

Abstract
First described in 1974, FG syndrome (FGS) is an X-linked multiple congenital anomaly/mental retardation (MCA/MR) disorder, characterized by high clinical variability and genetic heterogeneity. Five loci (FGS1-5) have so far been linked to this phenotype on the X chromosome, but only one gene, MED12, has been identified to date. Mutations in this gene account for a restricted number of FGS patients with a more distinctive phenotype, referred to as the Opitz-Kaveggia phenotype. We report here that a p.R28L (c.83G→T) missense mutation in CASK causes FGS phenotype in an Italian family previously mapped to Xp11.4-p11.3 (FGS4). The identified missense mutation cosegregates with the phenotype in this family and is absent in 1000 control X chromosomes of the same ethnic origin. An extensive analysis of CASK protein functions as well as structural and dynamic studies performed by molecular dynamics (MD) simulation did not reveal significant alterations induced by the p.R28L substitution. However, we observed a partial skipping of the exon 2 of CASK, presumably a consequence of improper recognition of exonic splicing enhancers (ESEs) induced by the c.83G→T transversion. CASK is a multidomain scaffold protein highly expressed in the central nervous system (CNS) with specific localization to the synapses, where it forms large signaling complexes regulating neurotransmission. We suggest that the observed phenotype is most likely a consequence of an altered CASK expression profile during embryogenesis, brain development, and differentiation.
Introduction
FG syndrome (FGS [MIM 305450]), also known as Opitz-Kaveggia syndrome,1 is a rare X-linked disorder characterized by multiple congenital anomalies and mental retardation (MCA/MR). Several families have been reported to date, increasing the phenotype variability.2–8
FGS is characterized by developmental delay, congenital hypotonia, complete or partial agenesis of the corpus callosum, characteristic face, relative macrocephaly, constipation (with or without anal/intestinal anomalies), and an unusual personality.1,2 However, FGS has a variable clinical presentation and clinical diagnosis may be difficult, especially in sporadic patients.9
FGS is genetically heterogeneous and five loci have so far been identified on the X chromosome. The FGS1 (MIM 305450), FGS3 (MIM 300406), and FGS4 (MIM 300422) loci have been mapped to Xq12-q22.1, Xp22.3, and Xp11.4-p11.3, respectively, by linkage analysis.6,10–12 The FGS2 (MIM 300321) locus was located at Xq11 or Xq28 by analyzing an X chromosome inversion [inv(X)(q11q28)].13,14 Recently, the FGS5 (MIM 300581) locus has been identified by detecting an Xq22.3 duplication in a Brazilian FGS patient by CGH array.15
Even more recently, a recurrent c.2881C→T (p.R961W) mutation in the MED12 gene (MIM 300188) at Xq13 has been shown to be responsible for FGS in 6 out of 45 families with the clinical diagnosis of Opitz-Kaveggia syndrome, including the only surviving affected male from the original Opitz-Kaveggia family.16 MED12 represents the first FGS gene identified. Its causal role appears to be restricted to the Opitz-Kaveggia phenotype, which seems to represent a specific phenotype within the broad spectrum of FGS.9 A different p.N1007S (c.3020A→G) missense mutation in MED12 has also been found in the original family with Lujan-Fryns syndrome (MIM 309520) and in another distinct family.17 In addition, mutations in UPF3B (MIM 300298), BRWD3 (MIM 300553), and FLNA (MIM 300017) genes have also been found in sporadic patients with clinical features overlapping the FGS phenotype.18–20 Most probably, FLNA corresponds to the FGS2 gene and the others should be considered novel putative FGS loci.21
Previously, we clinically and genetically characterized an Italian FGS family that included 31 members with 3 affected males and 2 obligate carriers.12 The affected males showed many clinical signs typical of FGS such as mental retardation, relative macrocephaly, congenital hypotonia, severe constipation, and behavioral disturbances.
By linkage analysis, we identified the FGS4 locus localized in Xp11.4-p11.3 between DXS8113 and sWXD805, corresponding to a 4.4 Mb region on the X chromosome.
Here, we describe CASK (MIM 300172) as the FGS4 gene mutated in this Italian FGS family. CASK maps to Xp11.4 and encodes a multidomain scaffold protein highly expressed in the central nervous system (CNS), but also, at lower levels, in epithelial cells and other tissues.22,23
In our FGS patients, we found a c.83G→T (p.R28L) CASK mutation that cosegregates with the phenotype. The mutation changes a highly conserved amino acid in the N-terminal domain of CASK homologous to calcium/calmodulin-dependent protein kinase II (CaM-kinase domain).22
We show that the p.R28L mutation did not interfere with the known interactions and functions involving CaM-kinase domain, but it may act by producing a partial skipping of the exon 2 as a result of improper recognition of exonic splicing enhancers (ESEs). The resulting aberrant CASK exon 2-skipped transcript is out of frame. We speculate that an altered CASK expression profile during embryogenesis and CNS development could be at the basis of the FGS4 phenotype.
Material and Methods
FGS4 Family Members
The clinical description of the patients was previously reported.12 In accordance with Italian law, an informed consent was obtained from all family members involved in this study.
Gene Selection and Primer Design
The 4.4 Mb region of the FGS4 locus between DXS8113 and sWXD805 was scanned on UCSC Genome Browser. Eleven known genes were identified. Accession number of the reference sequence, gene symbol, number of coding exons, and number of amplicons analyzed in mutation screening are listed in Table 1 (UCSC Human Genome Database; freeze March 2006).24,25 All oligonucleotides utilized in mutation screening and in all other PCR protocols were designed with web-based tool Primer3.26
Table 1.
Candidate Genes at FGS4 Locus
| Acc. Number | Gene Symbol | Gene | Exons | Amplicons | Status |
|---|---|---|---|---|---|
| NM_001123384 | BCOR | BCL6 interacting corepressor | 15 | 24 | A |
| NM_005765 | ATP6AP2 | ATPase, H+ transporting, lysosomal accessory protein 2 | 9 | 9 | A |
| NM_144970 | CXorf38 | chromosome X open reading frame 38 (hypothetical protein) | 7 | 6 | A |
| NM_004229 | MED14 | mediator complex subunit 14 | 31 | 31 | A |
| NM_001039590 | USP9X | ubiquitin specific peptidase 9, X-linked | 45 | 44 | A |
| NM_001356 | DDX3X | DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, X-linked | 17 | 17 | A |
| NM_022567 | NIX | nyctalopin | 2 | N/A | |
| NM_001126054 | CASK | calcium/calmodulin-dependent serine protein kinase | 27 | 27 | A |
| NM_001097579 | GPR34 | G protein-coupled receptor 34 | 3 | 4 | A |
| NM_080817 | GPR82 | G protein-coupled receptor 82 | 3 | 5 | A |
| NM_000240 | MAOA | monoamine oxidase A | 15 | N/A |
Abbreviations: A, analyzed; N/A, not analyzed by CMS.
Comparative Mutation Scanning
The major problem in identifying causative mutations in large genomic regions is the huge number of potential candidate mutations to be distinguished from polymorphisms or private variants.
For this study, we set up comparative mutation scanning (CMS) analysis, a DHPLC-based approach that performs, in a very short time, a comparison of each candidate mutation with all members of the family, an initial pool of controls, and (when necessary) a second larger pool of controls.
CMS analysis requires an accurate definition of DHPLC conditions. For each gene analyzed, we performed DHPLC optimization by designing oligonucleotide pairs to amplify each exon and its intronic flanking regions, carefully evaluating the GC content of primers and PCR products to facilitate the next temperature optimization. We utilized the same annealing temperature for all the exons, allowing different exons to be amplified on the same 96/384-well plate.
For this X-linked disease, each exon was first amplified with genomic DNA from a carrier female, an affected male, and a control male. PCR products were then analyzed by agarose gel electrophoresis to highlight any possible gene deletion/duplication. DHPLC conditions were optimized on a Wave 3500HT system with Navigator 1.6.4 software according to manufacturer's indications (Transgenomic Inc.). We utilized Rapid DNA methods with a 2-min-long gradient and a flow rate of 1.5 ml/min. For CASK, primer pairs, annealing temperature, length of amplicons, amplified exons, column temperature, and percentage of B solution at start gradient are listed in Table S1 available online. The same data can be requested from the authors for all the other genes at the FGS4 locus.
For each exon analyzed, different pooled DNAs were used in PCR reaction, differently assembled according to the mechanism of inheritance. The samples were dispensed into ready-to-use 96-well plates in which each row contained 12 mixes of pooled DNAs, as shown in Table S2.
PCR products were run on DHPLC under the optimized conditions. By using the CMS approach, we were able to distinguish rapidly between mutation and polymorphism. According to the mechanism of inheritance, heteroduplexes present in samples 2–5 and absent in samples 1 and 6–12 were indicative of a mutation (see Table S2). For the more frequent polymorphisms, the number of control chromosomes analyzed was sufficient to exclude these from further analysis, reducing time and cost of the mutation screening.
Each variation observed in patients and absent in controls was further characterized by direct sequencing, and its pathogenetic role was validated by analyzing at least 1000 control X chromosomes of the same ethnic origin.
FGS/XLMR Patient Recruitment
One hundred and one affected individuals (91 males and 10 females) with FGS/XLMR phenotype were collected worldwide with the support of several medical genetics centers. They were classified into three different groups: (1) 32 familial cases with linkage data overlapping the FGS4 locus (27 XLMR patients and 5 FGS patients); (2) 27 familial cases without linkage data; and (3) 42 sporadic FGS patients.
We received DNA and/or blood samples. For blood samples, DNA was extracted via a standard protocol.27 For the members of the FGS family under study and other Italian FGS patients, EBV-transformed lymphocyte cell lines were also established via standard procedures.28 An informed consent was obtained from all the human subjects involved in this study.
