

Abstract
Ectopia lentis is a genetically heterogeneous condition that is characterized by the subluxation of the lens resulting from the disruption of the zonular fibers. Patients with ectopia lentis commonly present with a marked loss in visual acuity in addition to a number of possibly accompanying ocular complications including cataract, myopia, and retinal detachment. We here describe an isolated form of ectopia lentis in a large inbred family that shows autosomal-recessive inheritance. We map the ectopia lentis locus in this family to the pericentromeric region on chromosome 1 (1p13.2-q21.1). The linkage region contains well more than 60 genes. Mutation screening of four candidate genes revealed a homozygous nonsense mutation in exon 11 of ADAMTSL4 (p.Y595X; c.1785T→G) in all affected individuals that is absent in 380 control chromosomes. The mutation would result in a truncated protein of half the original length, if the mRNA escapes nonsense-mediated decay. We conclude that mutations in ADAMTSL4 are responsible for autosomal-recessive simple ectopia lentis and that ADAMTS-like4 plays a role in the development and/or integrity of the zonular fibers.
Main Text
Ectopia lentis (EL) is a rare condition characterized by partial or complete displacement of the lens from its space. In the absence of traumatic injury, the occurrence of EL is predominantly attributed to hereditary systemic diseases, such as Marfan syndrome (MFS [MIM 154700]),1 homocystinuria (CBS [MIM 236200]),2 and Weill-Marchesani syndrome (WMS [MIM 277600; 608328]);3 however, EL can present as an isolated condition.4–6 Despite its clinical homogeneity, isolated EL is genetically heterogeneous with both autosomal-dominant (MIM 129600)5 and -recessive (MIM 225100) forms.7,8
Patients with EL commonly present with marked loss in visual acuity that varies with the degree of lens subluxation.9 Accompanying complications include myopia,10 retinal detachment,10 cataract,11 and glaucoma.12 Although the disruption of the zonular fibers supporting the lens has been established as the underlying pathology of EL,13 no comprehensive theory of the pathogenesis has emerged. The zonular fibers are microfibrillar structures that are not associated with elastin, but share common antigenic determinants with the elastin-associated microfibrils.14 Mutations in FBN1 (MIM 134797) encoding fibrillin have been reported to cause Marfan syndrome15 and were described in association with the autosomal-dominant form of Weill-Marchesani syndrome,3 both syndromes associated with EL.
To identify the gene responsible for an autosomal-recessive isolated EL, we recruited members of a previously described inbred family from Jordan with 15 affected individuals, from 4 related sibships.8 The diagnosis was based on ocular involvement without systemic manifestations. In all examined affected individuals, the corneas were clear, pupils were round and normal, and there were no areas of iris atrophy or positive iris transillumination. The displacement of the lens was variable among the examined affected individuals. Complications found in the affected family members include deprivation amblyopia, retinal detachment, and cataract. A recent clinical review of all affected family members showed a stable ocular condition, without any major changes. A detailed family history was obtained and a three-generation pedigree was constructed (Figure 1A). The participants in this study are from two related sibships with first cousin parents: four affected individuals and six of their unaffected relatives. Informed consent was obtained from all participating family members or their legal guardians, and the informed consent document was approved by the local IRB of Jordan University of Science and Technology, Irbid, Jordan. We also obtained 190 unrelated, ethnically matched anonymous DNA samples, which were used as controls.
Figure 1.

The Family Pedigree, Haplotype, and ADAMTSL4 Mutation
(A) Reduced pedigree of the family showing only participating members and their immediate relatives. The genotypes of eight microsatellite markers are shown with the arrows pointing to the crossover events that delineate the region of linkage.
(B) Sequence chromatograph showing the T→G transversion producing a premature stop codon TAG.
DNA was extracted from whole blood via standard procedures. A genome-wide scan for linkage was performed with the ABI PRISM linkage mapping set of fluorescent microsatellite DNA markers, version 2 (PE Biosystems, Foster City, CA). The regions of homozygosity were inspected and one region around the centromere of chromosome 1 was the most significant. To refine the region, all family members were genotyped with additional nonfluorescent microsatellite markers. The amplified products of the polymerase chain reaction (PCR) were analyzed on 6% acrylamide gel; 5 M urea and the bands detected by silver staining.16 The genome-wide linkage screen identified the pericentromeric region on chromosome 1 (1p13.2-q21.1) as the region of linkage and fine mapping identified a 35 Mb region between D1S1675 and D1S498 as the critical region, of which about 20 Mb are heterochromatic in nature (centromere and 1q12) and do not contain genes.
