs were eluted from GST-VP16, diluted in binding buffer, and incubated with GST-EBNA2, almost all of the HAT bound to GST-EBNA2. The reciprocal experiment yielded the same result. These data indicate that VP16 and EBNA2 acidic domains bind the same HAT(s).
Figure 1.
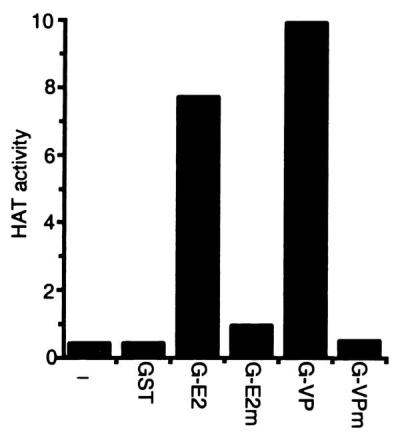
HAT activity from BJAB lysates binds specifically to the EBNA2 and VP16 acidic domains. The wild-type EBNA2 (E2) or VP16 (VP) or their null point mutant (E2m and VPm) acidic domains were fused to GST (G), and glutathione-Sepharose beads coated with fusion protein were used in pull downs with extracts from BJAB B cell lymphoma cells. The presence of HAT activity in proteins bound to beads was determined by liquid HAT assay. HAT activity was shown as 103 × count per minute. Results from one representative experiment of five similar experiments are shown.
Purification of the EBNA2-Bound HAT Results in Purification of p300/CBP.
To further characterize the EBNA2-associated HAT(s), three approaches were undertaken. First, because an EBNA2 and VP16 acidic domain-associated protein, p100 (15), was found in a HAT preparation (27), we considered the possibility that p100 might be the HAT that specifically bound to the EBNA2 and VP16 acidic domains. The potential intrinsic HAT activity of p100 was assayed by using recombinant 6×His-tagged p100 purified from Escherichia coli with Ni-NTA beads (15). However, His-tagged p100 on beads or after release from beads lacked intrinsic HAT activity. Furthermore, incubation of His-tagged p100 NTA beads with extracts of BJAB cells did not result in adsorption of detectable HAT activity to the p100 beads. Thus, p100 lacks intrinsic HAT activity and cannot bind HAT activity from lymphoblast extracts.
In a second series of experiments, we attempted to identify the size of the EBNA2- and VP16-associated HAT proteins. Some HATs such as hGCN5 and TAF250 retain activity after SDS/PAGE (28) and can be detected in an in-gel HAT assay (29). EBNA2- or VP16-bound HAT activities were eluted from beads, and in-gel HAT assays were run. No significant HAT activity could be detected under conditions that can detect hGCN5 or TAF250 (data not shown).
Having failed in specific attempts to identify the EBNA2- or VP16-associated HAT activity, we set out to biochemically characterize the activity. The HAT activity associated with EBNA2 was eluted from GST-EBNA2 beads with 0.2 M NaCl. A total of 5E7 cpm HAT activity was obtained from 2 liters of BJAB cell extracts. HAT activity could not be recovered after cation-exchange, size-exclusion, inorganic hydroxyapatite, or hydrophobic-interaction column chromatography, suggesting the HAT activity is quite labile. Most HAT activity precipitated with 10% saturated (NH4)2SO4 and could be recovered as soluble activity. However, (NH4)2SO4-precipitated HAT could not be purified further.
Velocity sedimentation and anion-exchange chromatography were compatible with recovery of most of the starting HAT activity, and these therefore were used for purification. Among 21 fractions collected from the bottom of a tube after velocity sedimentation in sucrose gradients, HAT activity was detected in fractions 16, 17, and 18, near the top of the gradients. The total HAT activity of these three fractions was 6E7 cpm, similar to the starting activity. P100 was readily detected in fractions 10–19 and was not more abundant in fractions 16–18, which had most of the HAT activity. Fractions 16, 17, and 18 were pooled and purified further by FPLC using a linear salt gradient elution from an anion-exchange column. A total of 2E7 cpm HAT activity was recovered in fractions 15–17 from the anion-exchange column. Histones H3 and H4 were the dominant substrates for the HAT(s) in these fractions.