Construction of Fusion Genes and Expression Plasmids
The sequences encoding the CaM-kinase domain of CASK (CASKwt: nt. 12–1003 of NM_003688; aa. 5–334) as well as the interacting domains of Mint-1 (MNT1: nt. 345–1336 of NM_001163; aa. 116–445), Caskin 1 (CSK1: nt. 1029–1496 of NM_020764; aa. 344–498), and Carom (CARM: nt. 1833–2223 of NM_014824; aa. 612–740) were amplified by RT-PCR from total human brain RNA (OriGene Technologies) with a thermostable DNA polymerase with proofreading activity (PfuTurbo DNA polymerase; Stratagene) and cloned in pBlueScriptSK vector. After automated full-length sequencing to confirm that no base errors occurred, each coding fragment was in-frame subcloned in pGBKT7 and pGADT7 yeast two-hybrid system vectors, in pGEX-2TK vector for bacterial expression of GST-fusion proteins, and in a modified pSG5 vector for expression in eukaryotic cells of proteins N-terminal labeled with the c-Myc epitope tag.
The p.R28L mutation under study (CASKmut) and the hypomorphic double mutant (p.S24D and p.V26L) (CASKsv)29 were obtained by site-specific direct mutagenesis of the wild-type coding sequence of the CaM-kinase domain of CASK cloned in the required vectors (QuikChange Site-Directed Mutagenesis Kit; Stratagene).
Yeast Two-Hybrid Assay and Library Screening
The pGBKT7-CASKwt and pGBKT7-CASKmut plasmids were separately transformed in S. cerevisiae AH109 strain by lithium acetate method, selected on Trp− plates, and used as baits. Conversely, pGADT7-MNT1, pGADT7-CSK1, and pGADT7-CARM plasmids were separately transformed in S. cerevisiae Y187 strain with the same method, selected on Leu− plates, and used as preys. According to manufacturer's indications (Clontech), the pTD1 and p53 control vectors as well as the pGBKT7 and pGADT7 empty vectors were also separately transformed in the specific yeast strain and utilized as positive and negative controls of interaction, respectively. Each bait was mated with prey lines and diploid cells were selected on Trp− and Leu− plates. The protein-protein binding was then detected by selecting positive clones on minimal Trp−, Leu−, His−, and Ade− plates, as a result of the activation of HIS3 and ADE2 reporter genes. As a further control of all tested interactions, plasmid vectors and yeast strains were also opportunely switched.
The screening of a human brain cDNA library cloned in pACT2 vector and pretransformed in S. cerevisiae Y187 strain was carried out according to manufacturer's specifications (Clontech). In brief, pGBKT7-CASKwt and pGBKT7-CASKmut plasmids separately transformed in S. cerevisiae AH109 strain were grown overnight in SD/Trp− liquid medium. After centrifugation, pellets of both bait strains were separately resuspended in 5 ml of the same liquid medium. For mating, each bait strain was combined with an aliquot of the pretransformed cDNA library in 50 ml of YPDA liquid medium and incubated at 30°C for 24 hr with slow shaking. The diploid strains were plated on high-stringency Trp−, Leu−, His−, and Ade− synthetic medium. His+ and Ade+ diploid colonies were patched on selective plates and assayed for β-galactosidase activity by colony-lift filter assay. Total DNA was extracted from His+, Ade+, and LacZ+ colonies, and pACT2 library plasmids were then rescued after transformation in XL1-blue competent cells (Stratagene). The pACT2 library plasmids trapped by using pGBKT7-CASKwt or pGBKT7-CASKmut plasmids as baits were tested for specificity by cotransformation into AH109 strain either alone, in combination with the pGBKT7-CASKwt or pGBKT7-CASKmut constructs, or in combination with negative and positive control plasmids. The cDNA inserts from verified CaM-kinase domain-interacting clones were characterized by direct sequencing.
In Vitro GST Pull-Down Assay
The in vitro GST pull-down assay was performed as previously described.30 In brief, the wild-type or mutant CaM-kinase domains of CASK were expressed in BL21 E. coli cells (Stratagene) as GST-fusion protein (pGEX-CASKwt and pGEX-CASKmut plasmids) and purified on glutathione-agarose beads. Conversely, the interacting domains of Mint-1 and Caskin 1 cloned in a modified pSG5 vector with c-Myc epitope tag to the N terminus (pSG5Myc-MNT1 and pSG5Myc-CSK1 plasmid) were transiently expressed in COS-7 eukaryotic cells. COS-7 cell protein extracts were then incubated with GST-CASKwt, GST-CASKmut, and GST-alone bead-bound fusion proteins. After extensive washing, samples were run on 10% SDS-polyacrylamide gel and analyzed by western blotting with an anti-Myc monoclonal antibody (Hybridoma 9E10).
In Vitro Phosphorylation Assay
Phosphorylation assay was performed as previously described.29
All the experiments were carried out in 50 mM Tris-HCl (pH 7.2) and 50 mM KCl buffer containing 2 mM EDTA or 4 mM MgCl2.
In brief, bead-bound GST-fusion proteins corresponding to the wild-type, the mutant (p.R28L), and the double hypomorphic mutant (p.S24D and p.V26L) CaM-kinase domain of CASK were incubated in equimolar quantity with 1 mM [γ-32P]ATP (specific activity: 2 × 107 cpm) (PerkinElmer) for 1 hr at 30°C with shaking. The reaction was stopped by adding an equal volume of 2× loading buffer (250 mM Tris-HCl [pH 6.8], 8 M urea, 8% SDS, 200 mM dithiothreitol, and 0.005% bromphenol blue), and after boiling, samples were separated by SDS-PAGE, Coomassie blue stained, and visualized on a phosphorimager (Typhoon; GE Healthcare). The incorporated radioactivity was measured with ImageQuant software (GE Healthcare) and, after background subtraction, values were normalized by comparison with protein quantities.
Molecular Dynamics Simulation
All MD simulations were performed with GROMACS (GROningen Machine for Chemical Simulations) v.3.2 and united-atom forcefield.31 The starting configuration of the CaM-kinase domain of CASK was obtained by crystallographic coordinates (PDB: 3c0h) with routines included in Visual Molecular Dynamics (VMD) package,32 also used to introduce the p.R28L substitution. For all MD simulations, the protein was placed in the center of a cubic box of approximate dimensions (8.48 × 8.48 × 8.48 nm3). The minimum distance between the protein and the edge of the box was 1.0 nm. All water molecules with their oxygen closer than 0.23 nm to any nonhydrogen protein atom or crystal water oxygen were removed. The box was full of equilibrated SPC (Single Point Charge) rigid water molecules. Initial velocities were taken from a Maxwellian distribution at 300 K. Simulations were performed keeping temperature and pressure coupled to external reference, with a Berendsen thermostat and barostat;33 coupling constants were 0.1 and 1.0 ps, respectively. All molecular groups (protein, water, and counterions) were coupled independently. Long-range electrostatic interactions were calculated with the Particle Mesh Ewald (PME) method with a 9 Å cut-off.34 The LINear Constraint Solver (LINCS) algorithm was used to restrain bond lengths.35 In order to relieve unfavorable nonbonded interactions with the added water molecules, each system was energy minimized by a steepest descent algorithm followed by a short 100 ps simulation during which the protein and nonhydrogen atoms were harmonically restrained with a force constant of 1000 kJ/mol·nm2. All restraints were then removed and each simulation was run for 2.5 ns saving coordinates and velocities every 5 ps for subsequent analysis. Typical CPU times on HP 6200 Workstation running Linux Kernel 2.6.3-7 were 1 day per ns. The simulated system comprised 3,083 protein atoms and 14,000 water molecules. Each simulation was stopped after 2.5 ns. As previously discussed, the first nanosecond of each simulation is treated as an equilibration period and is not considered in the analysis. To assess the stability of the simulations, the rmsd of protein atoms with respect to the X-ray structure, the solvent-accessible surface area, and the presence of secondary structure elements were monitored.
RT-PCR and Real-Time RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) from EBV-transformed lymphocyte cell lines from affected males, carrier females, and normal individuals from the FGS family as well as additional healthy male and female controls. RNAs were then retrotranscripted with SuperScript III RT (Invitrogen) and random primers, according to manufacturer's specifications. Single-strand cDNAs were used in later experiments.
To test any putative effect of the c.83G→T (p.R28L) mutation on splicing of CASK exon 2, different primer pairs were designed and utilized in RT-PCR and real-time RT-PCR experiments (Table S3).
An exon 1-specific primer (CASK16/F) was combined with an exon 3-specific primer (CASK268/R) to amplify a 253-bp-long fragment from the normal CASK transcript or a 140-bp-long fragment expected when CASK exon 2 is skipped. The exon 1-specific primer (CASK16/F) was also combined with a downstream primer in exon 8 (CASK744/R). The expected PCR products were 729 bp and 616 bp long, respectively.
RT-PCR was performed in a final volume of 20 μl containing 2 μl of cDNA, 1× PCR Buffer II (Applied Biosystems), 1 mM MgCl2, 1 mM dNTPs, 0.5 μM of each primer, and 0.5 U of AmpliTaq Gold DNA polymerase (Applied Biosystems). The cycling conditions consisted of a first step at 96°C for 7 min and 30 cycles of 30 s at 96°C, 1 min at 63°C, and 3 min plus 3 s/cycle at 68°C.
To quantify the expression level of the exon 2-skipped CASK transcript, further primers were designed to be specifically used in quantitative real-time RT-PCR experiments (Table S3).
To discriminate unskipped and exon 2-skipped isoforms of CASK transcripts, two additional primers were designed, the first overlapping exon 1-exon 2 (CASKsp1-2/F) junction and the second overlapping exon 1-exon 3 (CASKsp1-3/F). The normal CASK transcript was detected with CASKsp1-2/F and CASK268/R primer pair (amplified product 220 bp long), whereas the exon 2-skipped isoform of CASK transcript was detected by combining CASKsp1-3/F primer with a downstream primer in exon 5 (CASK398/R; amplified product 240 bp long). Total CASK transcripts were detected by using CASK2310/F and CASK2545/R primer pair (amplified product 236 bp long), designed in exons 25 and 26, respectively. GAPDH was amplified as housekeeping gene for data normalization.