Two-point and multipoint LOD score analysis was performed with MLINK of the FASTLINK computer program package17 and ALLEGRO,18,19 respectively. The genetic and physical map distances for the genome scan and the fine mapping markers were obtained from the Rutgers combined linkage-physical map (Build 35).20 The analysis was conducted assuming an autosomal-recessive mode of inheritance with full penetrance and no phenocopies. A disease allele frequency of 0.001 was used in the analysis. The allele frequency for all markers was assumed to be equal, because the true allele frequency for the population is unknown. A LOD score of 3 or higher was used to establish linkage. A maximum two-point LOD score of 3.3 was obtained for marker D1S534 at (θ) = 0 (Table 1) and a maximum multipoint LOD score of 4.4 occurred at markers D1S250, D1S467, and D1S189 (results not shown). Both the region of homozygosity and the 3-unit support interval are delineated by markers D1S1675 and D1S498.
Table 1.
Two-Point LOD Scores between EL and Chromosome 1p13.2-1q21.1 Polymorphic Markers
|
LOD Score at Recombination Fractions (θ) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Marker | Marker Position | θ0 | θ0.01 | θ0.02 | θ0.05 | θ0.1 | θ0.2 | θ0.3 |
| D1S1675 | 114.5 | - ∞ | 1.14 | 1.37 | 1.56 | 1.51 | 1.13 | 0.69 |
| D1S250 | 115.0 | 3.21 | 3.13 | 3.05 | 2.82 | 2.43 | 1.65 | 0.93 |
| D1S467 | 115.8 | 2.91 | 2.84 | 2.77 | 2.56 | 2.21 | 1.50 | 0.82 |
| D1S189 | 116.5 | 2.56 | 2.50 | 2.43 | 2.24 | 1.91 | 1.26 | 0.65 |
| D1S2744 | 117.1 | 3.21 | 3.14 | 3.06 | 2.84 | 2.46 | 1.71 | 0.98 |
| D1S534 | 119.5 | 3.30 | 3.18 | 3.11 | 2.88 | 2.50 | 1.72 | 0.98 |
| D1S3466 | 148.5 | 3.05 | 2.98 | 2.90 | 2.69 | 2.33 | 1.60 | 0.91 |
| D1S498 | 149.6 | −0.40 | 0.99 | 1.22 | 1.42 | 1.40 | 1.05 | 0.59 |
The critical region comprises more than 60 known and predicted genes. In silico investigation of genes in the critical region of linkage was performed to generate a priority list for sequencing. Four genes ANXA9 (MIM 603319), MAN1A2 (MIM 604345), ADAM30 (MIM 604779), and ADAMTSL4 (MIM 610113) were short listed and their genomic sequences were obtained from the public databases. Primers were designed to amplify all exons, splice sites, and the flanking sequences (Table S1 available online). Sequencing of the amplified fragments was initially performed for an obligate carrier, an affected individual from the same family, and a normal unrelated control. The allele frequency of identified variations was determined by genotyping 190 ethnically matched controls. Sequence analysis of the selected candidate genes revealed a nonsense mutation in exon 11:c.1785T→G (p.Y595X) of ADAMTSL4, which changes an evolutionary conserved tyrosine to a premature termination codon, resulting in a truncated protein of 594 amino acids, if a protein is made (Figure 1B). This mutation segregates with the phenotype in the examined family members and was not found in 380 ethnically matched control chromosomes.