The proteins in all fractions from the anion-exchange column were separated by SDS/PAGE and visualized by silver staining. A cluster of proteins with an apparent size greater than 220 kDa were enriched in fractions 15–17 containing HAT activity. Western blot analysis was used to test whether they were p300/CBP. P300/CBP sedimented in sucrose gradients and eluted from anion-exchange columns in complete correlation with the EBNA2-associated HAT activity. In sucrose gradients, HAT activity was mostly in fractions 16, 17, and 18 (Fig. 2A Upper), which correlated well with the presence of p300/CBP proteins in these three fractions, as shown by Western blot with mAb AC26, which recognizes both p300 and CBP (Fig. 2A Lower). When antibody specific to the large subunit of RNA polymerase II was used to blot the sucrose gradient fractions, RNA polymerase II could be detected in fraction 9 and not in fractions with HAT activity (data not shown). Thus, although some RNA polymerase II binds to the EBNA2 acidic domain, and p300 and CBP have been associated with RNA polymerase II holoenzyme preparations (30), the HAT and p300/CBP that bound to EBNA2 from BJAB extract is not associated with RNA polymerase II. The correlation between HAT activity and the presence of p300/CBP continued in anion-exchange chromatography. HAT activity peaked in fractions 15–17 (Fig. 2B Upper). Antibodies specific for both p300 and CBP as well as antibodies specific for p300 or CBP detected p300 and CBP in fractions 15–17, in parallel with the HAT activity (Fig. 2B Lower and data not shown). A 2-hr incubation with beads coated with antibody specific for both p300 and CBP depleted up to 70% of the HAT activity from fraction 15, whereas beads coated with antibody specific for p300 or CBP depleted 30–40% of the HAT activity and beads coated with control antibody had a minimal effect. Thus, p300 and CBP are the predominant EBNA2- and VP16-associated HAT activities and there appears to be similar activities attributable to p300 or CBP.
Figure 2.
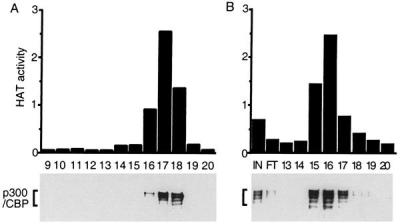
Purification of GST-EBNA2-associated HAT activity. BJAB cell extracts were incubated with GST-EBNA2 beads. The HAT activity was eluted from the beads with 0.2 M NaCl and loaded onto 10–30% sucrose gradient. Fractions containing HAT activity were pooled, applied to anion-exchange column, and eluted with a linear 50- to 300-mM NaCl gradient. Protein fractions from sucrose gradient (A) and UNO Q column (B) were separated by SDS/PAGE and identified by Western blot with anti-CBP/p300 antibody (Lower). The HAT activity of these fractions also was measured (Upper) and shown in 104 × count per minute. Fraction numbers are indicated just below the Upper parts of A and B.
Wild-Type EBNA2 and VP16 Acidic Domains Specifically Bind p300/CBP and PCAF.
To further explore the similarity between the EBNA2- and VP16-associated HATs, the proteins that bound to EBNA2 or VP16 acidic domain–GST fusion proteins were subjected to SDS/PAGE and probed with CBP/p300 antibody. GST-VP16, like GST-EBNA2, retained CBP/p300 from BJAB cell lysates (Fig. 3). EBNA2 and VP16 bound similar amounts of CBP/p300, consistent with the similar binding of HAT activity. Also consistent with HAT activity, neither CBP nor p300 bound to the null point mutant acidic domain–GST fusion proteins. These data indicate that p300 and CBP bind only to the wild-type VP16 and EBNA2 acidic domains in parallel with the HAT activity.
Figure 3.

The EBNA2 and VP16 acidic domains specifically bind p300, CBP, and PCAF. BJAB cell extracts were incubated with glutathione-Sepharose beads loaded with wild-type GST-EBNA2 or GST-VP16 acidic domains or with the null point mutants. The amount of p300, CBP, or PCAF that bound to the beads was assayed by SDS/PAGE followed by Western blot with anti-CBP/p300 antibody or with anti-PCAF antibody.
PCAF binds to p300 and CBP and also has intrinsic HAT (31). To test whether PCAF also binds specifically to the wild-type EBNA2 and VP16 acidic domains, the proteins bound to EBNA2 or VP16 acidic domain fusion proteins were examined by Western blot with PCAF antibody. As shown in Fig. 3, PCAF also bound specifically to the wild-type GST-EBNA2 and VP16 acidic domains. However, PCAF was not present in the anion-exchange column fractions 15–17 that contain most of the EBNA2- or VP16-associated HAT activity (data not shown). These data indicated that PCAF is among the HATs that can bind to EBNA2 but does not fractionate with the dominant HAT activity. Despite its absence from the p300 and CBP fractions, PCAF binding to the wild-type EBNA2 or VP16 fusion proteins could be direct or through p300 or CBP.
