

Abstract
Six infants in an Old Order Amish pedigree were observed to be affected with endocrine-cerebro-osteodysplasia (ECO). ECO is a previously unidentified neonatal lethal recessive disorder with multiple anomalies involving the endocrine, cerebral, and skeletal systems. Autozygosity mapping and sequencing identified a previously unknown missense mutation, R272Q, in ICK, encoding intestinal cell kinase (ICK). Our results established that R272 is conserved across species and among ethnicities, and three-dimensional analysis of the protein structure suggests protein instability due to the R272Q mutation. We also demonstrate that the R272Q mutant fails to localize at the nucleus and has diminished kinase activity. These findings suggest that ICK plays a key role in the development of multiple organ systems.
Introduction
Protein kinases belong to one of the largest and most functionally diverse gene families in eukaryotes, constituting approximately 1.7% of all human genes.1 By phosphorylating substrates, kinases can direct activity, localization, and overall function of numerous target proteins. Kinases are particularly important for signal transduction and coordination of complex cellular functions, as seen with the mitogen-activated protein (MAP) kinases and cyclin-dependent kinases (CDKs), which play a central role in regulating mammalian cell proliferation and division.2 Altered kinase activity or abnormal substrate phosphorylation has been implicated in monogenic diseases—“kinasopathies”—including endocrine disorders,3 cancers,4 immunodeficiencies,5 and cardiovascular diseases.6
Inherited skeletal dysplasias or osteochondrodysplasias are characterized by abnormal development, growth, and maintenance of the skeleton.7 Manifestations of skeletal dysplasias range from clinically undetectable to severe deformities and lethality.7 A group of lethal autosomal-recessive skeletal dysplasias are the short rib-polydactyly (SRP) syndromes,8 which include SRP type II or Majewski Syndrome (MIM: 263520).9
Herein we report a new syndrome, to our knowledge, comprising osteodysplasia, cerebral anomalies, and endocrine gland hypoplasia. A pedigree from an Old Order Amish community was identified as having six affected infants with this previously unreported multisystem, neonatal lethal condition, designated here as the endocrine-cerebro-osteodysplasia (ECO) syndrome. Because the Old Order Amish population reportedly have a high degree of consanguinity,10 this pedigree was ideal for autozygosity mapping of the putative molecular defect.11,12
We delineate the clinical features of ECO and report the first mutation, R272Q (c.1305G→A), within ICK, encoding intestinal cell kinase (ICK). Biochemical and immunocytochemical studies indicate function and localization deficits, implicating ICK as a key player in development of the central nervous, skeletal, and endocrine systems.
Material and Methods
Patients and Biological Materials
Two families from an Old Order Amish community were referred for genetic assessment and counseling, and all six affected infants were examined by one of the authors (V.S.). Photos, blood, and tissue samples were provided for research purposes, with ethics approval (from the Office of Research Ethics at the University of Western Ontario) and informed consent from participating parents. Peripheral blood and skin biopsy for DNA extraction was collected from five affected children, three parents, and two unaffected siblings and used for autozygosity mapping. The genealogy of the families affected with the disease was prepared through interviews and local Amish community records. DNA from umbilical cord blood and buccal swabs was extracted from 257 ECO-unaffected individuals born into the Old Order Amish community for determining the mutant-allele frequency in the community. Autopsies were performed on three patients (IV-1, IV-2, and IV-8 from pedigree) after 33, 29, and 23 weeks of gestation, respectively.
Histology
Routine tissue samples including liver, kidney, and central nervous system (CNS) from each of the three autopsy patients were collected for histopathological examination, formaldehyde fixed, paraffin embedded, sectioned, and then stained with hemolysin and eosin. Neuropathological examination was performed on formalin-fixed brain and spinal cord of each of the three autopsy patients.
Genotyping
DNA from five affected infants and five unaffected parents and siblings were genotyped for single-nucleotide polymorphisms (SNPs) with GeneChip Mapping 500K Array Set (Affymetrix, Santa Clara, CA, USA) at the London Regional Genomics Centre. 250 ng of double-stranded genomic DNA was digested with either Nsp or Sty, followed by adaptor ligation and PCR amplification with generic primers. PCR products were then purified, fragmented with DNaseI, labeled with terminal deoxytransferase, and finally hybridized to the Mapping 250K Nsp or Sty GeneChips. SNP genotypes were determined with the B-RLMM algorithm implemented in Affymetrix GTYPE software.
Autozygosity Mapping
Autozygosity (homozygosity) mapping was performed with Agilent GT v2.0 (Agilent Technologies, Santa Clara, CA, USA), which scans the genome for regions that are identical by descent. SNP allele frequencies from controls of European descent were used for estimating the two-point logarithm of the odds (LOD) scores for each SNP. Location scores, which are the summation of two-point LOD scores for a block of homozygous SNPs, were then calculated for providing a relative measure of likelihood that the region harbors the disease gene.13
Mutation Analysis
Eleven of the 36 positional candidate genes in the linked region on chromosome 6p were screened by genomic DNA sequencing in an affected individual, a parent, an unaffected sibling, and a non-Amish control. Candidate genes from genomic DNA were PCR amplified with primer pairs designed for all coding exons and intron-exon boundaries. All amplicons were then sequenced with the sequencing platform of the London Regional Genomics Centre Sequencing Facility.
Cosegregation of the exon 7 ICK mutant with disease in the pedigree was demonstrated with direct sequence analysis of all available family members. The target sequence (552 base pairs [bp]) was amplified with primers 5′ CTC ATT CCA TAC AGT GCC ACA and 3′ GAA TTA CAT GCC AAT TTT CAA AG, followed by electrophoresis purification on a 1.5% agarose gel and analysis on a ABI 3730 DNA Sequencer (Applied Biosystems, Mississauga, ON, Canada). A total of 13 family members, including the affected individuals, were studied.
SNaPshot and TaqMan assays were used for identifying allele frequency of the ICK variation in exon 7, c.1305G→A (GI: 156671211), within 257 Old Order Amish controls and 2855 ethnically diverse and healthy non-Amish controls, respectively. For SNaPshot, which is a rapid allele-specific genotyping method, the purified 552 bp amplicon (with the above primers) was subjected to ddNTP extension (SnaPShot, Applied Biosystems) with primer 5′ CAG TGG GAT CCC AAG AAA C and analyzed by ABI 3730 DNA Sequencer. TaqMan quantitative real-time PCR assays were performed with an ABI 7900 sequence detection system (Applied Biosystems) for providing allele discrimination with PCR primers (forward primer: 5′ GCT CCT GAG AGA CAT GCT TCA; reverse primer: 5′ AAG AAA ATG GAA GAA AAC CTG ACT AGC T) and two allele-specific TaqMan probes synthesized for detecting the ICK variation (allele G: 5′ VIC-CCC AAG AAA CGA CCA AC and mutant allele A: 5′ FAM-CCA AGA AAC AAC CAA C).
In Silico Analysis
Conservation of the ICK protein across species was determined with ClustalW, which is a multiple-sequence-alignment computer program, by initially creating a phylogenetic tree of the query sequence.14 Impact of the amino acid mutation (R→Q at residue 272) on ICK protein structure, function, and pathological implication was predicted with four online tools, namely PMUT,15 PolyPhen,16 SNPs3D,17 and SIFT.18
The crystal structure of human CDK2 in complex with isopentenyladenine (PDB ID: 2EXM), solved by Schulze-Gahmen et al.19, was used as a basis for modeling the ICK with and without the R→Q mutation at residue 272. For mimicking ICK, 2EXM was substituted at A183P and I186V. The resulting structure was visualized in the program PyMOL (v0.99, DeLano Scientific, San Francisco, CA, USA).20 With the Rosetta Design program,21 used for approximating the change in potential energy (in kilocalories) of the ICK structure with the R→Q mutation, side chains of nearby contacting amino acids were allowed to vary in conformation. Change in energy values (in kilocalories) was replicated in Eris server, which is a protein-stability prediction server that calculates the change in protein stability caused by mutations.22 Eris server has the added feature of allowing backbone motion of the amino acids, which is crucial for protein-stability estimation of small-to-large mutations.