To determine the expression pattern of ADAMTSL4, first-strand cDNA libraries from an adult and fetal multiple-tissue human panel were obtained commercially (Genemed Biotechnologies, Inc; South San Francisco, CA). We designed primers to amplify two fragments, 340 bp and 481 bp, from the full-length cDNA of ADAMTSL1 (Table S2). The 340 bp fragment detects the two major reported isoforms (a and b) whereas the 481 bp fragment detects only isoform a (full length). A 900 bp β-actin fragment was used as a positive control for gene expression. Amplification of first-strand cDNA from adult human tissue showed expression of ADAMTSL4 in colon, heart, leukocyte, liver, lung, skeletal muscle, spleen, testis, and placenta; expression was weaker in bone marrow, brain tissue, kidney, and pancreas (Figure 2). On the other hand, the expression study in fetal tissue revealed strong expression in heart, kidney, liver, lung, and skeletal muscle, but weaker expression in brain and skin (Figure 3). Expression of ADAMTSL4 has been shown to occur in human and mouse eye tissue.21
Figure 2.

Expression of ADAMTSL4 in cDNA
The top panel shows expression in the cDNA libraries from adult human tissues, whereas the bottom panel shows expression in fetal tissues. The 340 bp fragment detects the two major reported isoforms (a and b) whereas the 481bp fragment detects only isoform a (full length).
Figure 3.
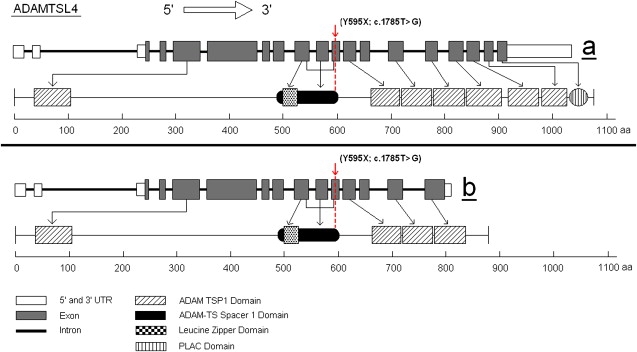
Diagram of ADAMTSL4 and Its Isoforms
Diagramatic presentation of ADAMTSL4, ADAMTSL4 and its two isoforms a (full length) and b. The protein domains are shown, as well as the position of the identified mutation that is present in the two major isoforms. Isoform b lacks three TSP1 domains and the PLAC domain.
The genomic structure of ADAMTSL4 was obtained from the public databases. The genomic sequence of ADAMTSL4 consists of 19 exons spanning about 11.5 kb on chromosome 1q21.2. The full-length mRNA is 4209 bp and encodes a 1074 amino acid protein (116.5 kDa), which displays a 76% homology with the mouse Adamtsl4 protein.21 Alternative transcriptional splice variants encoding different isoforms have been characterized, with two major isoforms, a (full length) and b. The mRNA of second isoform, b, encodes an 877 amino acid protein that lacks the last 200 amino acids (Figure 3). The 5′ untranslated region is 236 bp long and the translation initiation codon is in exon 3. The 3′ untranslated region is 740 bp long and follows of the termination codon in exon 19. The ADAMTS-like 4 comprises seven thrombospondin type 1 repeats (TSP1), one ADAM-TS Spacer 1 domain (that contains the reported mutation), a PLAC domain, and a leucine zipper domain (Figure 3). The protein is predicted to localize in the extracellular matrix. The investigation of an inbred Arabic family with autosomal-recessive isolated EL has revealed an inherently damaging p.Y595X nonsense mutation. The mutation would result in a truncated protein of 594 amino acids that lacks 6 of the 7 thrombospondin type 1 repeats, if it escapes nonsense-mediated mRNA decay. The mutation affects both major isoforms.
ADAMTSL4 belongs to a large superfamily containing 19 ADAMTS proteases22 and at least five ADAMTS-like proteins. They are believed to be anchored to the extracellular matrix (ECM) through interaction with matrix components via one or more TSP1 domains. The ADAMTS superfamily is involved in various biological processes including connective tissue organization, coagulation, inflammation, arthritis, angiogenesis, and cell migration.22 Mutations in ADAMTS10 (MIM 608990) have been implicated in the autosomal-recessive Weill-Marchesani syndrome, a connective-tissue disorder that includes EL in its phenotype.23 Mutations in FBN1 are responsible for the autosomal-dominant Weill-Marchesani syndrome.24 This suggests a role for ADAMTS10 in ECM and connective-tissue remodeling, and also suggests a significant interaction with fibrillin1, a major component of the zonular fibers.