EBNA2 Binds to the N- and C-Terminal Domains of p300 and Precipitates with p300 from 293T Cells in Which Both Proteins Are Overexpressed.
To identify the part of p300 that can interact with EBNA2, the amino-terminal, middle, carboxyl-terminal, and HAT domains of p300 were expressed as GST fusion proteins in E. coli and bound to glutathione beads. Ni-NTA bead, purified 6×His-tagged full-length wild-type EBNA2 was incubated with GST p300 fusion proteins. As shown in Fig. 4A, EBNA2 did not bind to either the p300 middle (p300M) or HAT domain (p300HAT), but approximately 20% of the EBNA2 bound to both the amino- (p300N) and carboxyl-terminal domains (p300C). Because both EBNA2 and p300 proteins are soluble recombinant proteins purified from E. coli, these data indicate that wild-type full-length EBNA2 can interact directly with p300. In interacting with the p300 amino and carboxyl termini, EBNA2 is similar to p53 (32).
Figure 4.

EBNA2 interacts with p300 in vitro and in vivo. (A) EBNA2 interacts with amino- and carboxyl-terminal of p300 in vitro. GST fusions to the p300 amino-terminal (amino acids 1–585, p300N), middle (amino acids 766–1571, p300M), carboxyl-terminal (amino acids 1572–2370, p300C), or HAT (amino acids 1195–1701, p300HAT) domains were expressed in E. coli, purified by using glutathione-Sepharose beads, and incubated with His-tagged full-length wild-type EBNA2 protein that has also been purified from E. coli. EBNA2 binding to GST-p300 beads was assayed by Western blot with an EBNA2-specific mAb (PE2) after SDS/PAGE of proteins that bound to the beads. The amount of protein used in the binding assays was five times that shown as input. (B) EBNA2 coimmune-precipitates with p300 from 293 T cells overexpressing both proteins. EBNA2, flag-tagged p300, or both were transfected into 293 T cells. Cell extracts were incubated with M2 beads to immune-precipitate flag-tagged p300, and EBNA2 was detected by Western blot with PE2 mAb. P300 was precipitated from 10 times the amount of lysate shown as input.
To study the interaction between p300 and EBNA2 in vivo, vectors expressing Flag-tagged p300 and EBNA2 were cotransfected into 293 T cells. M2 bead immune precipitation of the Flag-tagged p300 protein brought down about 20% of the p300 in the lysate (data not shown) and coimmune-precipitated about 3% of the EBNA2 as detected by Western blot with mAb to the EBNA2 acidic domain (Fig. 4B). Thus, about 15% of the EBNA2 in the cotransfected cells is associated with the Flag-tagged p300.
P300 (and CBP) Increase Endogenous LMP1 Expression Through EBNA2, and the HAT Activity of PCAF Potentiates p300's Effect.
One important function of EBNA2 is to transcriptionally activate and maintain expression of the EBV-encoded latent membrane protein 1 (LMP1) gene in latently infected cells. The ability of p300 and PCAF to coactivate EBNA2-mediated endogenous LMP1 expression therefore was evaluated by using Akata cells. Akata is an EBV-positive Burkitt's lymphoma cell line with a type I latency infection pattern in which EBNA2 and LMP1 are not expressed. Expression of wild-type full-length EBNA2 or p300 in Akata had little effect on LMP1 expression, whereas p300 coactivated EBNA2's induction of LMP1 expression in Akata cells (Fig. 5). The EBNA2 protein level was not affected by p300 cotransfection (data not shown). Cotransfection of PCAF and EBNA2 had no effect on LMP1 expression, consistent with the lack of effect of PCAF on EBNA2 acidic domain-mediated activation in 293 T cells (see below). However, when PCAF was cotransfected into Akata cells with EBNA2 and p300, PCAF reproducibly increased p300's coactivation of EBNA2-mediated LMP1 expression (Fig. 5). PCAF deletion mutants that lack HAT activity, PCAFΔ579–608 and PCAFΔ609–624, though expressed at a similar level as wild-type PCAF (data not shown), failed to increase p300's up-regulation of EBNA2-mediated transcription. Thus, p300 coactivates EBNA2-mediated endogenous LMP1 expression, and the HAT activity of PCAF is required for PCAF to potentiate p300's coactivation effect.