Real-time RT-PCR experiments were performed on ABI Prism 7900HT System (Applied Biosystems) with SYBR Green PCR Master Mix (Applied Biosystems) according to manufacturer's instructions. In order to prevent PCR carry-over contamination, Uracil N-glycosylase was used (Amperase UNG, Applied Biosystems). Each assay was performed in triplicate and results were normalized and analyzed with SDS2.1 software (Applied Biosystems).
DNA Sequencing
PCR products and cloned fragments were double-strand sequenced with BigDye Terminator sequencing chemistry (Applied Biosystems) and analyzed on an ABI 3130xL automatic DNA sequencer (Applied Biosystems).
Results
Mutation Screening
To select a candidate gene in the critical region of the FGS4 locus covering a 4.4 Mb region at Xp11.4-p11.3 between the markers DXS8113 and sWXD805,12 all known genes in this interval were systematically scanned for mutations with a new and specifically defined DHPLC-based approach that we named comparative mutation scanning (CMS) (for a complete description see Material and Methods). The FGS4 locus region contains at least 11 known genes (UCSC Human Genome Server, freeze March 2006) (Table 1).
CMS allows the rapid comparison of each sequence variation within all family members under study and a pool of normal controls, providing clear indications as to the variation type and distinguishing a unique variation from common polymorphisms without the necessity of extensive and very expensive direct sequencing of each variation observed.
Two genes (NIX and MAOA) were preliminarily excluded from the screening because the former is involved in complete congenital stationary night blindness (CSNB1 [MIM 310500]) and is expressed only in retina and kidney,36 and the latter (MAOA [MIM 309850]) is on the centromeric boundary of the disease interval, split by the marker sWXD805. Interestingly, MED14 gene maps at FGS4 locus37 and MED12, a member of Mediator complex, was previously reported to be mutated in Opitz-Kaveggia and Lujan-Fryns syndromes.16,17 Although MED14 escapes X-inactivation,37 it might represent a good candidate gene.
A total of 167 amplicons were analyzed by CMS. Polymorphic variants were identified in some of the screened genes, but these SNPs were not useful in narrowing the linkage interval.
We identified the p.R28L (c.83G→T) missense mutation in exon 2 of CASK (Figure 1). CASK encodes a 104 KDa calcium/calmodulin-dependent serine protein kinase, preferentially expressed in neuronal cells and involved in signal transduction at the synapses.
Figure 1.
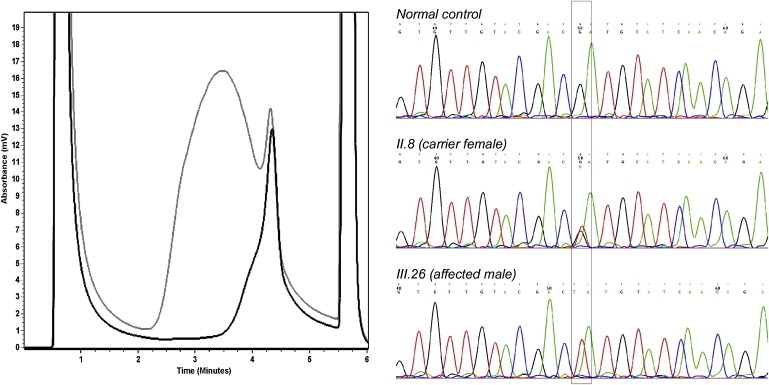
DHPLC Analysis and Characterization of the c.83G→T Mutation Identified in CASK
In the chromatogram on the left, the elution profile of a normal control (black curve) is compared with the mutant profile (gray curve). On the right, electropherograms from a normal control male, a carrier female (II.8), and an affected male (III.26) from pedigree12 are shown. The nucleotide change is boxed.
To verify that c.83G→T (p.R28L) mutation was present only in the three affected males and two carrier females, CASK exon 2 PCR products amplified in DNA samples from 22 family members were digested with Hpy99I. The variation fully cosegregated with the phenotype (Figure 2). In addition, it was absent in 1000 control X chromosomes of the same ethnic origin.
Figure 2.
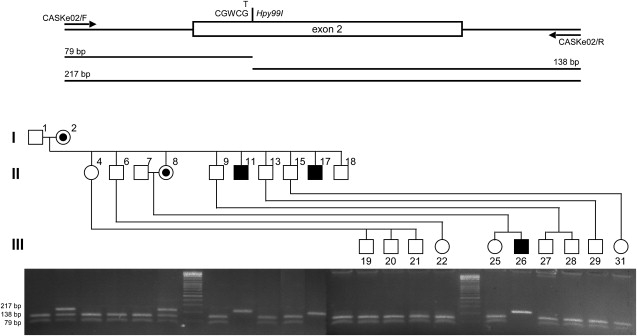
The c.83G→T Mutation Fully Cosegregates with the Phenotype in Our FGS Family
CASK exon 2 and intronic flanking regions were amplified by PCR in DNA samples from all the individuals in the pedigree and digested by Hpy99I. This restriction site is lost when the c.83G→T substitution is present. Agarose gel electrophoresis of Hpy99I-digested PCR products clearly indicates that the c.83G→T mutation is present only in the affected males (hemizygotes; 217-bp-long band) and carrier females (heterozygotes; 79-, 138-, and 217-bp-long bands) and is absent in all the other individuals from pedigree (79- and 138-bp-long bands).
By aligning the CaM-kinase domain from different hortologs of CASK, we observed that the c.83G→T transversion changes a basic and positively charged, highly conserved arginine residue at 28 position to a hydrophobic leucine residue (Figure 3).
Figure 3.
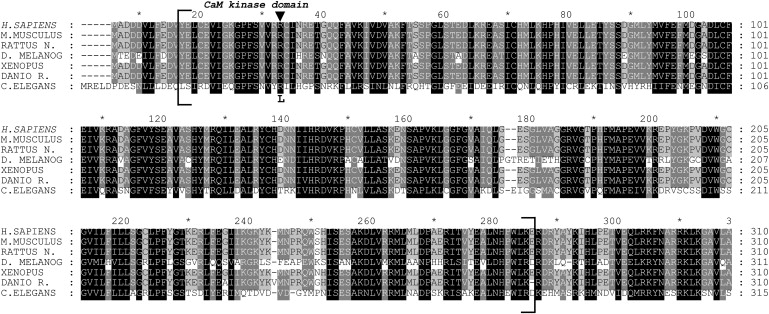
Alignment of CaM-Kinase Domain from Different Hortologs of CASK
The protein sequences of CASK from H. sapiens (NP_003679), M. musculus (NP_033936), R. norvegicus (NP_071520), D. melanogaster (NP_524441), X. tropicalis (NP_001016204), D. rerio (NP_694420), and C. elegans (NP_001024587) were aligned with Clustal W software.70 Amino acid conservation was highlighted with GeneDoc software. Only the N terminus region is shown and CaM-kinase domain of CASK is enclosed in square brackets. An arrow indicates the position of p.R28L substitution.
Taken together, this experimental evidence strongly suggested that the indentified missense mutation in CASK causes FGS in this family.
Patient Recruitment and Analysis
To test the involvement of CASK in FGS or XLMR phenotypes, a large cohort of FGS/XLMR patients was recruited worldwide. One hundred and one familial and sporadic FGS/XLMR patients were collected and analyzed for mutation in CASK by direct sequencing of all the 27 exons covering the coding sequence and of the upstream promoter region. No mutations were identified. This indicates that mutations in CASK represent a rare, if not a private, cause of FGS.
Functional Analysis of CASK Protein
Together with Velis and Mint-1 or Caskin 1, CASK forms two alternative tripartite protein complexes on the inner surface of the presynaptic membrane of the neurons.38–41 The N-terminal CaM-kinase domain of CASK participates in an evolutionarily conserved complex with Mint-1, also present in C. elegans, and in a mammalian-specific complex with Caskin 1.
By in vivo and in vitro protein-protein binding assays, we first evaluated whether the p.R28L substitution in CaM-kinase domain of CASK may interfere with the described interactions. To detect protein-protein binding in vivo, we used the yeast two-hybrid system. The CaM-kinase domain of CASK (residues 5–334) and the interacting domains of Mint-1 (residues 116–445) and Caskin 1 (residues 344–498) were expressed as fusion proteins with the DNA-binding domain and the transcription activation domain of GAL4, respectively. The p.R28L substitution was introduced by direct mutagenesis.
Protein-protein binding was then detected by selecting positive clones on minimal Trp−, Leu−, His−, and Ade− plates, as a result of the activation of the HIS3 and ADE2 reporter genes. The CaM-kinase domain of CASK interacts with the specific domains of Mint-1 and Caskin 1 in yeast, but these bindings were not modified by the p.R28L mutation identified in our patients (Figure 4A). The same result was also obtained by performing in vitro GST pull-down assays (Figure 4B).
Figure 4.
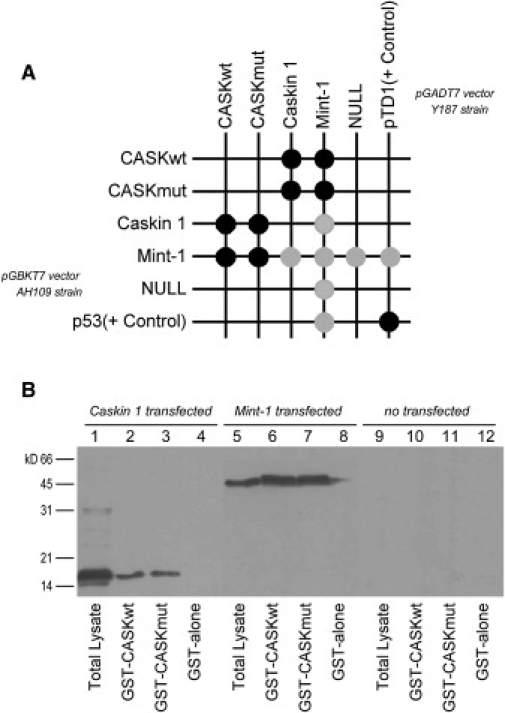
In Vivo and In Vitro Binding Assays of CaM-Kinase Domain of CASK with the Interacting Domains of Mint-1 and Caskin 1
(A) By yeast two-hybrid system, the CaM-kinase domain of CASK interacted with these partners and the p.R28L substitution did not modify binding properties. In the grid, strong and weak growth is indicated with black and gray disks, respectively. A mild autoactivation of reporter genes was observed for Mint-1. The p53 plasmid encoding murine p53 fused to GAL4 DNA-binding domain and the pTD1 plasmid encoding SV40 large T-antigen fused to GAL4 activating domain were used as positive control. pGBKT7 and pGADT7 empty plasmids were used as negative control.