Plasmids and Cell Culture
The Ultimate ORF Clone of human ICK cDNA (clone ID: IOH38087) was provided in the Gateway entry vector, pENTR221, containing a kanamycin-resistance cassette (Invitrogen, Carlsbad, CA, USA). The R→Q mutation was introduced into the wild-type ICK clone within pENT221 in vitro with the GeneTailor Site-Directed Mutagenesis System (Invitrogen). With Clonase II (Invitrogen) for aiding homologous recombination, the wild-type and mutant ICK cDNA was cloned directionally into the Gateway destination vector, pcDNA-DEST53, containing an N-terminal green fluorescent protein (GFP) tag and neomycin-resistance cassette. All clones were sequence verified. The plasmid pcDNA/GW-53/CAT, which contained an N-terminal GFP tag, neomycin-resistance cassette, and chloramphenicol-acetyltransferase (CAT) cassette, was provided as a vector control. HEK293 cells were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (GIBCO, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum.
Immunocytochemistry
For assessing nuclear localization of ICK constructs, HEK293 cells were grown on coverslips in six-well 35 mm dishes to 60%–70% confluency, followed by transfection with either wild-type (WT), R272Q mutant ICK-expression plasmid, or control vector containing a CAT cassette (4 μg DNA) by a calcium-phosphate-based method. 48 hr after transfection, cells were washed two times with PBS, fixed with 4% paraformaldehyde, and stained with Hoechst dye (2.5 μg/ml in PBS) (Sigma-Aldrich, Oakville, ON, Canada) on ice for 20 min. Cells were then washed three times with PBS and mounted on glass slides with PermaFluor Aqueous Mounting Medium (Fisher, Markham, ON, Canada). Images were captured with FITC and UV filter sets and 40× objective with a Leica (Deerfield, IL, USA) DMI6000B inverted fluorescence microscope, followed by image acquisition with the Leica Application Suite (LAS v. 2.8.1).
Protein Quantification
HEK293 cells were grown in 225 cm2 flasks until 60%–70% confluency was reached, followed by transfection with GFP-tagged expression constructs of either WT, R272Q mutant, or control vector containing a CAT cassette (96 μg DNA) by a calcium-phosphate-based method. 48 hr after transfection, cells were harvested in ice-cold PBS and lysed in lysis buffer (20 mmol/L Tris-HCl [pH = 7.4], 50 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 25 mmol/L NaF supplemented with phosphatase and protease inhibitors). The lysate was cleared by centrifugation. Cell lysates were precleared with immobilized protein-G beads (Fisher) for 3 hr at 4°C and then incubated with anti-GFP (3 μg) for 2 hr at 4°C, followed by incubation with immobilized protein-G beads (Fisher) overnight at 4°C. The beads were washed extensively with lysis buffer and then divided for immunoblotting and kinase assay.
GFP-immunoprecipitated beads of all three constructs were boiled in the SDS loading buffer for 5 min. Proteins were then resolved by 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Invitrogen). The membranes were blocked in TBS containing 0.1% Tween-20 and 5% fat-free dry milk for 1 hr and then incubated with anti-ICK (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. The membrane was then incubated with horseradish peroxidase-conjugated anti-goat secondary antibodies (1: 50,000, Santa Cruz Biotechnology) for 1 hr, followed by ICK protein visualization with enhanced chemiluminescence-detection Luminol reagent according to the manufacturer's instructions (Santa Cruz Biotechnology).
Kinase Assays
For assessing kinase activity, GFP-immunoprecipitated WT ICK, R272Q mutant ICK, and vector control were washed extensively in kinase assay buffer (50 mM HEPES [pH 7.5], 10 mM MgCl2, supplemented with 5 mM DTT, protease inhibitors and phosphatase inhibitors). The samples were then incubated with 5 μCi [γ-32P] ATP, 100 μM ATP, and 5 μg purified myelin basic protein (MBP) (Millipore, Billerica, MA, USA) at 30°C for 15 min in 50 μl kinase assay buffer. As a positive control, active MAPK 2 (Millipore) was incubated with 5 μCi [γ-32P] ATP, 100 μM ATP, and 5 μg MBP at 30°C for 10 min in 25 μl kinase assay buffer. Proteins in the reaction were separated by 15% SDS-PAGE. The gels were dried, and 32P was detected by autoradiography.
Statistical Analysis
Immunofluorescence localization data were analyzed with Pearson's chi-square test with SAS v9.1 (SAS Institute, Cary, NC, USA).
Results
Clinical and Pathological Features of ECO
Three affected individuals from two Old Order Amish families were originally reported as having a Majewski-hydrolethalus phenotype in 2004 (S. Bakker and V. Siu, 2004, Am. Soc. Hum. Genet., abstract). Three further infants were subsequently born, and the phenotype was further characterized (Figure 1A and Table 1).
Figure 1.
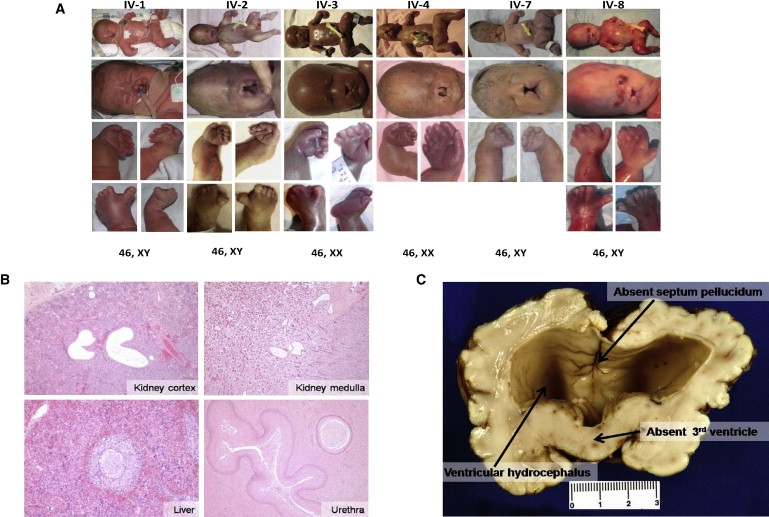
Clinical and Pathological Findings in ECO Patients
(A) Serial photographs of all six affected individuals (left to right from pedigree) showing full body with polydactyly and micromelia, craniofacial abnormalities including cleft lip and palate, bowed forearms, wide-gapped hallux (not shown for IV-4 and IV-7), and karyotype (top to bottom).
(B) Kidney cortex from patient IV-1 (top left) and medulla from patient IV-2 (top right) show cystically dilated tubules (hematoxylin and eosin staining [H&E], 40× magnification). Liver sections from the 29-week-gestated patient IV-2 (bottom left) illustrate the persistence of circular ductal plates (100× magnification). Squamous metaplasia is seen in the intraprostatic urethra from patient IV-1 (bottom right) (H&E, 40× magnification).
(C) Representative gross coronal view of the brain of patient IV-1, presenting with absent septum pellucidum, secondary to ventricular hydrocephalus, and fused thalami due to a lack of the third ventricle.