The ADAMTS-like subfamily comprises proteins homologous to the ADAMTS ancillary domains but are lacking the protease domain and hence lack catalytic activity. Mutations in ADAMTSL2 are responsible for the autosomal-recessive geleophysic dysplasia probably through the dysregulation of TGF-β signaling.25 The identification of the interaction of ADAMTSL4 and ECM proteins, including fibrillin, may shed some light on the pathogenetic mechanism relevant to EL. It has been suggested that defective connective tissue remodeling is the pathogenetic mechanisms behind autosomal-recessive Weill-Marchesani syndrome, a function that is performed by ADAMTS10 catalytic activity.23 ADAMTSL4 lacks this or similar catalytic activities, so the hypothesis would be that it plays a structural role in the assembly or integrity of ECM or both. Alternatively, its role may be in a signaling pathway similar to ADAMTSL2.25
The presence of only ocular manifestations in this family without systemic manifestations should not exclude an integral role of ADAMTSL4 in all microfibrillar fibers throughout the body. It would be hypothesized that the absence of ADAMTSL4 function is only apparent in the zonular fibers because these fibers experience slow turnover26 and as such, a temporal onset weakness in their structure is not seen elsewhere because of rapid turnover in other tissues. Alternatively, other ADAMTS-like proteins, or any other proteins for that matter, may compensate for the absence of ADAMTSL4 protein in nonocular tissue in affected individuals.
Acknowledgments
The authors wish to thank the family members for their gracious participation in this study. This study was supported by generous funds from The Shafallah Center for Children with Special Needs and its sibling the Shafallah Medical Genetics Center, Doha, Qatar (D.A., A.K., H.E.-S.). H.E.-S. was supported by the “Chaire Internationale de Recherche, Blaise Pascal, de l'etat et de la Règion d'Ile-de-France,” which is managed with further support by the “Fondation de l'Ecole Normale Supèrieure,” as well as start-up funds from the University of Iowa during the initial linkage studies.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Isoforms, https://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/
NCBI Genome Browser, http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9606
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Krupin T. Marfan syndrome, lens subluxation, and open-angle glaucoma. J. Glaucoma. 1999;8:396–399. [PubMed] [Google Scholar]
- 2.Hayasaka S., Asano Y., Tateda H., Hoshi K., Koga Y. Lens subluxation in homocystinuria. A case report. Acta Ophthalmol. (Copenh.) 1984;62:425–431. doi: 10.1111/j.1755-3768.1984.tb08422.x. [DOI] [PubMed] [Google Scholar]
- 3.Faivre L., Dollfus H., Lyonnet S., Alembik Y., Megarbane A., Samples J., Gorlin R.J., Alswaid A., Feingold J., Le Merrer M. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am. J. Med. Genet. 2003;123A:204–207. doi: 10.1002/ajmg.a.20289. [DOI] [PubMed] [Google Scholar]
- 4.Vanita V., Singh J.R., Singh D., Varon R., Robinson P.N., Sperling K. A recurrent FBN1 mutation in an autosomal dominant ectopia lentis family of Indian origin. Mol. Vis. 2007;13:2035–2040. [PubMed] [Google Scholar]
- 5.Jaureguy B.M., Hall J.G. Isolated congenital ectopia lentis with autosomal dominant inheritance. Clin. Genet. 1979;15:97–109. doi: 10.1111/j.1399-0004.1979.tb02033.x. [DOI] [PubMed] [Google Scholar]
- 6.Sachdev N.H., Coroneo M.T., Wakefield D., Hennessy M.P. Isolated ectopia lentis: potential role of matrix metalloproteinases in fibrillin degradation. Arch. Ophthalmol. 2004;122:111–114. doi: 10.1001/archopht.122.1.111. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz C., Rivas F., Villar-Calvo V.M., Serrano-Lucas J.I., Cantu J.M. Familial simple ectopia lentis. A probable autosomal recessive form. Ophthalmic Paediatr. Genet. 1986;7:81–84. doi: 10.3109/13816818609076113. [DOI] [PubMed] [Google Scholar]