(B) For GST pull-down assays, the interacting domains of Caskin 1 and Mint-1, N-terminal labeled with c-Myc epitope tag, were transiently expressed in COS-7 cells. Protein extracts from Caskin 1-transfected cells (lanes 1–4), Mint-1-transfected cells (lanes 5–8), and untransfected cells (lanes 9–12) were incubated with GST bead-bound fusion proteins: GST-CASKwt, GST-CASKmut, and GST alone. Western blotting analysis with an anti-Myc monoclonal antibody did not reveal differences in binding properties of CaM-kinase domain of CASK as a result of p.R28L substitution.
The multidomain protein Carom was also reported to bind the CaM-kinase domain of CASK in epithelial cell junctions.42 This protein is also expressed in brain. The interaction with the CaM-kinase domain of CASK was not shown to be modified by p.R28L mutation in an in vivo protein-protein binding assay (data not shown).
To verify whether as-yet-uncharacterized CASK interactions may be affected by the p.R28L missense mutation in the CaM-kinase domain, we also utilized the yeast two-hybrid system to screen a human brain cDNA library using as bait only the CaM-kinase domain of CASK (wild-type and mutant). Although novel putative interactors for the CaM-kinase domain of CASK were identified (unpublished data), these interactions were not specifically modified when using the mutant CaM-kinase domain of CASK as bait. Altogether, these results suggest that the observed phenotype is not determined by the alteration of the gross binding property of CaM-kinase domain as a consequence of the p.R28L substitution in CASK protein.
Phosphorylation regulates CASK activity. The Cdk5-dependent phosphorylation of CASK dynamically controls its localization to the synapses and the interaction between CASK and liprin-α in developing synapses, promoting synaptogenesis and neurotransmitter vesicle trafficking.43,44 More recently, crystallographic studies revealed that the CaM-kinase domain of CASK is a constitutively active kinase domain, able to phosphorylate itself and at least the neurexin-1 CASK-protein partner.29
Interestingly, the p.R28L substitution identified in FGS patients is located very close to the nucleotide-binding pocket of the CaM-kinase domain. By in vitro autophosphorylation assay, we tested whether the p.R28L mutation could interfere with ATP binding. We compared the autophosphorylation of the CaM-kinase domain (wild-type and mutant), purified as GST-fusion proteins, with the hypomorphic mutant of CaM-kinase domain reported by Mukherjee et al.29 in which the serine at position 24 and the valine at position 26, both involved in ATP binding, were mutated to aspartic acid and leucine, respectively. The p.R28L mutation only weakly reduced the autophosphorylation levels of CASK CaM-kinase domain, which did not have statistical significance (Figure 5).
Figure 5.
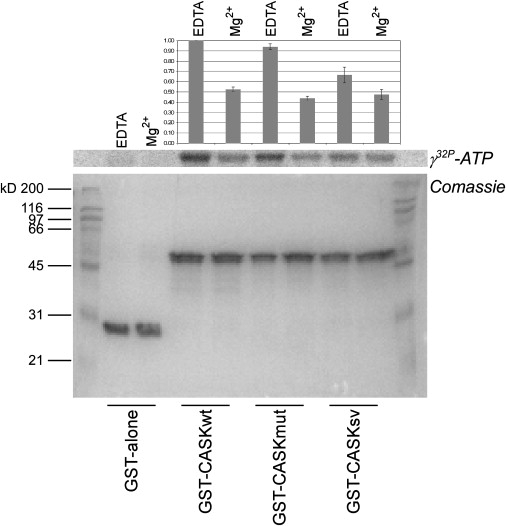
In Vitro Autophosphorylation Assay of CASK CaM-Kinase Domain
All the experiments were carried out as described.29 Equimolar quantities of purified GST bead-bound fusion proteins (GST alone, GST-CASKwt, GST-CASKmut, and GST-CASKsv) were incubated with [γ-32P]ATP (specific activity: 2 × 107 cpm) in Tris-KCl buffer under two different conditions: 2 mM EDTA or 4 mM Mg2+. The panel (from bottom) depicts Coomassie blue stain of SDS gel and related autoradiogram. Normalized values of incorporated radioactivity were reported in the upper bar graph where error bars indicate ±SD. When compared with the wild-type (CASKwt) and hypomorphic mutant (CASKsv) of CASK CaM-kinase domain, p.R28L mutation (CASKmut) did not seem to generate significant variations in autophosphorylation of CaM-kinase domain.
Molecular Dynamics Simulations
In order to investigate potential modifications in the protein structure and dynamics induced by p.R28L substitution, we performed molecular dynamics (MD) simulation both on wild-type and mutant CaM-kinase domain of CASK starting from crystallographic coordinates (PDB: 3c0h).29 Trajectories of 2.5 ns were sufficient to obtain stable conformations for both wild-type and mutant structures (see Material and Methods and Figure 6A). By comparing the structural features of the two MD-simulated proteins, they appeared as α/β structures highly similar in the secondary structure as well as in the spatial organization (Figure 6B). Small differences in the secondary structure were localized only in the segment 51-54 (yellow colored in Figure 6B). These four residues were coiled in the wild-type while they were in a turn in the p.R28L mutant protein. In the CASK CaM-kinase domain structures containing AMPPNP, the 5′AMP portion of the AMPPNP ligand showed an almost superimposable conformation for wild-type and mutant (Figure 6C). The environment of the ligand-binding site appears only slightly different by comparing wild-type versus mutant CaM-kinase domain of CASK (Figure 6D) and the hydrogen bonding network is substantially similar (Figure 6D and Table S4) to that previously reported.29 Only few hydrogen bonds are lacking in the p.R28L mutant CaM-kinase domain of CASK. Consequently, persistence time of the ligand in the correct geometry to perform its catalytic action could be slightly decreased. This observation may justify the small reduction experimentally observed in the autophosphorylation assay.
Figure 6.
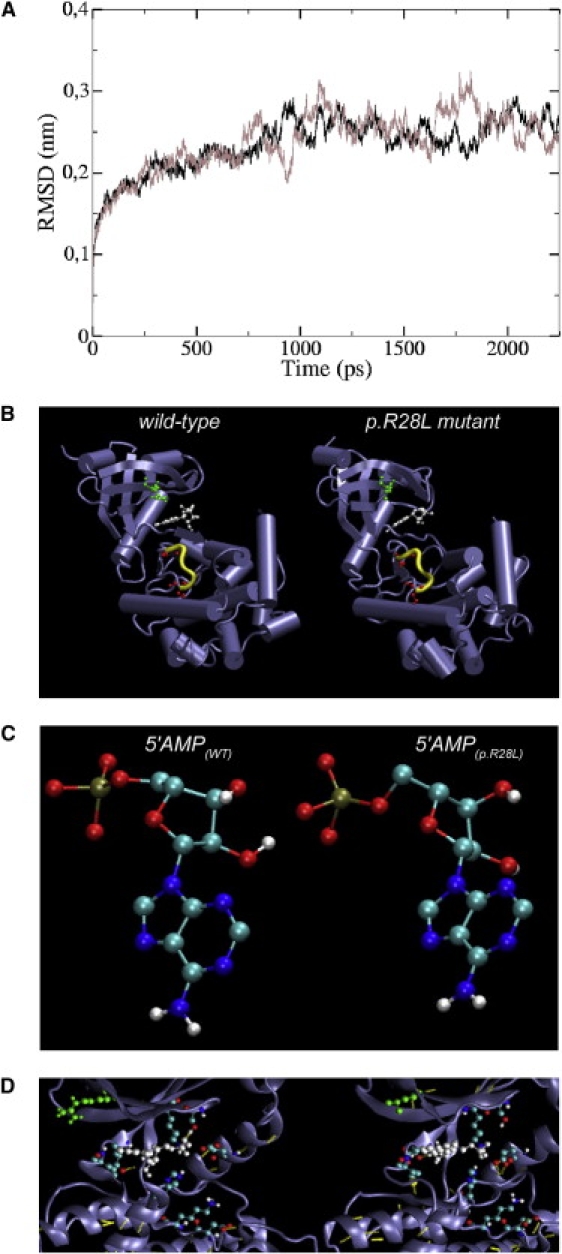
Molecular Dynamics Simulation
(A) Structural drift of wild-type (black line) and p.R28L mutant CaM-kinase domain of CASK (gray line) is shown as root mean square deviation of all atoms starting from crystallographic coordinates after energy minimization.
(B) Comparison between 3D structures of the wild-type (left) and p.R28L mutant (right) CaM-kinase domain of CASK obtained from the 2.5 ns MD simulations at 300 K. The residue at position 28, in which the mutation occurs, was colored in green, the AMPPNP ligand was colored in white, and the serine residues at position 147 and 151, targets of autophosphorylation activity,29 were colored in red. The residues 51–54 that showed small differences in secondary structure were highlighted in yellow.
(C) The conformation adopted by the 5′AMP portion of the AMPPNP ligand is shown for wild-type (left) and p.R28L mutant (right).
(D) A detailed view of the nucleotide-binding pocket of CASK CaM-kinase domain in complex with AMPPNP is shown. The color scheme used in (B) is conserved. In addition, side chains of residues involved in AMPPNP binding and H bonds (yellow dots) are shown.