Table 1.
Clinical and Pathological Descriptiona
| Clinical Features |
Affected Individuals (year of birth) |
|||||
|---|---|---|---|---|---|---|
| IV-1 (2004) | IV-2 (2005) | IV-3 (2005) | IV-4 (2006) | IV-7 (2002) | IV-8 (2003) | |
| Age at delivery (weeks) | 33 | 29 | 24 | 25 | 34 | 23 |
| Sex | male | male | female | female | male | male |
| Karyotype | 46, XY | 46, XY | 46, XX | 46, XX | 46, XY | 46, XY |
| Height40 | 40 cm (3rd percentile) | 37.3 cm (10th percentile) | 29 cm (75th percentile) | 32.5 cm (10th percentile) | 33.1 cm (90th percentile) | |
| Weight40 | 2250 grams (90th percentile) | 1400 grams (90th percentile) | 675 grams (25th percentile) | 897 grams (90th percentile) | 1871 grams (10th percentile) | 667 grams (90th percentile) |
| Head circumference40 | 36.5 cm (>97th percentile) | 31.7 cm (>97th percentile) | 24 cm (20th percentile) | 28 cm (3rd percentile) | 23.3 cm (90th percentile) | |
| Autopsy | + | + | − | − | − | + |
| Oral | ||||||
| Cleft palate | midline | midline | notch in alveolar ridge | midline | midline | midline |
| Cleft lip | bilateral | median | median | bilateral | median | median |
| Presence of premaxilla | + | − | − | +, tiny | − | − |
| Prominent upper lip region | − | + | − | + | − | − |
| Hypoplastic/absent epiglottis | + | − | + | |||
| Hypoplastic/absent larynx | + | − | + | |||
| Facial | ||||||
| Midface hypoplasia | + | + | + | + | + | + |
| Hypoplastic eyes | + | + | − | + | present; right: hypoplastic, left: cystic | cystic |
| Retinal dysplasia | − | − | +, cataracts | |||
| Deep-set eyes | + | + | + | + | + | |
| Fused eyelids | + | + | + | + | ||
| Hypotelorism | + | |||||
| Flat and wide nasal bridge | + | + | + | + | + | + |
| Dysplastic and low-set ears | + | + | dysplastic | + | + | |
| Micrognathia | + | + | + | + | + | + |
| Excess skin below chin | + | + | − | − | + | + |
| Teeth | − | two in lower jaw | − | − | − | − |
| Skeletal | ||||||
| Dolicocephalic | − | + | − | − | + | + |
| Prominent xyphoid | − | + | − | − | − | − |
| Polydactyly (postaxial) | 4 limbs | 4 limbs | 3 limbs | 3 limbs | 3 limbs | 4 limbs |
| Syndactyly | + | + | + | + | + | + |
| Brachydactyly | + | + | + | + | + | + |
| Single transverse palmar crease | unilateral | bilateral | unilateral | |||
| Ulnar deviation of hands | + | + | + | + | + | + |
| Bowing of forearms (radius and ulna) | + | + | + | + | + | + |
| Bowing of lower legs (fibula and tibia) | + | − | − | − | − | + |
| Hitch-hikers' thumbs | − | − | − | − | − | unilateral |
| Abducted hips | + | + | + | + | + | + |
| Wide gap between first and second toe | + | + | unilateral-right | + | + | + |
| Talipes equinovaris | − | − | − | − | − | − |
| Chest width | broad with wide-spaced nipples | − | − | − | narrow | − |
| Micromelia | + | + | + | + | + | + |
| bRadiography | ||||||
| Abnormal long bones (radius, ulna, tibia, fibula) | short diaphysis | |||||
| Short and incurved ulnae | + | |||||
| Short and ovoid tibiae | + | |||||
| Abnormal humerus | short diaphysis | |||||
| Abnormal femur | short and ovoid | |||||
| Abnormal Ilium | + | |||||
| Abnormal/hypoplastic acetabular roof | + | |||||
| Central nervous system | ||||||
| Evidence of holoprosencephaly | failure of separation of diencephalic elements | semilobar, monoventricular, fusion of basal ganglia and thalami | diencephalic agenesis | |||
| Corpus callosum | + | agenesis | hypoplastic | |||
| Absence of septum pellucidum | + | + | ||||
| Hydrocephalus (ventriculomegaly) | communicating | + | ||||
| Dysmorphic cerebral aqueduct | + | stenosis | ||||
| Olfactory bulbs | + | absent | ||||
| Cerebral cortex malformation | frontal and occipital regions | focal polymicrogyria | rudimentary sulcation | |||
| Brainstem malformation | + | + | small brainstem | |||
| Cerebellar abnormalities | hemorrhagic, dysmorphic peduncles | small cerebellum | ||||
| Hippocampus agenesis | + | + | ||||
| Leptomeningeal glioneruronal heterotopia | + | |||||
| Spinal cord malformation | + | − | ||||
| Endocrine system | ||||||
| Pituitary gland | absent | not identified | ||||
| Adrenal glands | hypoplastic | absent | hypoplastic | |||
| Other | ||||||
| Pulmonary hypoplasia | + | |||||
| Gastrointestinal anomalies | tube-like stomach | small stomach | − | |||
| Squamous metaplasia of bladder | + | − | − | |||
| External genitalia | microphallus, hypoplastic scrotum | microphallus, hypoplastic scrotum | normal | normal | sex reversal; hypoplastic labia majora | present, microphallus |
| Cryptorchidism | − | bilateral | − | − | − | − |
| Polyhydramnios | − | − | − | − | present | − |
Note: Blank cells indicate that information was unavailable.
Information gathered from attending-physician reports.
Report from International Skeletal Dysplasia Registry (Cedars-Sinai Medical Center) for Patient IV-1.
The infants have had a high birth weight (90th percentile)23 in four out of six times that this was measured. Some of the excess weight may be attributed to the excess fluid associated with severe hydrocephalus. In all of the pregnancies in which antenatal ultrasound was performed, multiple anomalies including ventriculomegaly, cleft lip, and shortened limbs were observed in the second trimester. Polyhydramnios occurred in one pregnancy. Spontaneous onset of labor occurred in two of the pregnancies at 33–34 weeks gestation. One infant (IV-1) showed no spontaneous respirations at birth, was intubated for a day, then extubated and taken home for palliative care. He died on day 3. The second infant (IV-7) had severe hydrocephalus, requiring drainage of 350 ml of cerebrospinal fluid from the ventricles to enable vaginal delivery. The baby died during birth. The other pregnancies were induced between 23 and 29 weeks gestation because of the severity of the malformations. Integrated prenatal screening was undertaken in one pregnancy and was screen positive for increased risk of Trisomy-21, with a low unconjugated estriol of 0.18 MoM and a low alphafetoprotein of 0.48 MoM. Adrenal hypoplasia found in this infant (IV-8) may potentially account for the low prenatal estriol levels.
The face shows a wide nasal bridge and flattened nasal tip with median cleft lip in four of the six cases and a small premaxilla with bilateral cleft lip and palate in the remaining two cases. There is swelling of the tissue derived from the maxillary prominences. The eyes are small and sunken, with a cystic component observed in two cases. As expected, the eyelids were fused in the three infants born between 23 and 25 weeks gestation, but they were also fused in the infant born at 29 weeks (eyelids normally remain fused until approximately 26 weeks gestation)23. Ears show varying degrees of dysplasia. The lower lip is deficient laterally, and the chin is small. One infant (IV-2) had two congenital supernumerary teeth at 29 weeks gestation.