Figure 5.

P300 coactivates LMP1 expression mediated by EBNA2 in Akata EBV-infected but EBNA2-negative cells, and PCAF potentiates the activation. Akata cells were transfected with 10 μg of SG5-EBNA2, 10 μg of SG5-p300, 20 μg of pCI-PCAF, or 20 μg of HAT-negative PCAF mutants Δ579–608 and Δ609–624 protein expression vectors. Empty vector was used to balance the transfections to 40 μg of total DNA when necessary. Whole-cell extracts were Western-blotted with the S12 anti-LMP1 mAb.
P300 and CBP (but Not PCAF) Coactivate GAL4-EBNA2 or GAL4-VP16 Acidic Domain-Mediated Transcription.
The effects of p300 on EBNA2 and VP16 acidic domain-mediated activation were tested by transfecting GAL4-EBNA2 or GAL4-VP16 acidic domain fusion into 293 T cells along with a plasmid containing a luciferase reporter with 5×GAL4 DNA-binding sites and increasing amounts of p300 expression plasmid (Fig. 6A). GAL4-EBNA2 activated reporter gene expression less than 2-fold, whereas p300 alone had a minimal effect. Cotransfection with 1, 2, or 4 μg of p300 expression vector DNA resulted in 10-, 24-, or 134-fold activation, respectively, over GAL4-EBNA2 acidic domain alone. Similar results were obtained when GAL4-EBNA2 and p300 were cotransfected in BJAB cells (data not shown), indicating that GAL4-EBNA2 coactivation by p300 is not cell type-specific. In contrast to small activating effects of GAL4-EBNA2 alone, GAL4-VP16 activated reporter gene expression more than 100-fold. Despite the high level of GAL4-VP16-mediated activation, cotransfection with 1, 2, or 4 μg of p300 expression vector DNA increased GAL4-VP16 acidic domain-mediated luciferase activity 3.5-, 13-, or 30-fold, respectively, over that with GAL4-VP16 alone.
Figure 6.

The effect of p300, CBP, or PCAF overexpression on GAL4-EBNA2 (GE) or GAL4-VP16 (GV) acidic domain-mediated transactivation of a GAL4-dependent promoter in 293 T cells. Various amounts of p300 expression vector (A) or 3.5 μg of CBP expression vector (B) was transfected into 293 T cell with 0.2 μg of GAL4-EBNA2 or 0.01 μg of GAL4-VP16 expression plasmids and 0.5 μg of reporter plasmid. The amount of cotransfected p300 DNA is indicated (in μg) in the figure. The fold activation induced by p300/CBP is relative to that induced by GAL4-EBNA2 or GAL4-VP16 alone. In C, the effect of 4 μg of p300 expression vector on coexpressed GAL4-EBNA2 acidic domain-mediated transactivation is compared with that of p300 HAT-negative mutants p300Δ1472–1522 or Δ1603–1653 in 293 T cells. The extent of cotransactivation by p300 mutants is shown relative to that by wild-type p300. In D, the effect of PCAF on GAL4-EBNA2- or GAL4-VP16-mediated activation of a GAL4-responsive promoter is assayed as described in A above. For all experiments in this figure, vector DNA was used to balance the different amounts of DNA used in different transfections. Luciferase activity was adjusted to the transfection efficiency represented by β-galactosidase activity. The results shown are the average of four to five independent experiments, with error bars representing SE.
CBP also coactivated the GAL4-dependent luciferase reporter when CBP expression vector was cotransfected into 293T cells with GAL4-EBNA2 or GAL4-VP16 expression vectors. Transfection of 3.5 μg of CBP expression plasmid activated GAL4-EBNA2 or GAL4-VP16 acidic domain-mediated luciferase activation about 10-fold and 4-fold, respectively (Fig. 6B). Although CBP appears to be less active than p300 at the single concentration of expression plasmid used in those studies, we have not compared the changes in p300 and CBP levels that result from these transfections, and the apparent difference could be due to suboptimal CBP expression.