To exclude that the p.R28L substitution in the CaM-kinase domain of CASK could cause some structural hindrance to ligand access, we also performed MD simulations in the absence of the ligand. The spatial organization of the wild-type and mutant proteins were very similar (e.g., the activation segment29), excluding the presence of marked structural and/or energetic barriers in the mutant CASK protein (data not shown).
Functional Analysis of CASK Transcript
Point mutations in the coding regions of genes are commonly assumed to exert their effects by altering single amino acids in the encoded proteins. However, nonsense, missense, and even translationally silent mutations can inactivate genes by inducing the splicing machinery to skip the mutant exons.45
To evaluate whether the c.83G→T (p.R28L) missense mutation found in exon 2 of CASK may modify the consensus sequence of exonic splicing enhancers (ESEs) interfering with the correct splicing of the exon, the wild-type and mutant sequence of CASK exon 2 were scanned with ESEfinder, a software tool used to identify putative ESEs responsive to the human SR proteins SF2/ASF, SC35, SRp40, and SRp55.46 A cluster of five overlapping ESE motifs was clearly identified in the 5′ half of the wild-type sequence of CASK exon 2, including two SF2/ASF motifs, two SRp55 motifs, and one SRp40 motif. With the mutant sequence, the same cluster lost one SF2/ASF motif and acquired a third SRp55 motif, overlapping the others and presenting a higher score (Figure 7).
Figure 7.
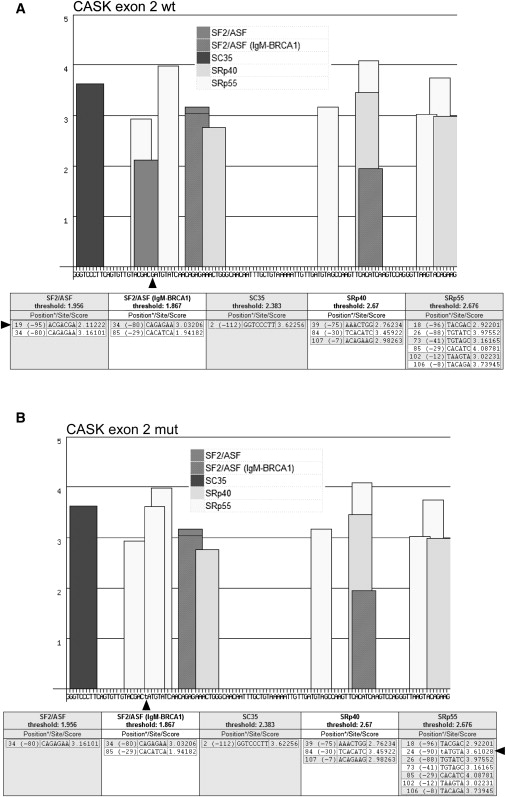
Analysis of the 113-bp-Long Coding Sequence of CASK Exon 2 via ESEfinder Software
As suggested by the authors,46 default threshold values were used. The bar graphs and tables below depict the ESE motifs for different SR proteins (SF2/ASF, SC35, SRp40, and SRp55) found in wild-type (A) and c.83G→T mutant (B) CASK exon 2 sequence. In the tables, both positions (∗) from 5′ end (through 1) and 3′ end (through −1) are given. The arrows indicate the position of nucleotide substitution and ESE motif lost or acquired as a result of the change.
To verify in a natural context whether the identified missense mutation may affect an ESE function by altering the splicing of exon 2 of CASK, we used EBV-transformed lymphocyte cell lines from affected, carrier, and normal individuals from the FGS family to test by RT-PCR any putative effect on the splicing in vivo.
CASK is normally expressed in lymphocytes. With a specific primer pair (CASK16/F and CASK268/R) covering a 253-bp-long cDNA region that includes exons 1–3 (Table S3 and Figure 8A), a 140-bp-long exon 2-skipped transcript was detected, at a low level, in the proband (III.26), in the two affected maternal uncles (II.11 and II.17, data not shown), and in the carrier grandmother (I.2), but was undetectable in the carrier mother (II.8) as well as in normal individuals from pedigree and controls (Figure 8A). To exclude the possibility that the exon 2-skipped PCR product could be the expression of incomplete and nonfunctional splicing products, we combined the primer in exon 1 (CASK16/F) with a downstream primer in exon 8 (CASK744/R) to amplify a 729-bp-long cDNA CASK fragment (Table S3 and Figure 8B). A specific 616-bp-long exon 2-skipped PCR product was likewise observed, at low level, in the same samples (Figure 8B).
Figure 8.
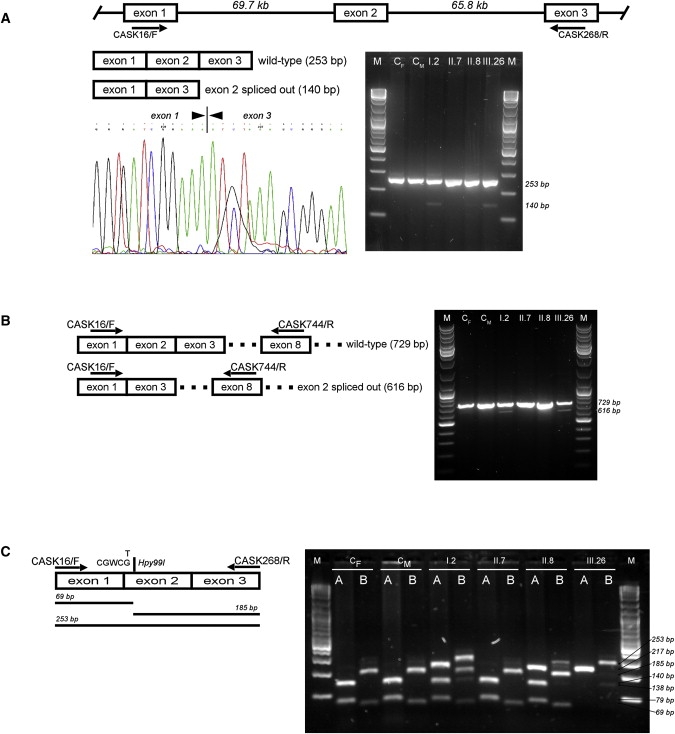
RT-PCR Analysis of CASK Transcripts
(A) The CASK cDNA fragment covering exons 1–3 was amplified by RT-PCR. The expected 253-bp-long PCR product was observed in all samples analyzed by agarose gel electrophoresis (right). cDNA samples are indicated with respect to their position on pedigree (Figure 2). CM and CF indicate control male or female, respectively. A 140-bp-long PCR product, corresponding to the CASK exon 2-skipped form, was observed, at low levels, only in the carrier female I.2 and the affected male III.26. The same result was also obtained with the other affected males (II.11 and II.17) from pedigree (data not shown). The band was undetectable in the other carrier female (II.8), mother of III.26. The skipping of CASK exon 2 was confirmed by direct sequencing of 140-bp-long PCR fragment (left).
(B) Different primer pairs were used to confirm the CASK exon-2 skipped transcript in the same sample panel. The 616-bp-long PCR product corresponding to the CASK exon-2 skipped transcript form was observed only in the carrier female I.2 and the affected male III.26.
(C) Hpy99I digestion of CASK exon 2 and its intronic flanking regions, as previously depicted in Figure 2, is combined with the Hpy99I digestion of CASK cDNA fragment covering exons 1–3. For the same sample panel, agarose gel electrophoresis of Hpy99I-digested genomic (A) and cDNA (B) PCR products are shown. For carrier females (I.2 and II.8), heterozygosity is confirmed (lanes A), but cDNA PCR products (lanes B) present a different digestion pattern in which 253- and 185-bp-long fragments show inverted intensity of the bands. This indicates that the wild-type allele is preferentially used by II.8 in CASK transcription.
Because no skewed X-inactivation was previously observed for carrier females of pedigree [I.2 (53:47) and II.8 (55:45)],12 we investigated whether both alleles contributed to the CASK transcription in carrier females. The genomic region of CASK exon 2 and the 253-bp-long cDNA fragment covering exons 1–3 of CASK transcript were amplified in the same sample panel and digested with Hpy99I (Figure 8C). Interestingly, we observed that the mother of proband (II.8) preferentially utilized the wild-type allele for CASK transcription, justifying the fact that CASK exon 2-skipped transcript was previously undetectable.
To better quantify the exon 2-skipped CASK transcript and the expression level of CASK, two additional primers were designed. The first overlapped exon 1-exon 2 junction (CASKsp1-2/F) and was able to amplify exclusively the normal CASK transcript when combined with a downstream primer in exon 3 (CASK268/R). The second overlapped exon 1-exon 3 junction (CASKsp1-3/F) and was able to amplify specifically the exon 2-skipped CASK transcript when combined with a downstream primer in exon 5 (CASK398/R). A third primer pair covering a region between exons 25-26 (CASK2310/F and CASK2545/R) was used to detect the total quantity of CASK transcripts (Table S3).
By real-time RT-PCR, we confirmed that the exon 2-skipped transcript was differently expressed in affected males and carrier females but was not detectable in either unaffected individuals from the pedigree or in controls (Figure 9A). In fact, the CT value for the CASK exon 2-skipped specific PCR product was always >30 for normal individuals from pedigree and controls but was significantly reduced in affected males and carrier females (Figure 9B). After data normalization, the CASK exon 2-skipped transcripts were estimated to be approximately 3%–6% of the unskipped CASK transcript (Figure 9C). The exon 2-skipped transcript is out-of-frame with a possible effect on CASK protein levels. Alternatively, by using the first methionine downstream of the skipped exon 2, a truncated protein without the first 67 amino acids could be produced. This methionine does not conform to Kozak consensus, and neither qualitative nor quantitative differences were observed by western blotting analysis on protein extracts from lymphocyte cell lines from affected, carrier, and normal individuals (data not shown). However, we cannot exclude that this aberrant CASK transcript may be increased in different cell types or brain regions during embryogenesis, brain development, and differentiation. Interestingly, Sfrs1 and Sfrs6 genes encoding SF2/ASF and SRp55, respectively, show a different expression pathway in adult mouse brain with a similar low expression of Sfrs1 gene in different brain regions compared with the very high expression of Sfrs6 gene in cerebral cortex, hippocampal regions, olfactory bulb, and striatum-like amigdalar nuclei (Allen Institute for Brain Science, Mouse Brain Atlas). Temporally and spatially differentiated expression profiles have also been annotated for both genes in mouse embryo at different stages of CNS development (Eurexpress II; MouseBrain Gene Expression Map [BGEM]).