The upper limbs are markedly shortened, with a characteristic bowing of the forearms, ulnar deviation of the hands, and postaxial polydactyly. X-rays reveal angulation of the diaphyses. Palmar creases are abnormal. Usually the index fingers have only one interphalangeal crease, whereas the other digits lack at least one, if not both, interphalangeal creases. There is severe brachydactyly of the digits on the hands, as well as syndactyly involving various combinations of digits two to six. The hips are abducted, with the thighs held at 90 degrees to the lower legs. There is a very wide gap between the halluces and the second toes, reminiscent of atelosteogenesis type 2. On X-rays, the proximal metaphyses of the femur and tibia are widened. Overall, the radiologic findings are most consistent with a diagnosis of Majewski syndrome.
Chest tends to be broad, with widely spaced nipples only in patient IV-1. No congenital heart defect was found in any of the three autopsies. The adrenal glands were hypoplastic in two cases and absent in one case.
Four genotypic males have all had varying degrees of abnormal differentiation of the external genitalia. Patient IV-7, born at 34 weeks, had apparent sex reversal, with unfused urogenital folds and microphallus. Position of the urethral orifice was not determined, and no autopsy was done for assessing for testes. Patients IV-1, IV-2, and IV-8 had microphallus, hypoplastic scrotum with no rugae, prominent scrotal raphe, and bilateral cryptorchidism. Although the cryptorchidism may not be significant in the context of prematurity (two were delivered at 24 and 28 weeks), IV-1 was delivered at 33 weeks gestation and had first-degree hypospadias. The urethral opening was present at the tip of the microphallus in the other two infants. The two genotypic females had unfused urogenital folds. Born at 24 weeks gestation, IV-3 had unusually prominent labia majora, whereas IV-4 (25 weeks gestation) had normal external female genitalia.
Tissue specimens examined from autopsies of patients IV-1, IV-2, and IV-8 (Figure 1B) showed cystically dilated tubules to a variable extent in both the medulla and the cortex of the kidney, as well as persistence of circular ductal plates in the liver. In addition, patient IV-8 had squamous metaplasia of the urethra and periurethral glands in the prostate.
Neuropathology findings were extensive, with the main features being evidence of holoprosencephaly, hypoplastic or absent corpus callosum, agenesis of the pituitary, and cerebral cortex malformations. Patients IV-1 (Figure 1C) and IV-8 had hydrocephalus in the form of ventricular dilatation, absence of the third ventricle, dysmorphic septum pellucidum, and malformed diencephalic elements. It is conceivable that the apparent overgrowth of the medial diencephalic structures led to the obliteration of the third ventricle, initiating secondary hydrocephalus, followed by the rupture or absence of the septum pellucidum. In patient IV-2, there is a development of a semilobar holoprosencephaly, which indicates an earlier initiation of the genetic defect resulting in hypertrophic diencephalic elements.
Overall, affected individuals had ventricular hydrocephalus, midline cleft lip and palate, abnormal bone development manifesting as micromelia, bowing of the long bones, postaxial polydactyly, hypoplastic adrenal and pituitary glands, and ambiguous genitalia. Many of the malformations observed involve a defect of apoptosis, especially the cleft lip and palate, syndactyly, prolonged persistence of fusion of the eyelids, and unfused urogenital folds.
Autozygosity Mapping of a Candidate Locus on Chromosome 6p
Pedigree analysis (Figure 2) of affected individuals revealed that the syndrome followed an autosomal-recessive inheritance pattern, with two parental consanguineous matings (III-1 with III-2 and III-3 with III-4). Genome-wide autozygosity mapping performed on five ECO-affected infants and three of the four parents from the pedigree unambiguously mapped ECO to 6p12.2–p12 with a location score of 462, the summation of two-point LOD data,13,24 which spans over 1.8 Mb of the chromosome (Figure 3A). This region in chromosome 6p is bound by SNPs rs2397061 and rs627217 and is comprised of 397 genotyped contiguous SNPs (Figure 3B). Autozygosity-based linkage was confirmed by haplotype analysis of the SNP genotypes within the candidate region at chromosome 6p (Figure 2).
Figure 2.
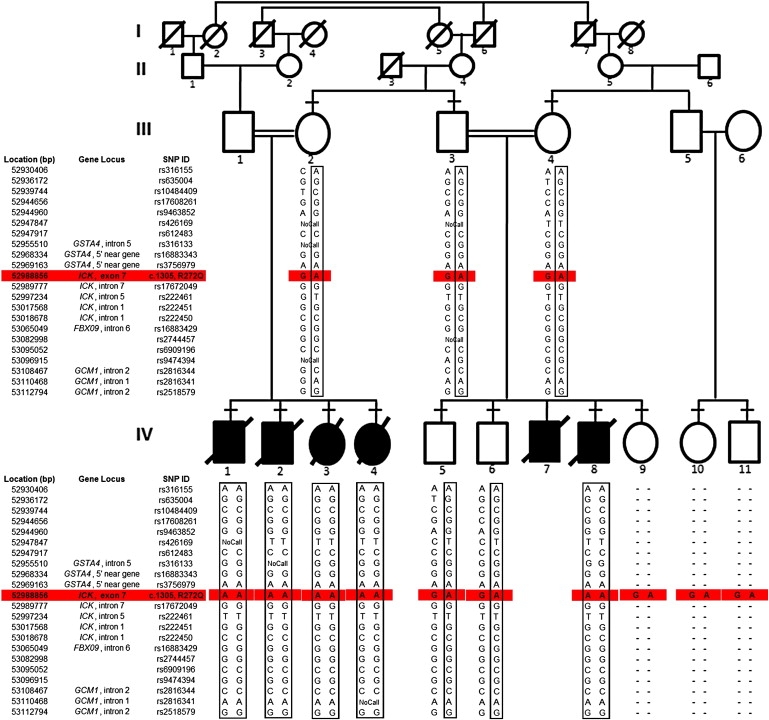
SNP-Based Genotyping of ECO-Affected Pedigree Identified a Homozygous Region in Chromosome 6p
ECO in a consanguineous Old Order Amish pedigree. The inheritance of the disease follows an autosomal-recessive mode of transmission. Affected individuals are shown as blackened squares (male) and circles (female). Diagonal lines across symbols indicate deceased individuals. A consanguineous marriage is shown by a double line between two individuals. Confirmation of autozygosity-based linkage was performed by observing the SNP-based genotyping within the candidate region on 6p12.2–p12. A portion of the homozygous region within patients is shown in boxes. The ICK variation at the coding sequence 1305 (G→A, accession number: NM_016513) that causes a R→Q change at residue 272 in the protein was analyzed (see Figure 3C for more details) and highlighted in red. Horizontal dashes above symbols indicate individuals who underwent DNA analysis, and hyphens indicate nongenotyped markers.
Figure 3.