To evaluate the extent to which the p300 effect on EBNA2- and VP16-mediated transcription activation is due to its HAT activity, two p300 deletion mutants that lack HAT activity were transfected into 293 T cells with GAL4-EBNA2 or GAL4-VP16 and the 5×GAL4 site reporter plasmids. Wild-type p300 expression plasmid increased GAL4-EBNA2 and GAL4-VP16 activity more than 100- and 30-fold, respectively (Fig. 6 A and C). The HAT deletion mutants, p300Δ1472–1522 and p300Δ1603–1653, had 20–50% of wild-type p300 activity (Fig. 6C). However, both p300 mutants were expressed at much lower levels than wild-type p300 (data not shown). Because EBNA2 and VP16 coactivation by p300 is dependent on the amount of p300 expressed (Fig. 6A), most of the p300 coactivation in 293 T cells is not due to p300 HAT activity.
To test whether PCAF is involved in EBNA2 and VP16 acidic domain-mediated transcription activation, a PCAF expression vector was cotransfected into 293 T cells with expression vector for GAL4-EBNA2 or GAL4-VP16 and a reporter plasmid. Different amounts of PCAF expression vector were tested, but PCAF could activate only GAL4-EBNA2- or GAL4-VP16-mediated transcription 1- to 2-fold (Fig. 6D). Thus, PCAF does not appear to contribute significantly to GAL4-EBNA2 or GAL4-VP16 transcription activation in 293 T cells.
Discussion
Under physiological conditions, transcription is believed to be positively affected by the balance of histone acetylase over deacetylase activities near transcriptional start sites (18, 19). These experiments were initiated to determine whether the transcriptionally activating EBNA2 and VP16 acidic domains can specifically bind HAT(s) and to identify the HAT(s). Indeed, substantial HAT activity bound to the EBNA2 or VP16 acidic domains and the binding was highly specific for the wild-type acidic domains. Although the EBNA2- and VP16-associated protein p100 is similar to HAT in binding to the wild-type and not to the transcriptionally inactive null point mutant acidic domains (15) and p100 peptides were present in a purified HAT preparation (27), E. coli-expressed p100 lacked HAT activity and p100 did not copurify with the EBNA2- and VP16-associated HAT activity on sucrose gradients or on ion-exchange chromatography. Thus, p100 is not a significant component of the EBNA2- or VP16-bound HAT activity.
Three other proteins, p300, CBP, and PCAF, that have intrinsic HAT activity bound specifically to the EBNA2 or VP16 wild-type acidic domains, but not to the EBNA2 or VP16 null mutant acidic domains. Most of the EBNA2 or VP16 acidic domain bound HAT activity fractionated by sucrose gradient and anion-exchange chromatography with p300 and CBP and not with PCAF. Indeed, full-length wild-type EBNA2 bound specifically to both the N- and C-terminal domains of p300, and immune precipitation of p300 resulted in coimmune precipitation of EBNA2. Thus, p300 and CBP are the predominant EBNA2 and VP16 acidic domain-associated HATs.
The PCAF that binds to EBNA2 and VP16 acidic domains could be associated with p300 or even CBP because p300 can directly bind EBNA2 and PCAF is tightly associated with p300 and CBP (31). However, PCAF did not remain associated with p300 and CBP through our HAT purification and is not a component of the principal, purified HAT activity. We have not determined whether PCAF purifies with p300/CBP in velocity sedimentation, but PCAF is not detectable in any of the fractions from the ion-exchange column. Nevertheless, PCAF overexpression potentiates p300's coactivation of EBNA2-mediated LMP1 expression. This potentiation requires p300 expression and the intrinsic HAT activity of PCAF, because wild-type PCAF is inactive in the absence of p300 coexpression and HAT null mutants of PCAF cannot potentiate.