Figure 9.
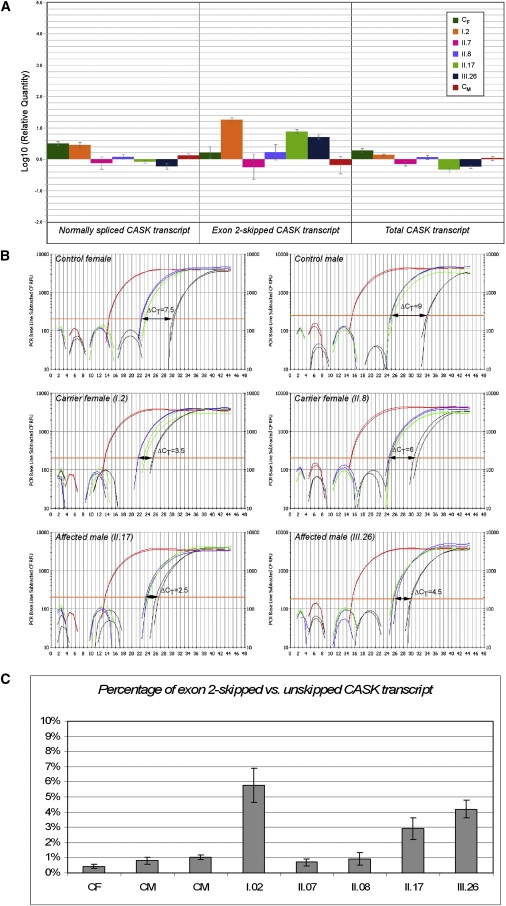
Relative Quantification of CASK Expression
The analyzed cDNA samples are indicated with respect to their position on pedigree (Figure 2). CM and CF indicate control male or female, respectively.
(A) The bar graph shows relative quantification of the expression of different CASK transcripts. The expression of the exon 2-skipped transcript (center), amplified with CASKsp1-2/F and CASK268/R primer pair, is significantly increased in I.2 (carrier female) as well as in II.17 and III.26 (affected males). For II.8 (carrier female), this aberrant transcript is undetectable, even considering the wide interval of error bar ±MSD). Expression levels of CASK (right) detected with the CASK2310/F and CASK2545/R primer pair are reduced in the affected males (II.17 and III.26). The expression of the unskipped CASK transcript (left), specifically amplified with CASKsp1-3/F and CASK398/R primer pair, is increased in carrier female II.8.
(B) Amplification plots describing the kinetics of PCR reactions for different targets and samples are presented. Differently colored curves correspond to GAPDH (red), unskipped CASK transcript (blue), CASK exon 2-skipped transcript (black), and total CASK transcript (green). The ΔCT obtained by subtracting the CT values of the unskipped and exon 2-skipped CASK forms is shown.
(C) After normalization, expression levels of the unskipped CASK transcript were compared to those of the exon 2-skipped form. For each sample, the bar graph shows the percentage accounting for the CASK exon 2-skipped transcript ranging between 3% and 6% in affected males (II.17 and III.26) and carrier female (I.2).
Discussion
Since the first description in 1974,1 an increasing number of familial and sporadic cases of FG syndrome have been diagnosed worldwide, expanding the phenotype spectrum of this X-linked MCA/MR disorder.
Recently, missense mutations in the MED12 gene at Xq13 were shown to be responsible for FG/Opitz-Kaveggia syndrome and Lujan-Fryns syndrome,16,17 most likely representing allelic X-linked disorders. Three other genes, UPF3B, BRWD3, and FLNA, have also been found to be mutated in patients with FG phenotype.18–21 However, despite recent progress, the molecular defect in a large part of FGS patients remains elusive.
Here, we identify CASK as a novel FGS gene mutated in a previously reported Italian FGS family linked to Xp11.4-p11.3.12
CASK is a member of the membrane-associated guanylate kinase (MAGUK) family. MAGUKs are characterized by at least three canonical domains that from the N terminus include PDZ, SH3, and guanylate kinase (GK) domains. CASK is the only MAGUK that contains an additional large N-terminal domain with homology to calcium/calmodulin-dependent protein kinase II (CaM-kinase domain).22,47 In brain, CASK localizes to synapses where it interacts with a large array of protein partners with which it forms large signaling complexes regulating trafficking, targeting, and signaling of ion channels in neurotransmission.47–49 In addition, CASK can also translocate to the nucleus where it interacts with the transcription factor TBR1 regulating the expression of genes involved in cortical development such as RELN.50–52
Different animal models clearly suggest that CASK plays an important role in embryogenesis, brain development, and differentiation, representing a candidate gene for syndromic as well as nonsyndromic X-linked mental retardation.53–57
In C. elegans, the CASK hortologous Lin-2 is essential for vulva development,54 whereas in D. melanogaster, Caki null mutants show behavioral and neurotransmitter-release abnormalities.56,57 In mammals, Cask knockout mice die on day 1 after birth and do not show major developmental abnormalities apart from a partially penetrant cleft palate syndrome, suggesting that Cask is required for mouse survival.53
We characterized a p.R28L (c.83G→T) missense mutation in exon 2 of CASK segregating with the FGS phenotype in the family under study (Figures 1 and 2), which alters a highly conserved amino acid in the CaM-kinase domain unique to CASK (Figure 3). Usually, an altered protein function should be expected as a result of a missense mutation. An extensive analysis of the CASK protein was carried out, but no apparent interaction effects caused by the p.R28L substitution were demonstrated (Figure 4). In addition, no marked structural effects caused by the p.R28L substitution were observed after an extensive analysis of the trajectories obtained by MD simulations performed on the CASK CaM-kinase domain (wild-type and mutant) (Figure 6). We cannot exclude that this allele is selective in its effects and cannot bind to as-yet-unknown CASK partners. Neither can we exclude modifications in other properties. Conversely, we identified an aberrant out-of-frame CASK transcript in which exon 2 is skipped, most probably as a consequence of an altered recognition of ESE motifs by SR proteins such as SF2/ASF or SRp55 (Figures 7 and 8). In FGS4 lymphoblastoid cell lines, we detected exon 2 skipping in approximately 3%–6% of total CASK transcripts by quantitative RT-PCR (Figure 9). Although we were unable to investigate this effect in other patient tissues, we speculate a dosage-dependent pathogenetic effect related to the temporally and spatially specific expression profiles of SR proteins during embryogenesis.
A large set of RNA-binding proteins, such as SR proteins, show a regionally restricted expression profile in developing mouse brain.58 ESE motifs are clustered and significantly enriched within exons compared with introns or pseudo exons. In addition, differences in ESE motif frequency distribution have been reported between exons with weak and strong splice sites for both 5′ and 3′ splice sites.59 It is intriguing to observe that exons with weak 5′ splice sites and exons with weak 3′ splice sites were shown to have more SRp55 and SF2/ASF motifs, respectively.59 The enrichment in SRp55-specific ESE consensus in the 5′ half of CASK exon 2 resulting from the identified c.83G→T transversion could weaken the 5′ splice site of the exon, possibly justifying the partially penetrant splice mutation in our FGS patients. A silent p.D107D (c.321C→T) mutation in the ATP6AP2 (MIM 300556) gene that similarly produced an inefficient inclusion of the exon 4 by altering the recognition of an ESE motif was previously reported in patients with XLMR and epilepsy.60
The selection between different splice sites is the result of interactions involving the cooperative binding of trans-acting splicing proteins to cis-acting sequence elements in the pre-mRNA, and the regulation engages multiple positively and negatively acting factors with precise qualitative and quantitative ratios.61,62 Brain has the highest proportion (>40%) of alternative spliced genes that can influence neurophysiology through spatial and temporal alterations in the expression of different classes of proteins.61,63,64 Different types of neurological diseases have been associated with defects in splicing.63
Altogether, our results suggest that the phenotype observed in the FGS family under study should be considered a cis-acting splicing disorder interfering in the normal CASK expression profile during embryogenesis, brain development, and differentiation.
A dosage-dependent CASK involvement in X-linked mental retardation has previously been reported. By X chromosome-specific CGH array, Froyen et al.65 indentified a 15.5 Mb duplication in a male and a 3.2 Mb deletion in a female, both involving CASK. Interestingly, the distal break-point for the female was located within CASK, suggesting that CASK haploinsufficiency might cause the condition. Subsequently, another 4 Mb deletion encompassing CASK was also reported in a Japanese female, showing marked mental retardation with microcephaly, dysmorphic features, and cerebellar atrophy.66 By quantitative RT-PCR analysis, CASK expression was shown to be significantly reduced in this patient compared with controls.
More recently, a novel X-linked brain malformation syndrome caused by mutations in CASK has been reported, preferentially affecting females and characterized by severe mental retardation, microcephaly, disproportionate pontine, and cerebellar hypoplasia.67 Heterozygous loss-of-function mutations of CASK were identified in four females, whereas a partially penetrant splice mutation was present in the only affected male presenting a more severe phenotype, who died at 2 weeks. Interestingly, the reported p.K305K (c.915G→A) silent mutation in the affected male gave rise to the skipping of the exon 9 in approximately 20% of transcripts in in vitro splicing assay.67
Unlike the affected females described by Najm et al.67 in which de novo mutations inactivated one copy of CASK determining haploinsufficiency, females from this FGS family carrying the p.R28L missense CASK mutation were healthy with only very mitigated clinical signs in the proband's mother (II.8).12 In females carrying mutations in X-linked genes, phenotypic variability can be modulated by dosage compensation as observed in X-linked disorders characterized by male lethality.68,69 Females are mosaics of two cell populations with respect to X-linked gene expression. The effect of the p.R28L mutant CASK allele in the carrier females is most probably balanced by the X chromosome inactivation. This is in agreement with the observation that the two carrier females of the FGS family under study differently utilized the wild-type and mutant CASK allele for transcription (Figure 8C).