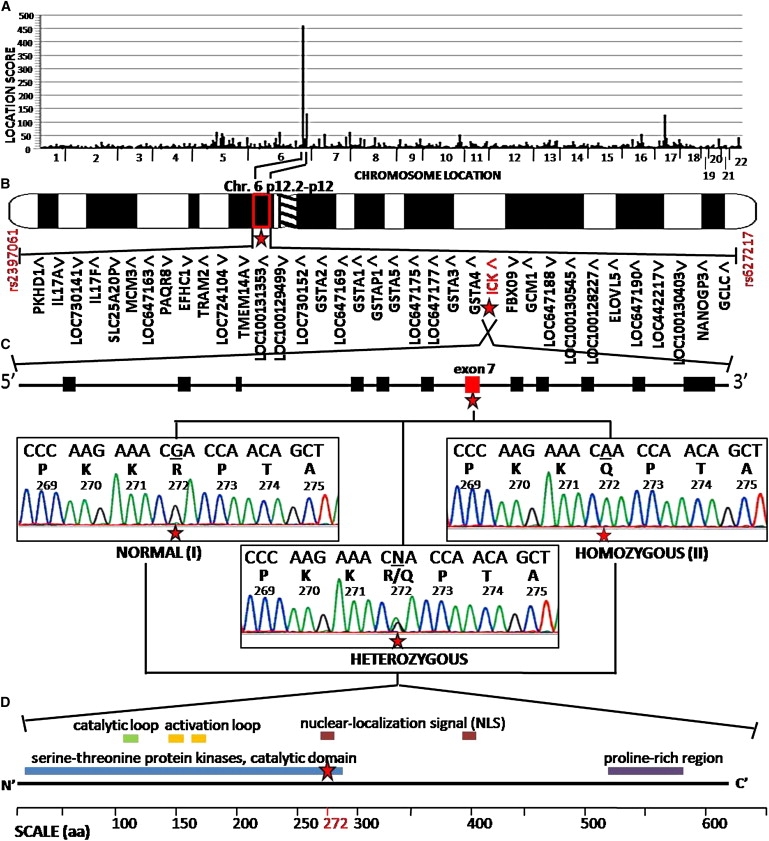
Autozygosity Mapping of ECO-Affected Pedigree Identified an Amino Acid Change, R272Q, in ICK
(A) Autozygosity mapping with SNP genotypes from 500,000 SNP microarrays across the chromosomes (x axis) yielded SNP haplotype location scores (y axis), with the highest peak on chromosome 6. Location score is the summation of the point LOD scores representing the likelihood of observing contiguous homozygosity in all five affected individuals of each SNP within the region of homozygosity.13
(B) Expanded view of the candidate locus shows the peak to be on chromosome 6p12.2–p12 (indicated by a star), which is defined by SNPs rs2397061 and rs627217 and consists of 36 candidate genes, including ICK (indicated in red with a star), distributed over 1.8 Mb of the genome. Transcriptional direction is indicated by arrowheads.
(C) The genomic structure of ICK gene consists of 12 coding exons with a nonsynonymous nucleotide change in exon 7 (indicated in red with a star) that alters the amino acid of arginine to glutamine at residue 272 (R272Q). DNA sequence analysis of ICK exon 7 from genomic DNA of a normal (I, top left tracing) individual, an affected (II, top right tracing) individual, and a R272Q heterozygote (bottom middle tracing). For each tracing, a normal nucleotide sequence is shown in the top line of letters, with single-letter amino acid codes and codon numbers beneath. The position of the mutated nucleotides is indicated by the star.
(D) The domain structure of ICK protein consists of the protein serine-threonine kinase catalytic domain, a catalytic loop, two activation loops, two nuclear-localization-signal sites, and a proline-rich region, from the N-terminal to C-terminal end. The amino acid (aa) 272 (indicated in red) lies within the nuclear-localization signal (indicated with a star).
Identification of the Causative Mutation, R272Q, in ICK
The candidate interval harbored a total of 36 known and hypothetical genes. We prioritized sequencing of candidate genes on the basis of the following criteria: (1) gene involvement in other single-gene disorders, (2) the role and tissue specificity of the encoded protein, and (3) the number of exons per gene, for feasibility reasons. On the basis of this priority list, exons of 11 candidate genes were directly sequenced from the genomic DNA of one of each of the following: affected individuals, unaffected siblings, parents, and non-Amish controls. This sequencing led to the discovery of numerous DNA sequence variations (Table S1 available online). In an autosomal-recessive mode of inheritance, the disease causing the variation would be due to a homozygous genotype observed only in the affected individual, and the parent would be an obligate carrier of the causative allele. The only such variation that also affected the gene and/or its gene product was in ICK. The ICK gene, composed of 12 exons, had a nonsynonymous nucleotide change c.1305G→A in exon 7 (Figure 3C) in the DNA of the affected individual. This nucleotide change results in an amino acid change from arginine to glutamine at residue 272 (R272Q), which lies within the nuclear-localization-signal domain of ICK (Figure 3D). DNA sequencing further demonstrated that this alteration was homozygous in all five affected individuals, whereas the phenotypically unaffected parents and siblings were heterozygotes, consistent with the predicted autosomal-recessive pattern of inheritance. Complete linkage of ECO to the R272Q mutation (at chromosome 6p) within the pedigree was observed, with a two-point linkage LOD score of 3.61 (at recombination fraction = 0).
In Silico Analysis Demonstrates R272 to Be Conserved and Required for Protein Stability
The ICK protein belongs to the CDK subfamily, which is within the CMGC serine-threonine protein kinase family.1 R272 in human ICK is conserved in the CMGC group of kinases, which includes the CDK, MAP kinase, glycogen synthase kinase 3 (GSK3), and CDC-like kinase (CLK) families. ICK homologs were identified across phylogeny, ranging from Homo sapiens to Candida albicans. Using ClustalW protein sequence alignment, we observed R272 to be conserved in all 84 known orthologous ICK homologs (a representative set is shown in Figure 4A). The conservation of R272 suggests that mutations in this amino acid are not well tolerated, given that it may be involved in crucial aspects of structure and function. A previous study of ICK that mutagenized several residues, including R272, suggested that the arginine is required to create a functionally and structurally stable conformation by forming ionic bonds with glutamic acid at residue 189.25 There is presently no three-dimensional (3D) model of ICK because it has not yet been crystallized. However, CDK2 and ICK share very high amino acid homology within the critical functional domain of interest. For homology-based modeling of ICK in PyMol,20 the 3D region harboring the nuclear-localization signal of CDK2 was substituted at only 2 of the 35 residues (residues 183 and 186). As visualized in Figure 4B, when arginine is mutated to glutamine, the bond with glutamic acid is disrupted such that the glutamic acid rotates its side chain so that it is no longer buried in the protein but becomes surface exposed, suggesting a cause for protein instability. Instability was confirmed with thermodynamic values acquired from Rosetta21 and Eris,22 two discrete protein-structure programs. Both Rosetta and Eris, which predict protein-packing energy changes, predicted that the R272Q mutant protein was less stable (by 5.1 and 3.4 kCal/mol, respectively) compared to the wild-type protein. In addition, four different bioinformatic programs predicted a deleterious effect of R272Q mutation on the ICK protein (“pathological,” “probably damaging,” “affected protein function,” and “deleterious” from PMUT, PolyPhen, SIFT, and SNPs3D, respectively).
Figure 4.
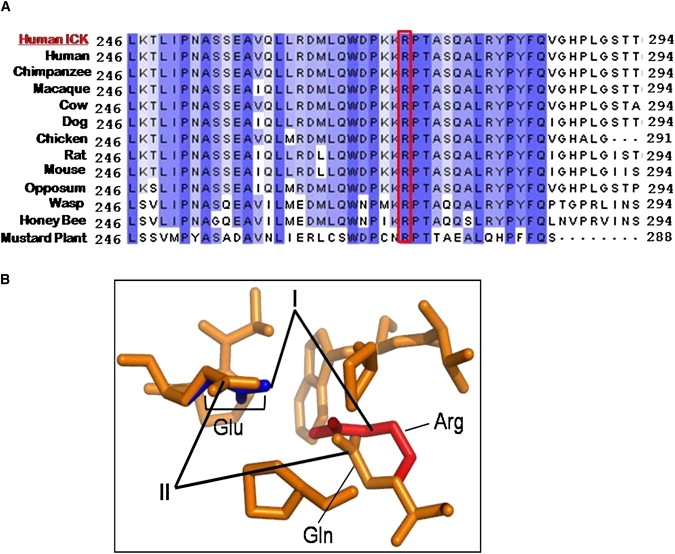
ICK Protein Analysis Demonstrates that R272 Is Highly Conserved and that the R272Q Mutation Alters Protein Structure
(A) Multiple alignments demonstrate that R272 residue is highly conserved across a representative set of species-specific ICK homologs. A ClustalW analysis of the ICK region encompassing the mutation site at residue 272 (highlighted in red) in aligned homologs with multiple divergent sequences is shown. The residues shaded in blue indicate amino acids that are similar between homologs.