The HAT activities of p300, CBP, and PCAF are likely to be important for EBNA2 activation of promoters, including the promoter for the EBV oncogene, LMP1. In EBV-transformed B lymphocytes, EBNA2 is highly associated with a cellular transcription factor, RBP-Jκ (4, 5). RBP-Jκ brings EBNA2 to viral and cellular promoters, including the LMP1 promoter (4, 5). RBP-Jκ can have repressive effects, and a negative transcriptional regulatory domain of RBP-Jκ binds to SMRT and CIR, which associate with histone deacetylase complexes (33, 34). Another site in the LMP1 promoter can bind Sin3A, which also associates with histone deacetylase (35). Further, evidence for the importance of these histone deacetylase activities in repressing transcription from the LMP1 promoter comes from the observation that treatment of EBV-infected Burkitt's lymphoma cells with histone deacetylase inhibitors results in induction of LMP1 expression even in the absence of EBNA2 (35). Thus, EBNA2 is likely to need to counter the repressive effect of RBP-Jκ and Sin3A by bringing p300, CBP, and PCAF HATs to the LMP1 promoter. Indeed, PCAF potentiation of p300 coactivation of LMP1 expression in Akata cells required PCAF HAT activity. The participation of PCAF HAT activity in potentiating p300 coactivation of EBNA2-mediated LMP1 expression is likely to be an important biological effect because of the critical role of EBNA2 in regulating LMP1 expression.
EBNA2 is also critical for the earliest effects of EBV in turning on viral and cellular gene expression upon EBV infection of resting human B lymphocytes (3). EBNA2-mediated regulation of the LMP1 and c-myc (36) promoters is critical for initiation and maintenance of EBV-mediated primary B lymphocyte transformation. Thus, coactivation of EBNA2-mediated transcription by p300 and CBP and potentiation by PCAF are likely to be critical to EBV initial transformation of resting primary B lymphocytes as well as for maintenance of the transformed state.
EBNA2 and VP16 are likely to recruit p300 and CBP not only for their intrinsic HAT activities, but also for their role in bridging to other transcriptional factors. As noted above, p300 bridging to PCAF potentiates p300 coactivation of EBNA2-mediated LMP1 expression in Akata cells, and this requires PCAF HAT activity. Further, in 293 T cells, expression of p300 mutants lacking intrinsic HAT activity coactivates transcription mediated by the binding of the EBNA2 or VP16 acidic domains to a minimal promoter with five upstream GAL4-binding sites. Coactivation of the EBNA2 and VP16 acidic domains by p300 proteins that lack intrinsic HAT activity indicates that this p300 effect in 293 T cells is through bridging to other positively acting transcription factors. Such bridging effects also have been observed with more complex promoters under more physiologically relevant cell types. For example, the HAT activity of p300 is unnecessary for Tat- or MyoD-mediated transcription of the HIV long terminal repeat and p21 promoters (20, 37).
EBNA2 interaction with p300/CBP could be important in the ability of EBV to cause resting B lymphocytes to enter cell cycle, to proliferate perpetually, and to slow differentiation. These effects could be pleiotropic because p300 and CBP appear to be important for E2F1-mediated (38), NF-κB-mediated (39, 40), or p53-mediated (41, 42) transcriptional activation. In lymphocytes, E2F1 and NF-κB can effect c-myc transcription (43, 44) and E2F1-mediated transcription is important for G1 transition to S phase (45), whereas p53-mediated transcription is important for cell cycle arrest (46). In interacting with p300/CBP, EBNA2 may sequester p300/CBP for EBNA2-mediated promoter activations or may be brought by p300 to promoters that are regulated by these other transcription factors. While this paper was in preparation, CBP and PCAF were shown to have a 2-fold effect on EBNA2 activation of a c-myc promoter in murine embryo fibroblasts and immunoprecipitation of p300 resulted in the coprecipitation of EBNA2 expressed in these cell (47), consistent with a role for p300, CBP, or PCAF in the effects of EBNA2 on the c-myc promoter.
CBP, p300, and PCAF coactivate or potentiate transcription mediated by EBV EBNA2 and herpes simplex virus VP16 acidic domains. Both of these viral transactivators normally function at the outset of virus infection in G0 cells. These acidic domains are likely to have evolved to take advantage of the HAT and scaffolding properties of p300, CBP, and PCAF so as to efficiently regulate viral and cell gene expression. In interacting with p300, CBP, and PCAF, these herpes virus trans-activators have incorporated strategies in common with adenovirus E1A (25), SV40 T antigen (48), HPV-16 E6 (49, 50), HIV Tat (37, 51), and HTLV Tax (52), which appear to use p300, CBP, and PCAF for their intrinsic HAT activities, as scaffolds for bridging to other transcription factors and as transcriptionally limiting coactivators whose sequestration from p53 or other factors may have cell cycle entry, growth promoting, or survival effects.