Our results and literature data suggest that mildly hypomorphic alleles could be a model for CASK mutations in males, albeit representing very rare occurrences. CASK is part of a complex and growing protein-protein network with important functions in brain but also in other tissues. Therefore, missense mutations altering CASK protein functions should be expected in males, presumably associated with clinical variability, but with severe mental retardation as a common trait.
Compared to the affected individuals described by Najm et al.,67 our FGS patients shared only a few clinical signs (Table S5).12 Severe mental retardation, a similar dysmorphic appearance, and a mildly abnormal EEG were present in our surviving affected males. The main differences included macrocephaly versus microcephaly and the absence of evident anatomical abnormality in brain MRI, with the exception of a persistent cavum septum/vergae.
The differences in the observed phenotypes are probably related to the complex and as yet incompletely clarified role of CASK in embryogenesis and CNS development.
CASK must have a key role in regulating mammalian development, given its high level of expression during embryogenesis.23,56 The genetic defects that we and others identified in CASK further highlight this point.65–67 In the embryonic period, approximately 20% of CASK proteins are present in the nuclei of neurons in cerebral cortex and other brain regions.50 When associated with TBR1 and CINAP, CASK regulates the expression of different genes such as RELN and NR2b, most likely playing an important role in cerebral cortex development.51,52 Likewise, the growing protein-protein network established by the cytoplasmic fraction of the multidomain CASK protein seems to be important in synaptic interaction and formation, protein trafficking, and synaptic targeting.47 All these functions are most probably modulated by CASK phosphorylation in a manner that needs to be further elucidated.29,44
Supplemental Data
Supplemental Data include five tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Allen Institute for Brain Science, Mouse Brain Atlas, http://www.brain-map.org/
ClustalW software, http://www.ebi.ac.uk/Tools/clustalw2/index.html
Entrez Nucleotide database at NCBI, http://www.ncbi.nlm.nih.gov/sites/entrez?db = nuccore
ESEfinder software, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process = home
Eurexpress, a Transcriptome Atlas Database for Mouse Embryo, http://www.eurexpress.org/
GeneDoc software, http://www.nrbsc.org/
GROMACS software version 3.2, http://www.gromacs.org/
MouseBrain Gene Expression Map (BGEM), http://www.stjudebgem.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/entrez?db = omim
PDB Protein DataBank, http://www.pdb.org/pdb/home/home.do
Primer3, http://primer3.sourceforge.net
UCSC Genome Browser, http://genome.ucsc.edu/
Visual Molecular Dynamics (VMD) package, http://www.ks.uiuc.edu/
Acknowledgments
The authors are grateful to the family members for their participation and help. We thank Michele D'Avanzo who first clinically evaluated the family. We are indebted to Agatino Battaglia, Hans van Bokhoven and Euro-MRX consortium, Sylvain Briault, Livia Garavelli, Alfredo Orrico, and Maria M. Rinaldi for their assistance in providing us with DNA samples. We also acknowledge Anna Cuomo for sequencing, Manuela Dionisi at the Telethon-SUN core facility of Mutation Detection, and Catherine Fischer for the English language revision. The authors have no disclosures and no conflicts of interest. C.E.S. was supported, in part, by a grant from NICHD (HD 26202) and a grant from the South Carolina Department of Disabilities and Special Needs (SCDDSN). V.N. was supported by grants from Telethon (TIGEM-TNP42TELC), Ministero dell'Istruzione dell'Università e della Ricerca (MIUR: PRIN 2006), Ministero della Salute (d.lgs 502/92), and Ricerca d'Ateneo. This paper is dedicated to the memory of Francesco Piluso, 1931–2005.
References
- 1.Opitz J.M., Kaveggia E.G. Studies of malformation syndromes of man 33: the FG syndrome. An X- linked recessive syndrome of multiple congenital anomalies and mental retardation. Z. Kinderheilkd. 1974;117:1–18. doi: 10.1007/BF00439020. [DOI] [PubMed] [Google Scholar]
- 2.Opitz J.M., Richieri-da Costa A., Aase J.M., Benke P.J. FG syndrome update 1988: note of 5 new patients and bibliography. Am. J. Med. Genet. 1988;30:309–328. doi: 10.1002/ajmg.1320300132. [DOI] [PubMed] [Google Scholar]
- 3.Romano C., Baraitser M., Thompson E. A clinical follow-up of British patients with FG syndrome. Clin. Dysmorphol. 1994;3:104–114. [PubMed] [Google Scholar]
- 4.Thompson E.M., Baraitser M., Lindenbaum R.H., Zaidi Z.H., Kroll J.S. The FG syndrome: 7 new cases. Clin. Genet. 1985;27:582–594. doi: 10.1111/j.1399-0004.1985.tb02043.x. [DOI] [PubMed] [Google Scholar]
- 5.Neri G., Blumberg B., Miles P.V., Opitz J.M. Sensorineural deafness in the FG syndrome: report on four new cases. Am. J. Med. Genet. 1984;19:369–377. doi: 10.1002/ajmg.1320190219. [DOI] [PubMed] [Google Scholar]
- 6.Graham J.M., Tackels D., Dibbern K., Superneau D., Rogers C., Corning K., Schwartz C.E. FG syndrome: report of three new families with linkage to Xq12-q22.1. Am. J. Med. Genet. 1998;80:145–156. [PubMed] [Google Scholar]
- 7.Graham J.M., Superneau D., Rogers R.C., Corning K., Schwartz C.E., Dykens E.M. Clinical and behavioral characteristics in FG syndrome. Am. J. Med. Genet. 1999;85:470–475. [PubMed] [Google Scholar]
- 8.Battaglia A., Chines C., Carey J.C. The FG syndrome: report of a large Italian series. Am. J. Med. Genet. A. 2006;140:2075–2079. doi: 10.1002/ajmg.a.31302. [DOI] [PubMed] [Google Scholar]
- 9.Lyons M.J., Graham J.M., Neri G., Hunter A.G., Clark R.D., Rogers R.C., Moscarda M., Boccuto L., Simensen R., Dodd J. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J. Med. Genet. 2008;46:9–13. doi: 10.1136/jmg.2008.060509. [DOI] [PubMed] [Google Scholar]
- 10.Dessay S., Moizard M.P., Gilardi J.L., Opitz J.M., Middleton-Price H., Pembrey M., Moraine C., Briault S. FG syndrome: linkage analysis in two families supporting a new gene localization at Xp22.3. Am. J. Med. Genet. 2002;112:6–11. doi: 10.1002/ajmg.10546. [DOI] [PubMed] [Google Scholar]
- 11.Briault S., Hill R., Shrimpton A., Zhu D., Till M., Ronce N., Margaritte-Jeannin P., Baraitser M., Middleton-Price H., Malcolm S. A gene for FG syndrome maps in the Xq12-q21.31 region. Am. J. Med. Genet. 1997;73:87–90. [PubMed] [Google Scholar]
- 12.Piluso G., Carella M., D'Avanzo M., Santinelli R., Carrano E.M., D'Avanzo A., D'Adamo A.P., Gasparini P., Nigro V. Genetic heterogeneity of FG syndrome: a fourth locus (FGS4) maps to Xp11.4-p11.3 in an Italian family. Hum. Genet. 2003;112:124–130. doi: 10.1007/s00439-002-0863-7. [DOI] [PubMed] [Google Scholar]
- 13.Briault S., Odent S., Lucas J., Le Merrer M., Turleau C., Munnich A., Moraine C. Paracentric inversion of the X chromosome [inv(X)(q12q28)] in familial FG syndrome. Am. J. Med. Genet. 1999;86:112–114. doi: 10.1002/(sici)1096-8628(19990910)86:2<112::aid-ajmg4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Briault S., Villard L., Rogner U., Coy J., Odent S., Lucas J., Passage E., Zhu D., Shrimpton A., Pembrey M. Mapping of X chromosome inversion breakpoints [inv(X)(q11q28)] associated with FG syndrome: a second FG locus? Am. J. Med. Genet. 2000;95:178–181. doi: 10.1002/1096-8628(20001113)95:2<178::aid-ajmg17>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Jehee F.S., Rosenberg C., Krepischi-Santos A.C., Kok F., Knijnenburg J., Froyen G., Vianna-Morgante A.M., Opitz J.M., Passos-Bueno M.R. An Xq22.3 duplication detected by comparative genomic hybridization microarray (Array-CGH) defines a new locus (FGS5) for FG syndrome. Am. J. Med. Genet. A. 2005;139:221–226. doi: 10.1002/ajmg.a.30991. [DOI] [PubMed] [Google Scholar]
- 16.Risheg H., Graham J.M., Clark R.D., Rogers R.C., Opitz J.M., Moeschler J.B., Peiffer A.P., May M., Joseph S.M., Jones J.R. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007;39:451–453. doi: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz C.E., Tarpey P.S., Lubs H.A., Verloes A., May M.M., Risheg H., Friez M.J., Futreal P.A., Edkins S., Teague J. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 2007;44:472–477. doi: 10.1136/jmg.2006.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unger S., Mainberger A., Spitz C., Bahr A., Zeschnigk C., Zabel B., Superti-Furga A., Morris-Rosendahl D.J. Filamin A mutation is one cause of FG syndrome. Am. J. Med. Genet. A. 2007;143:1876–1879. doi: 10.1002/ajmg.a.31751. [DOI] [PubMed] [Google Scholar]
- 19.Field M., Tarpey P.S., Smith R., Edkins S., O'Meara S., Stevens C., Tofts C., Teague J., Butler A., Dicks E. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am. J. Hum. Genet. 2007;81:367–374. doi: 10.1086/520677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarpey P.S., Raymond F.L., Nguyen L.S., Rodriguez J., Hackett A., Vandeleur L., Smith R., Shoubridge C., Edkins S., Stevens C. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Opitz J.M., Smith J.F., Santoro L. The FG syndromes (Online Mendelian Inheritance in Man 305450): perspective in 2008. Adv. Pediatr. 2008;55:123–170. doi: 10.1016/j.yapd.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 22.Hata Y., Butz S., Sudhof T.C. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevenson D., Laverty H.G., Wenwieser S., Douglas M., Wilson J.B. Mapping and expression analysis of the human CASK gene. Mamm. Genome. 2000;11:934–937. doi: 10.1007/s003350010170. [DOI] [PubMed] [Google Scholar]