- 8.Al-Salem M. Autosomal recessive ectopia lentis in two Arab family pedigrees. Ophthalmic Paediatr. Genet. 1990;11:123–127. doi: 10.3109/13816819009012957. [DOI] [PubMed] [Google Scholar]
- 9.Casper D.S., Simon J.W., Nelson L.B., Porter I.H., Lichtenstein S.B. Familial simple ectopia lentis: a case study. J. Pediatr. Ophthalmol. Strabismus. 1985;22:227–230. doi: 10.3928/0191-3913-19851101-06. [DOI] [PubMed] [Google Scholar]
- 10.Noble K.G., Bass S., Sherman J. Ectopia lentis, chorioretinal dystrophy and myopia. A new autosomal recessive syndrome. Doc. Ophthalmol. 1993;83:97–102. doi: 10.1007/BF01206207. [DOI] [PubMed] [Google Scholar]
- 11.Cruysberg J.R., Pinckers A. Ectopia lentis et pupillae syndrome in three generations. Br. J. Ophthalmol. 1995;79:135–138. doi: 10.1136/bjo.79.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagi L.R., Walton D.S. Anterior axial lens subluxation, progressive myopia, and angle-closure glaucoma: recognition and treatment of atypical presentation of ectopia lentis. J. AAPOS. 2006;10:345–350. doi: 10.1016/j.jaapos.2006.01.218. [DOI] [PubMed] [Google Scholar]
- 13.Bjerrum K., Kessing S.V. Congenital ectopia lentis and secondary buphthalmos likely occurring as an autosomal recessive trait. Acta Ophthalmol. (Copenh.) 1991;69:630–634. doi: 10.1111/j.1755-3768.1991.tb04851.x. [DOI] [PubMed] [Google Scholar]
- 14.Streeten B.W., Licari P.A. The zonules and the elastic microfibrillar system in the ciliary body. Invest. Ophthalmol. Vis. Sci. 1983;24:667–681. [PubMed] [Google Scholar]
- 15.Tsipouras P., Del Mastro R., Sarfarazi M., Lee B., Vitale E., Child A.H., Godfrey M., Devereux R.B., Hewett D., Steinmann B. Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital contractural arachnodactyly to the fibrillin genes on chromosomes 15 and 5. The International Marfan Syndrome Collaborative Study. N. Engl. J. Med. 1992;326:905–909. doi: 10.1056/NEJM199204023261401. [DOI] [PubMed] [Google Scholar]
- 16.Bassam B.J., Caetano-Anolles G., Gresshoff P.M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 1991;196:80–83. doi: 10.1016/0003-2697(91)90120-i. [DOI] [PubMed] [Google Scholar]
- 17.Cottingham R.W., Jr., Idury R.M., Schaffer A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 18.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 19.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 20.Kong X., Murphy K., Raj T., He C., White P.S., Matise T.C. A combined linkage-physical map of the human genome. Am. J. Hum. Genet. 2004;75:1143–1148. doi: 10.1086/426405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchner D.A., Meisler M.H. TSRC1, a widely expressed gene containing seven thrombospondin type I repeats. Gene. 2003;307:23–30. doi: 10.1016/s0378-1119(03)00423-2. [DOI] [PubMed] [Google Scholar]
- 22.Porter S., Clark I.M., Kevorkian L., Edwards D.R. The ADAMTS metalloproteinases. Biochem. J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dagoneau N., Benoist-Lasselin C., Huber C., Faivre L., Megarbane A., Alswaid A., Dollfus H., Alembik Y., Munnich A., Legeai-Mallet L. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am. J. Hum. Genet. 2004;75:801–806. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faivre L., Gorlin R.J., Wirtz M.K., Godfrey M., Dagoneau N., Samples J.R., Le Merrer M., Collod-Beroud G., Boileau C., Munnich A. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J. Med. Genet. 2003;40:34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Goff C., Morice-Picard F., Dagoneau N., Wang L.W., Perrot C., Crow Y.J., Bauer F., Flori E., Prost-Squarcioni C., Krakow D. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 2008 doi: 10.1038/ng.199. in press. Published online August 1, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanssen E., Franc S., Garrone R. Synthesis and structural organization of zonular fibers during development and aging. Matrix Biol. 2001;20:77–85. doi: 10.1016/s0945-053x(01)00122-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.