Acknowledgments
We are grateful to Dr. Vasily Ogryzko from Dr. Yoshihiro Nakatani's lab and Dr. Richard Goodman for providing critical reagents. We thank Jianxin Zhou in Dr. David Allis' lab for advice on the in-gel HAT assay and David Livingston for helpful discussions. This research was supported by Grant CA47006 from the National Cancer Institute of the U.S. Public Health Service.
Abbreviations
- EBV
Epstein–Barr virus
- EBNA2
EBV nuclear protein 2
- HAT
histone acetyltransferase
- GST
glutathione S-transferase
References
- 1.Wang F, Gregory C D, Rowe M, Rickinson A B, Wang D, Birkenbach M, Kikutani H, Kishimoto T, Kieff E. Proc Natl Acad Sci USA. 1987;84:3452–3456. doi: 10.1073/pnas.84.10.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen J I, Wang F, Mannick J, Kieff E. Proc Natl Acad Sci USA. 1989;86:9558–9562. doi: 10.1073/pnas.86.23.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfieri C, Birkenbach M, Kieff E. Virology. 1991;181:595–608. doi: 10.1016/0042-6822(91)90893-g. [DOI] [PubMed] [Google Scholar]
- 4.Grossman S R, Johannsen E, Tong X, Yalamanchili R, Kieff E. Proc Natl Acad Sci USA. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henkel T, Ling P D, Hayward S D, Peterson M G. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 6.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman S R. J Virol. 1995;69:253–262. doi: 10.1128/jvi.69.1.253-262.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen J I, Kieff E. J Virol. 1991;65:5880–5885. doi: 10.1128/jvi.65.11.5880-5885.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen J I, Wang F, Kieff E. J Virol. 1991;65:2545–2554. doi: 10.1128/jvi.65.5.2545-2554.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong X, Yalamanchili R, Harada S, Kieff E. J Virol. 1994;68:6188–6197. doi: 10.1128/jvi.68.10.6188-6197.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yalamanchili R, Harada S, Kieff E. J Virol. 1996;70:2468–2473. doi: 10.1128/jvi.70.4.2468-2473.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harada S, Yalamanchili R, Kieff E. J Virol. 1998;72:9948–9954. doi: 10.1128/jvi.72.12.9948-9954.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen J I. Proc Natl Acad Sci USA. 1992;89:8030–8034. doi: 10.1073/pnas.89.17.8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stringer K F, Ingles C J, Greenblatt J. Nature (London) 1990;345:783–786. doi: 10.1038/345783a0. [DOI] [PubMed] [Google Scholar]
- 14.Lin Y S, Ha I, Maldonado E, Reinberg D, Green M R. Nature (London) 1991;353:569–571. doi: 10.1038/353569a0. [DOI] [PubMed] [Google Scholar]
- 15.Tong X, Drapkin R, Yalamanchili R, Mosialos G, Kieff E. Mol Cell Biol. 1995;15:4735–4744. doi: 10.1128/mcb.15.9.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong X, Drapkin R, Reinberg D, Kieff E. Proc Natl Acad Sci USA. 1995;92:3259–3263. doi: 10.1073/pnas.92.8.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cress W D, Triezenberg S J. Science. 1991;251:87–90. doi: 10.1126/science.1846049. [DOI] [PubMed] [Google Scholar]
- 18.Workman J L, Kingston R E. Annu Rev Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 19.Kornberg R D, Lorch Y. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 20.Puri P L, Sartorelli V, Yang X J, Hamamori Y, Ogryzko V V, Howard B H, Kedes L, Wang J Y, Graessmann A, Nakatani Y, Levrero M. Mol Cell. 1997;1:35–45. doi: 10.1016/s1097-2765(00)80005-2. [DOI] [PubMed] [Google Scholar]
- 21.Korzus E, Torchia J, Rose D W, Xu L, Kurokawa R, McInerney E M, Mullen T M, Glass C K, Rosenfeld M G. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- 22.Kuo M H, Zhou J, Jambeck P, Churchill M E, Allis C D. Genes Dev. 1998;12:627–639. doi: 10.1101/gad.12.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Liu L, Berger S L. Genes Dev. 1998;12:640–653. doi: 10.1101/gad.12.5.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J S, Galvin K M, See R H, Eckner R, Livingston D, Moran E, Shi Y. Genes Dev. 1995;9:1188–1198. doi: 10.1101/gad.9.10.1188. [DOI] [PubMed] [Google Scholar]