- 24.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karolchik D., Kuhn R.M., Baertsch R., Barber G.P., Clawson H., Diekhans M., Giardine B., Harte R.A., Hinrichs A.S., Hsu F. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 27.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. John Wiley and Sons, Inc.; New York: 1994. Current Protocols in Molecular Biology, Volume 1. [Google Scholar]
- 28.Bolton B.J., Spurr N.K. B-lymphocytes. In: Freshney R.I., Freshney M.G., editors. Culture of Immortalized Cells. Wiley-Liss; New York: 1996. pp. 283–297. [Google Scholar]
- 29.Mukherjee K., Sharma M., Urlaub H., Bourenkov G.P., Jahn R., Sudhof T.C., Wahl M.C. CASK functions as a Mg2+-independent neurexin kinase. Cell. 2008;133:328–339. doi: 10.1016/j.cell.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piluso G., Mirabella M., Ricci E., Belsito A., Abbondanza C., Servidei S., Puca A.A., Tonali P., Puca G.A., Nigro V. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J. Biol. Chem. 2000;275:15851–15860. doi: 10.1074/jbc.M000439200. [DOI] [PubMed] [Google Scholar]
- 31.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 32.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Di Nola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 34.Luty B.A., Tironi I.G., van Gunsteren W.F. Lattice-sum methods for calculating electrostatic interactions in molecular simulations. J. Chem. Phys. 1995;103:3014–3021. [Google Scholar]
- 35.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 36.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 37.Yoshikawa H., Fujiyama A., Nakai K., Inazawa J., Matsubara K. Detection and isolation of a novel human gene located on Xp11.2-p11.4 that escapes X-inactivation using a two-dimensional DNA mapping method. Genomics. 1998;49:237–246. doi: 10.1006/geno.1998.5246. [DOI] [PubMed] [Google Scholar]
- 38.Butz S., Okamoto M., Sudhof T.C. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- 39.Borg J.P., Straight S.W., Kaech S.M., de Taddeo-Borg M., Kroon D.E., Karnak D., Turner R.S., Kim S.K., Margolis B. Identification of an evolutionarily conserved heterotrimeric protein complex involved in protein targeting. J. Biol. Chem. 1998;273:31633–31636. doi: 10.1074/jbc.273.48.31633. [DOI] [PubMed] [Google Scholar]
- 40.Borg J.P., Lopez-Figueroa M.O., de Taddeo-Borg M., Kroon D.E., Turner R.S., Watson S.J., Margolis B. Molecular analysis of the X11-mLin-2/CASK complex in brain. J. Neurosci. 1999;19:1307–1316. doi: 10.1523/JNEUROSCI.19-04-01307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabuchi K., Biederer T., Butz S., Sudhof T.C. CASK participates in alternative tripartite complexes in which Mint 1 competes for binding with caskin 1, a novel CASK-binding protein. J. Neurosci. 2002;22:4264–4273. doi: 10.1523/JNEUROSCI.22-11-04264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohno H., Hirabayashi S., Kansaku A., Yao I., Tajima M., Nishimura W., Ohnishi H., Mashima H., Fujita T., Omata M. Carom: a novel membrane-associated guanylate kinase-interacting protein with two SH3 domains. Oncogene. 2003;22:8422–8431. doi: 10.1038/sj.onc.1206996. [DOI] [PubMed] [Google Scholar]
- 43.Olsen O., Moore K.A., Fukata M., Kazuta T., Trinidad J.C., Kauer F.W., Streuli M., Misawa H., Burlingame A.L., Nicoll R.A. Neurotransmitter release regulated by a MALS-liprin-alpha presynaptic complex. J. Cell Biol. 2005;170:1127–1134. doi: 10.1083/jcb.200503011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samuels B.A., Hsueh Y.P., Shu T., Liang H., Tseng H.C., Hong C.J., Su S.C., Volker J., Neve R.L., Yue D.T. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56:823–837. doi: 10.1016/j.neuron.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 46.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsueh Y.P. The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 2006;13:1915–1927. doi: 10.2174/092986706777585040. [DOI] [PubMed] [Google Scholar]
- 48.Biederer T., Sudhof T.C. CASK and protein 4.1 support F-actin nucleation on neurexins. J. Biol. Chem. 2001;276:47869–47876. doi: 10.1074/jbc.M105287200. [DOI] [PubMed] [Google Scholar]
- 49.Chetkovich D.M., Bunn R.C., Kuo S.H., Kawasaki Y., Kohwi M., Bredt D.S. Postsynaptic targeting of alternative postsynaptic density-95 isoforms by distinct mechanisms. J. Neurosci. 2002;22:6415–6425. doi: 10.1523/JNEUROSCI.22-15-06415.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsueh Y.P., Wang T.F., Yang F.C., Sheng M. Nuclear translocation and transcription regulation by the membrane- associated guanylate kinase CASK/LIN-2. Nature. 2000;404:298–302. doi: 10.1038/35005118. [DOI] [PubMed] [Google Scholar]
- 51.Wang G.S., Hong C.J., Yen T.Y., Huang H.Y., Ou Y., Huang T.N., Jung W.G., Kuo T.Y., Sheng M., Wang T.F. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron. 2004;42:113–128. doi: 10.1016/s0896-6273(04)00139-4. [DOI] [PubMed] [Google Scholar]
- 52.Wang T.F., Ding C.N., Wang G.S., Luo S.C., Lin Y.L., Ruan Y., Hevner R., Rubenstein J.L., Hsueh Y.P. Identification of Tbr-1/CASK complex target genes in neurons. J. Neurochem. 2004;91:1483–1492. doi: 10.1111/j.1471-4159.2004.02845.x. [DOI] [PubMed] [Google Scholar]
- 53.Atasoy D., Schoch S., Ho A., Nadasy K.A., Liu X., Zhang W., Mukherjee K., Nosyreva E.D., Fernandez-Chacon R., Missler M. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. USA. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoskins R., Hajnal A.F., Harp S.A., Kim S.K. The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development. 1996;122:97–111. doi: 10.1242/dev.122.1.97. [DOI] [PubMed] [Google Scholar]
- 55.Laverty H.G., Wilson J.B. Murine CASK is disrupted in a sex-linked cleft palate mouse mutant. Genomics. 1998;53:29–41. doi: 10.1006/geno.1998.5479. [DOI] [PubMed] [Google Scholar]
- 56.Lopes C., Gassanova S., Delabar J.M., Rachidi M. The CASK/Lin-2 Drosophila homologue, Camguk, could play a role in epithelial patterning and in neuronal targeting. Biochem. Biophys. Res. Commun. 2001;284:1004–1010. doi: 10.1006/bbrc.2001.5050. [DOI] [PubMed] [Google Scholar]
- 57.Zordan M.A., Massironi M., Ducato M.G., Te Kronnie G., Costa R., Reggiani C., Chagneau C., Martin J.R., Megighian A. Drosophila CAKI/CMG protein, a homolog of human CASK, is essential for regulation of neurotransmitter vesicle release. J. Neurophysiol. 2005;94:1074–1083. doi: 10.1152/jn.00954.2004. [DOI] [PubMed] [Google Scholar]
- 58.McKee A.E., Minet E., Stern C., Riahi S., Stiles C.D., Silver P.A. A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev. Biol. 2005;5:14. doi: 10.1186/1471-213X-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J., Smith P.J., Krainer A.R., Zhang M.Q. Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res. 2005;33:5053–5062. doi: 10.1093/nar/gki810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramser J., Abidi F.E., Burckle C.A., Lenski C., Toriello H., Wen G., Lubs H.A., Engert S., Stevenson R.E., Meindl A. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum. Mol. Genet. 2005;14:1019–1027. doi: 10.1093/hmg/ddi094. [DOI] [PubMed] [Google Scholar]
- 61.Grosso A.R., Gomes A.Q., Barbosa-Morais N.L., Caldeira S., Thorne N.P., Grech G., von Lindern M., Carmo-Fonseca M. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008;36:4823–4832. doi: 10.1093/nar/gkn463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matlin A.J., Clark F., Smith C.W. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 63.Licatalosi D.D., Darnell R.B. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 64.Yeo G., Holste D., Kreiman G., Burge C.B. Variation in alternative splicing across human tissues. Genome Biol. 2004;5:R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Froyen G., Van Esch H., Bauters M., Hollanders K., Frints S.G., Vermeesch J.R., Devriendt K., Fryns J.P., Marynen P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum. Mutat. 2007;28:1034–1042. doi: 10.1002/humu.20564. [DOI] [PubMed] [Google Scholar]
- 66.Hayashi S., Mizuno S., Migita O., Okuyama T., Makita Y., Hata A., Imoto I., Inazawa J. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am. J. Med. Genet. A. 2008;146A:2145–2151. doi: 10.1002/ajmg.a.32433. [DOI] [PubMed] [Google Scholar]
- 67.Najm J., Horn D., Wimplinger I., Golden J.A., Chizhikov V.V., Sudi J., Christian S.L., Ullmann R., Kuechler A., Haas C.A. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008 doi: 10.1038/ng.194. in press. Published online August 10, 2008. [DOI] [PubMed] [Google Scholar]
- 68.Franco B., Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr. Opin. Genet. Dev. 2006;16:254–259. doi: 10.1016/j.gde.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 69.Morleo M., Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J. Med. Genet. 2008;45:401–408. doi: 10.1136/jmg.2008.058305. [DOI] [PubMed] [Google Scholar]
- 70.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.