(B) A magnified look at the 3D region surrounding residue 272 in ICK according to PyMOL modeling, such that the normal (I) protein is superimposed on the R272Q mutant (II) protein. This modeling predicts that the arginine (Arg, in red) and glutamic acid (Glu, in blue) form an ionic pair because of close proximity, in normal or wild-type protein (indicated by I). By comparison, the R272Q mutant (indicated by II) is a basic polar to neutral polar substitution and predicts a change in structure such that the glutamine (Gln, in orange) and glutamic acid (Glu, in orange) can no longer ion pair, leaving the glutamic acid exposed to the surface of the protein rather than buried within the 3D structure.
Mutation Screening in Amish and Non-Amish Populations Suggests R272Q to Be a Private Mutation
The frequency of the A allele in ICK at exon 7, c.1305 G→A, was assessed within members in the Old Order Amish community as well as individuals from other ethnic groups. Genotype analysis of 257 healthy Old Order Amish controls demonstrated no AA homozygote but identified five heterozygote GA carriers outside the ECO-affected family. The A allele frequency in the Old Order Amish community was 0.97%; thus, ∼1 in 10,000 Old Order Amish subjects would be predicted to have the AA genotype. TaqMan-based genotyping of the ICK mutation in an additional 2855 individuals from six ethnic groups demonstrated the complete absence of the A allele, indicating that this mutation was specific to the Amish.
Nuclear Localization Affected by ICK R272Q Mutation
GFP-tagged ICK wild-type overexpressed in HEK293 cells localized predominately to the nucleus, whereas the GFP-tagged ICK R272Q mutant localized predominantly to the cytoplasm (Figure 5A). Therefore, this single R272Q point mutation was sufficient to cause a loss of nuclear localization. Statistical evidence in a bar graph is provided in Figure S1.
Figure 5.
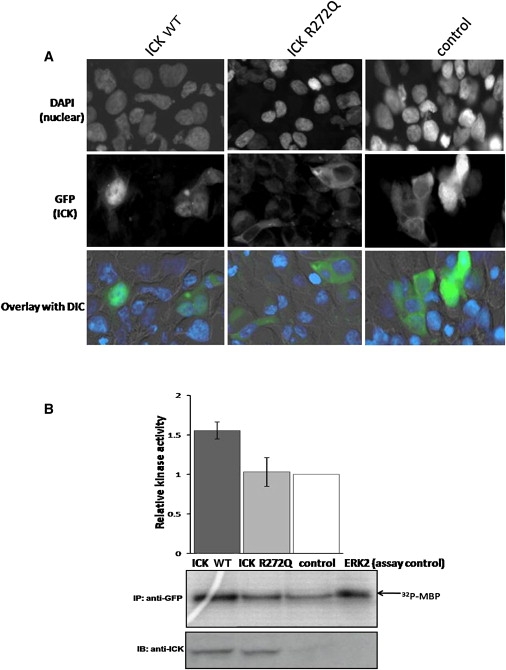
Altered Subcellular Localization and Protein Activity of the R272Q ICK Mutation
(A) Subcellular localization of wild-type and mutant ICK is mainly nuclear and cytoplasmic, respectively. HEK293 cells were transfected for 48 hr with wild-type ICK (ICK WT), mutant ICK (ICK R272Q), or vector control tagged with GFP and analyzed by immunofluorescence microscopy at 40× magnification. Transfected cells were identified by GFP fluorescence (green), and nuclei were stained with DAPI (blue). The signals obtained for DAPI and GFP are shown separately (rows 1 and 2), and an overlay of both fluorescence signals with differential interference contrast (DIC) is shown in row 3. We counted 502, 501, and 501 transfected cells per construct of wild-type ICK, mutant ICK, and control, respectively. The difference in subcellular localization is statistically significant (chi-square p value = 3.9 × 10−97), such that the wild-type and the mutant ICK protein localizes in the nucleus of 71.9% and 9.78% of the transfected cells, respectively. In addition, blinded counting was performed in a small subset of the whole, and the overall results of the much larger cell numbers were representative of this blinded sample.
(B) In vitro kinase assays were performed with MBP as substrate with anti-GFP immunoprecipates obtained from HEK293 transfected cells and active-purified ERK2 as a positive control (top panel). HEK293 cells were transfected for 48 hr with GFP-tagged ICK WT, ICK R272Q, or vector control followed by anti-GFP immunoprecipitation. Bar graphs indicate means ± standard deviations from three sets of experiments, showing the relative kinase activities normalized to vector control quantitated by densitometry. The autoradiograph shows results of one experiment demonstrating incorporation of 32P into MBP. The upper blot shows results of a representative experiment demonstrating the levels of MBP phosphorylation. The lower blot, developed with anti-ICK, demonstrates equal amounts of ICK WT and ICK R272Q.
Loss of Kinase Activity of ICK with R272Q Mutation
We studied kinase activity in the presence of the R272Q mutation. GFP-labeled ICK wild-type and R272Q mutant were overexpressed in HEK293 cells prior to isolation via immunoprecipitation, and subsequent kinase-activity assays were performed for phosphotransferase activity. Incorporation of 32P into the exogenous substrate, MBP, was catalyzed by ICK wild-type, but no such incorporation was detected in the presence of the ICK R272Q mutant (Figure 5B). The lower panel for Figure 5B indicates that equal amounts of ICK protein were assayed for kinase activity. Bands for the ICK constructs were observed specifically, providing solid evidence for specificity of the ICK antibody. Overall, the assay for 32P incorporation suggests that the R272Q mutation significantly impairs kinase activity.
Discussion
Rare congenital disorders provide new information on the biological pathways in human organogenesis.26 We report in an Old Order Amish community a previously unidentified and phenotypically distinct syndrome, ECO, whose clinical manifestations occur early in development and include dysplasias of skeletal, cerebral, and endocrine systems resulting in neonatal mortality. In affected patients, we identified a homozygous missense mutation, R272Q, in the kinase ICK. R272 was highly conserved across species and kinase family members. Moreover, the protein carrying the mutation was predicted to change the conformation of the structure, as visualized in PyMol, and to cause protein instability, as predicted by the change in energy values in the programs Rosetta Design and Eris. The R272Q mutation impairs nuclear localization and kinase activity. The ICK R272Q mutation underlying the ECO syndrome implicates intestinal cell kinase as central to the development of several organ systems in humans.