- 25.Eckner R, Ewen M E, Newsome D, Gerdes M, DeCaprio J A, Lawrence J B, Livingston D M. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 26.Brownell J E, Allis C D. Curr Opin Genet Dev. 1996;6:176–184. doi: 10.1016/s0959-437x(96)80048-7. [DOI] [PubMed] [Google Scholar]
- 27.Attisano L, Lewis P N. J Biol Chem. 1990;265:3949–3955. [PubMed] [Google Scholar]
- 28.Mizzen C A, Yang X J, Kokubo T, Brownell J E, Bannister A J, Owen-Hughes T, Workman J, Wang L, Berger S L, Kouzarides T, et al. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- 29.Brownell J E, Allis C D. Proc Natl Acad Sci USA. 1995;92:6364–6368. doi: 10.1073/pnas.92.14.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neish A S, Anderson S F, Schlegel B P, Wei W, Parvin J D. Nucleic Acids Res. 1998;26:847–853. doi: 10.1093/nar/26.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X J, Ogryzko V V, Nishikawa J, Howard B H, Nakatani Y. Nature (London) 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 32.Grossman S R, Perez M, Kung A L, Joseph M, Mansur C, Xiao Z X, Kumar S, Howley P M, Livingston D M. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- 33.Kao H Y, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner C R, Evans R M, Kadesch T. Genes Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh J J, Zhou S, Chen L, Young D B, Hayward S D. Proc Natl Acad Sci USA. 1999;96:23–28. doi: 10.1073/pnas.96.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sjoblom-Hallen A, Yang W, Jansson A, Rymo L. J Virol. 1999;73:2983–2993. doi: 10.1128/jvi.73.4.2983-2993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm G W, Kempkes B. J Virol. 1999;73:4481–4484. doi: 10.1128/jvi.73.5.4481-4484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benkirane M, Chun R F, Xiao H, Ogryzko V V, Howard B H, Nakatani Y, Jeang K T. J Biol Chem. 1998;273:24898–24905. doi: 10.1074/jbc.273.38.24898. [DOI] [PubMed] [Google Scholar]
- 38.Trouche D, Kouzarides T. Proc Natl Acad Sci USA. 1996;93:1439–1442. doi: 10.1073/pnas.93.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerritsen M E, Williams A J, Neish A S, Moore S, Shi Y, Collins T. Proc Natl Acad Sci USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perkins N D, Felzien L K, Betts J C, Leung K, Beach D H, Nabel G J. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 41.Lill N L, Tevethia M J, Eckner R, Livingston D M, Modjtahedi N. J Virol. 1997;71:129–137. doi: 10.1128/jvi.71.1.129-137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu W, Shi X L, Roeder R G. Nature (London) 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 43.Hiebert S W, Lipp M, Nevins J R. Proc Natl Acad Sci USA. 1989;86:3594–3598. doi: 10.1073/pnas.86.10.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duyao M P, Kessler D J, Spicer D B, Sonenshein G E. Curr Top Microbiol Immunol. 1990;166:211–220. doi: 10.1007/978-3-642-75889-8_27. [DOI] [PubMed] [Google Scholar]
- 45.Lavia P, Jansen-Durr P. BioEssays. 1999;21:221–230. doi: 10.1002/(SICI)1521-1878(199903)21:3<221::AID-BIES6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz D, Rotter V. Semin Cancer Biol. 1998;8:325–336. doi: 10.1006/scbi.1998.0095. [DOI] [PubMed] [Google Scholar]
- 47.Jayachandra S, Low K G, Thlick A E, Yu J, Ling P D, Chang Y, Moore P S. Proc Natl Acad Sci USA. 1999;96:11566–11571. doi: 10.1073/pnas.96.20.11566. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Eckner R, Ludlow J W, Lill N L, Oldread E, Arany Z, Modjtahedi N, DeCaprio J A, Livingston D M, Morgan J A. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel D, Huang S M, Baglia L A, McCance D J. EMBO J. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmermann H, Degenkolbe R, Bernard H U, O'Connor M J. J Virol. 1999;73:6209–6219. doi: 10.1128/jvi.73.8.6209-6219.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marzio G, Tyagi M, Gutierrez M I, Giacca M. Proc Natl Acad Sci USA. 1998;95:13519–13524. doi: 10.1073/pnas.95.23.13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bex F, Yin M J, Burny A, Gaynor R B. Mol Cell Biol. 1998;18:2392–2405. doi: 10.1128/mcb.18.4.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]