Although ECO is, to our knowledge, a distinct new syndrome, it does share some clinical features with previously reported syndromes, such as Majewski syndrome and hydrolethalus. Majewski syndrome, described in 1971, maps to chromosome 4 and is a lethal form of neonatal dwarfism characterized by short ribs, micromelia, polysyndactyly, median cleft lip, polycystic kidneys, ambiguous genitalia, and hypoplastic epiglottis, larynx, and lungs.27 Hydrolethalus, described in 1981, maps to chromosome 11q24.2 and is a neonatal lethal autosomal-recessive syndrome characterized by multiple congenital anomalies including hydramnios, hydrocephalus, a “keyhole” defect of the occipital bone, low-set ears, and midline malformations, such as heart and brain defects, cleft lip or palate, an abnormally shaped nose or jaw, incomplete lung development, and abnormal genitalia.28 The causative genes for both Majewski syndrome and hydrolethalus have yet to be identified,29 but neither locus overlaps with 6p12.2–p12 harboring ICK. A transitional clinical condition, pseudotrisomy 13 (MIM 264480), consists of characteristics found in both Majewski syndrome and hydrolethalus, including holoprosencephaly, hydrocephalus, polydactyly, heart defects, and facial anomalies consistent with the trisomy 13 condition without any chromosomal defects.30 Another clinical condition that bears some similarity to Majewski syndrome and hydrolethalus was reported in 1992, uniting two distinct genetic conditions such that the two conditions are suspected to be causally related.31 “Majewski hydrolethalus” was characterized by short limbs and ribs and abnormal tibiae consistent with Majewski syndrome as well as hydraminos, hydrocephalus, and keyhole-shaped occipital bone consistent with hydrolethalus syndrome.32 Again, the causative gene is unknown. Another distinct yet related condition with an unknown causative gene is the autosomal-recessive Mohr-Majewski syndrome (MIM: 258860).33
Hierarchical cluster analysis reordered these six related diseases on the basis of clinical features such that ECO had the closest clinical description to the Majewski and Mohr-Majewski syndromes (Figure 6). However, Majewski and Mohr-Majewski have the presence of short ribs and cardiac defects, respectively, which are features that are not observed in ECO-affected infants. Despite the absence of overlap of loci from mapping studies, DNA sequencing of ICK in individuals with Majewski and hydrolethalus syndromes might help to establish whether these disorders are allelic or whether ECO truly represents a distinct and unique disorder.
Figure 6.

Schematic of Overlapping Features between Six Disorders, including ECO
The grid shows organ-system involvement, with darkened cells indicating the presence of the subphenotype for each disease. Prevalence of disorder is in a white-to-black gradient system in which a frequency of 0% is white, 50% is gray, and 100% is black. The reordering of the disorders was the result of hierarchical cluster analysis that clustered the disease on the basis of similarities in organ-system involvement. The diseases included are (from top to bottom) Hydrolethalus, Pseudotrisomy 13, Majewski Hydrolethalus, Mohr-Majewski, Majewski syndrome, and ECO (highlighted in red).
ICK was initially cloned from the human intestinal crypt with degenerate primers specific for MAP kinases.34,35 The designation ICK appears to be a misnomer, given that ICK is ubiquitously expressed in adult human tissues.34,35 Alternatively, this protein has sometimes been called MAK-related kinase (MRK) in previous reports.35,36 ICK has a variant, ICKb (accession number AAH35807); however, ICKb is 337 amino acids shorter with an alternative C terminus, such that ICKb is predominantly in the cytoplasm.25 ICK shares 38% to 40% identity to the catalytic domains of CDKs, which are involved in cell-cycle transition, and MAP kinases, regulators of cell-cycle entry.25 ICK is activated by dual phophorylation of the TDY motif (Y-159 and T-157), such that it autophosphorylates at Y-159 and can be phosphorylated by human kinase cell-cycle-related kinase (CCRK), while being deactivated by protein phosphatase 5 (PP5).37 It may be speculated that because the residue 272 mutation of ICK prevents nuclear localization, it hinders phosphorylation of the ICK by the nuclear-residing CCRK, which is ultimately required for kinase activity of ICK.
A previous in vitro study of ICK coincidentally mutagenized residue 272 from arginine to alanine (R272A). Like R272Q, this non-naturally occurring R272A mutant showed impaired nuclear localization and kinase activity.25 As determined by in vitro studies, ICK can phosphorylate exogenous substrates, such as myelin basic protein25,36 and Scythe, an antiapoptotic protein required during mammalian development.38 Scythe (BAT3) is a nuclear protein that is implicated in apoptosis.39 Scythe knockout mice display brain heteropia (abnormal migration), hydrocephalus, and dilated and hypoplastic kidneys as well as lung abnormalities, leading to perinatal death.39 In addition to endocrine hypoplasia, syndactyly, cleft lip, and cleft palate, ECO-affected infants also develop phenotypes observed with Scythe deficiency. Because ICK is involved in phosphorylation and activation of Scythe, it is presumable that an ICK mutation would include defects observed with Scythe inactivation. Overall, ICK seems to be involved in cell-cycle regulation and apoptosis during mammalian development. Other evidence that ICK has an effect on embryological development is the observed presence of ICK mRNA in placental tissue,35 although specific experiments will need to be performed for confirmation. Previous work36 has suggested that ICK has a role in development of the myocardium, but because ECO syndrome is a multiorgan disorder, this kinase must have a role in development of other tissues and organ systems. Interestingly, no cardiac anomalies have been observed in ECO syndrome patients.
Thus, we have characterized a previously unreported human syndrome—ECO—in which mutated ICK is functionally impaired. This single point mutation at residue 272 not only affects structure, but also function, such that it can no longer localize properly, which has detrimental downstream effects and ultimately leads to abnormal fetal development. These findings suggest that ICK plays a key role in the development of multiple organ systems.
Supplemental Data
Supplemental Data include one table and one figure and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW alignments, http://www.clustal.org
Eris Server, http://troll.med.unc.edu/eris/login.php
London Regional Genomics Centre, http://www.lrgc.ca
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
PolyPhen, http://coot.embl.de/PolyPhen/
PyMol, http://pymol.sourceforge.net
Rosetta Design, http://rosettadesign.med.unc.edu
SNPs3D, http://www.SNPs3D.org
Acknowledgments
We acknowledge the generous cooperation of the families participating in this study. We acknowledge the excellent technical assistance of Rebecca Provost, Rachel Rollings, Caroline O'Neil, and Roger Dewar. R.A.H. is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Edith Schulich Vinet Canada Research Chair (Tier I) in Human Genetics and the Jacob J. Wolfe Distinguished Medical Research Chair. P.L. was supported by the Canadian Institutes of Health Research (CIHR) Scriver Family MD/PhD studentship award and the HSFO Vascular Training Program. This work was supported by a team grant from the CIHR (CTP-79853, MOP-37854), the Heart and Stroke Foundation of Ontario, Genome Canada through the Ontario Genomics Institute, and grants from the Children's Health Research Institute, the Lawson Health Research Institute, and the Garrod Association.
References
- 1.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Lew J. MAP kinases and CDKs: Kinetic basis for catalytic activation. Biochemistry. 2003;42:849–856. doi: 10.1021/bi0269761. [DOI] [PubMed] [Google Scholar]
- 3.Budhiraja S., Singh J. Protein kinase C beta inhibitors: A new therapeutic target for diabetic nephropathy and vascular complications. Fundam. Clin. Pharmacol. 2008;22:231–240. doi: 10.1111/j.1472-8206.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- 4.Xing M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer. 2005;12:245–262. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]
- 5.Bolen J.B. Protein tyrosine kinases in the initiation of antigen receptor signaling. Curr. Opin. Immunol. 1995;7:306–311. doi: 10.1016/0952-7915(95)80103-0. [DOI] [PubMed] [Google Scholar]
- 6.Wilson F.H., Disse-Nicodeme S., Choate K.A., Ishikawa K., Nelson-Williams C., Desitter I., Gunel M., Milford D.V., Lipkin G.W., Achard J.M. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 7.Hurst J.A., Firth H.V., Smithson S. Skeletal dysplasias. Semin. Fetal Neonatal Med. 2005;10:233–241. doi: 10.1016/j.siny.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Norgard M., Yankowitz J., Rhead W., Kanis A.B., Hall B.D. Prenatal ultrasound findings in hydrolethalus: Continuing difficulties in diagnosis. Prenat. Diagn. 1996;16:173–179. doi: 10.1002/(SICI)1097-0223(199602)16:2<173::AID-PD821>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 9.Chen H., Yang S.S., Gonzalez E., Fowler M., Al Saadi A. Short rib-polydactyly syndrome, Majewski type. Am. J. Med. Genet. 1980;7:215–222. doi: 10.1002/ajmg.1320070214. [DOI] [PubMed] [Google Scholar]
- 10.Hostetler J.A. Johns Hopkins University Press; Baltimore: 1993. Amish Society. [Google Scholar]
- 11.Lander E.S., Botstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 12.Rauch A., Thiel C.T., Schindler D., Wick U., Crow Y.J., Ekici A.B., van Essen A.J., Goecke T.O., Al-Gazali L., Chrzanowska K.H. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 13.Puffenberger E.G., Hu-Lince D., Parod J.M., Craig D.W., Dobrin S.E., Conway A.R., Donarum E.A., Strauss K.A., Dunckley T., Cardenas J.F. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc. Natl. Acad. Sci. USA. 2004;101:11689–11694. doi: 10.1073/pnas.0401194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins, D., Thompson, J., and Gibson, T. (2007). Clustal: Multiple Sequence Alignment (http://www.clustal.org).
- 15.Ferrer-Costa C., Gelpi J.L., Zamakola L., Parraga I., de la Cruz X., Orozco M. PMUT: A web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 16.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue P., Melamud E., Moult J. SNPs3D: Candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulze-Gahmen U., Brandsen J., Jones H.D., Morgan D.O., Meijer L., Vesely J., Kim S.H. Multiple modes of ligand recognition: Crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopentenyladenine. Proteins. 1995;22:378–391. doi: 10.1002/prot.340220408. [DOI] [PubMed] [Google Scholar]
- 20.DeLano W.L. DeLano Scientific; Palo Alto, CA: 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 21.Das R., Baker D. Macromolecular modeling with rosetta. Annu. Rev. Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 22.Yin S., Ding F., Dokholyan N.V. Eris: An automated estimator of protein stability. Nat. Methods. 2007;4:466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 23.Moore K.L. WB Saunders Company; Toronto: 1977. The Developing Human. [Google Scholar]
- 24.Strauss K.A., Puffenberger E.G., Huentelman M.J., Gottlieb S., Dobrin S.E., Parod J.M., Stephan D.A., Morton D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 25.Fu Z., Schroeder M.J., Shabanowitz J., Kaldis P., Togawa K., Rustgi A.K., Hunt D.F., Sturgill T.W. Activation of a nuclear Cdc2-related kinase within a mitogen-activated protein kinase-like TDY motif by autophosphorylation and cyclin-dependent protein kinase-activating kinase. Mol. Cell. Biol. 2005;25:6047–6064. doi: 10.1128/MCB.25.14.6047-6064.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peltonen L., Uusitalo A. Rare disease genes–Lessons and challenges. Genome Res. 1997;7:765–767. doi: 10.1101/gr.7.8.765. [DOI] [PubMed] [Google Scholar]
- 27.Urioste M., Martinez-Frias M.L., Bermejo E., Jimenez N., Romero D., Nieto C., Villa A. Short rib-polydactyly syndrome and pericentric inversion of chromosome 4. Am. J. Med. Genet. 1994;49:94–97. doi: 10.1002/ajmg.1320490118. [DOI] [PubMed] [Google Scholar]
- 28.Salonen R., Herva R., Norio R. The hydrolethalus syndrome: Delineation of a “new”, lethal malformation syndrome based on 28 patients. Clin. Genet. 1981;19:321–330. doi: 10.1111/j.1399-0004.1981.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 29.Visapaa I., Salonen R., Varilo T., Paavola P., Peltonen L. Assignment of the locus for hydrolethalus syndrome to a highly restricted region on 11q23–25. Am. J. Hum. Genet. 1999;65:1086–1095. doi: 10.1086/302603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dincsoy M.Y., Salih M.A., al-Jurayyan N., al Saadi M., Patel P.J. Multiple congenital malformations in two sibs reminiscent of hydrolethalus and pseudotrisomy 13 syndromes. Am. J. Med. Genet. 1995;56:317–321. doi: 10.1002/ajmg.1320560321. [DOI] [PubMed] [Google Scholar]
- 31.Neri G., Gurrieri F., Genuardi M. Oral-facial-skeletal syndromes. Am. J. Med. Genet. 1995;59:365–368. doi: 10.1002/ajmg.1320590317. [DOI] [PubMed] [Google Scholar]
- 32.Sharma A.K., Phadke S., Chandra K., Upreti M., Khan E.M., Naveed M., Agarwal S.S. Overlap between Majewski and hydrolethalus syndromes: A report of two cases. Am. J. Med. Genet. 1992;43:949–953. doi: 10.1002/ajmg.1320430609. [DOI] [PubMed] [Google Scholar]
- 33.Rosing B., Kempe A., Berg C., Kahl P., Knopfle G., Gembruch U., Geipel A. Orofaciodigital syndrome Type IV (Mohr-Majewski): Early prenatal diagnosis in siblings. Ultrasound Obstet. Gynecol. 2008;31:457–460. doi: 10.1002/uog.5285. [DOI] [PubMed] [Google Scholar]
- 34.Togawa K., Yan Y.X., Inomoto T., Slaugenhaupt S., Rustgi A.K. Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J. Cell. Physiol. 2000;183:129–139. doi: 10.1002/(SICI)1097-4652(200004)183:1<129::AID-JCP15>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 35.Yang T., Jiang Y., Chen J. The identification and subcellular localization of human MRK. Biomol. Eng. 2002;19:1–4. doi: 10.1016/s1389-0344(02)00002-3. [DOI] [PubMed] [Google Scholar]
- 36.Abe S., Yagi T., Ishiyama S., Hiroe M., Marumo F., Ikawa Y. Molecular cloning of a novel serine/threonine kinase, MRK, possibly involved in cardiac development. Oncogene. 1995;11:2187–2195. [PubMed] [Google Scholar]
- 37.Fu Z., Larson K.A., Chitta R.K., Parker S.A., Turk B.E., Lawrence M.W., Kaldis P., Galaktionov K., Cohn S.M., Shabanowitz J. Identification of yin-yang regulators and a phosphorylation consensus for male germ cell-associated kinase (MAK)-related kinase. Mol. Cell. Biol. 2006;26:8639–8654. doi: 10.1128/MCB.00816-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardina P.J., Clark T.A., Shimada B., Staples M.K., Yang Q., Veitch J., Schweitzer A., Awad T., Sugnet C., Dee S. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desmots F., Russell H.R., Lee Y., Boyd K., McKinnon P.J. The reaper-binding protein scythe modulates apoptosis and proliferation during mammalian development. Mol. Cell. Biol. 2005;25:10329–10337. doi: 10.1128/MCB.25.23.10329-10337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fenton T.R. A new growth chart for preterm babies: Babson and Benda's chart updated with recent data and a new format. BMC Pediatr. 2003;3:13. doi: 10.1186/1471-2431-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.