

Abstract
Previous genome-wide association (GWA) studies typically focus on single-locus analysis, which may not have the power to detect the majority of genuinely associated loci. Here, we applied pathway analysis using Affymetrix SNP genotype data from the Wellcome Trust Case Control Consortium (WTCCC) and uncovered significant association between Crohn Disease (CD) and the IL12/IL23 pathway, harboring 20 genes (p = 8 × 10−5). Interestingly, the pathway contains multiple genes (IL12B and JAK2) or homologs of genes (STAT3 and CCR6) that were recently identified as genuine susceptibility genes only through meta-analysis of several GWA studies. In addition, the pathway contains other susceptibility genes for CD, including IL18R1, JUN, IL12RB1, and TYK2, which do not reach genome-wide significance by single-marker association tests. The observed pathway-specific association signal was subsequently replicated in three additional GWA studies of European and African American ancestry generated on the Illumina HumanHap550 platform. Our study suggests that examination beyond individual SNP hits, by focusing on genetic networks and pathways, is important to unleashing the true power of GWA studies.
Main Text
Genome-wide association (GWA) studies have been successfully employed to interrogate the genetic structure of common and complex diseases.1 Through single-marker association tests, several diseases, including Crohn disease (CD [MIM 266600])2 and type 2 diabetes (T2D [MIM 125853]),3 have been studied by multiple groups and have revealed tens of confirmed susceptibility loci. Initial insights from comparative analysis of different GWA studies on the same disease suggest that the most significant SNPs in one study may not necessarily show up as the most significant SNPs in another study. Furthermore, SNPs that are genuinely associated with disease may not be identified by any GWA study because of the small effect sizes of the SNPs and the lack of power of any individual study. For example, of the 32 confirmed susceptibility loci for CD, 21 of them were not implicated in any given GWA study but achieved significance only after combining three GWA studies and following up with replication.2 Similar observations have been made for other diseases, in which genuine susceptibility loci do not rank as top in any study but manifest moderate levels of significance across multiple studies.3,4 Given that most individual GWA studies are probably underpowered to detect all but the biggest effect sizes, the “most significant SNPs” approach is less capable of separating the majority of truly associated signals from background noise.
Rather than focusing on individual top SNPs from GWA studies, we hypothesized that pathway-based approaches, which jointly consider multiple variants in interacting or related genes in the same pathway, might complement the “most significant SNPs” approach for interpreting data from such studies.5 The pathway-based approach ranks all genes by their statistical significance and examines whether a group of related genes have modest yet consistent deviation from what is expected by chance, simulating a string of interconnected “needles in a haystack.” The technical difficulty of the pathway-based approach lies in the representation of each gene by multiple SNPs, the handling of linkage disequilibrium (LD) between SNPs, and the proper adjustment of different sizes of genes.6 We have previously proposed an approach that appropriately addresses these issues by using a procedure that is analogous to gene set enrichment analysis.7
In brief, in our pathway-based association approach, we associate each SNP to the overlapping gene or genes, or its closest gene in the genome if it does not overlap with a gene. For each gene, we assigned the highest statistic value (chi-square value) among all SNPs mapped to the gene as the statistic value of the gene. For all the N genes that are represented by SNPs in the GWA study, we sorted their statistic values from the largest to the smallest, denoted by r(1), …, r(N). For any given gene set S composed of NH genes, we then calculated a weighted Kolmogorov-Smirnov-like running sum statistic8 that reflects the overrepresentation of genes within the set S at the top of the entire ranked list of genes in the genome,
where and p (default value = 1) is a parameter that gives higher weight to genes with extreme statistic values. To adjust for differences in gene size (hence the different number of SNPs located within or nearby each gene), as well as the linkage disequilibrium between SNPs within the same gene, we conducted a two-step correction procedure. In the first step, we permuted the disease labels of all samples, thereby ensuring the same number of individuals in each phenotype group for case-control studies. During each permutation (denoted by π), we repeated the calculation of enrichment score as described above as ES(S, π). In the second step, we calculated normalized enrichment score (NES) as a Z score, defined as (ES(S) − mean(ES(S, π)))/SD(ES(S, π)), so that different gene sets are directly comparable to each other. The p value can be calculated from the π permutations, and a false discovery rate (FDR) procedure can be used to control the fraction of expected false positive findings below a certain threshold.9
In the present study, several adjustments were made in our software implementation: (1) We used the latest RefSeq gene annotated in UCSC genome browser10 (as of September 2008) to define the genomic region for each gene in the NCBI 36 genome assembly; the “genomic span” for each gene corresponds to the entire RefSeq transcript region, as well as the 20 kb up/downstream of the transcript. (2) We have used the Cochran-Armitage trend test for the single-marker association test and used the chi-square values as the test statistic for each SNP. (3) We used an updated version of the Gene Ontology11 level 4 annotation data sets, as well as updated version of the BioCarta database and the KEGG database12 (as of September 2008) for our pathway collection.
This pathway-based association approach requires the use of individual-level genotype data for all SNPs in a given GWA study rather than summary statistics only. To test our pathway-based association approach on CD, we accessed the raw genotype data on the CD case cohort and on the 1958 birth control cohort from the Wellcome Trust Case Control Consortium (WTCCC), which were generated using the Affymetrix GeneChip Mapping 500K array sets with ∼500,000 SNP markers.13 Per WTCCC recommendations as specified in the “exclusion-list-05-02-2007.txt” file, 24 control subjects as well as 257 CD cases were not used in study. Therefore, 1748 CD patients and 1480 control subjects of European ancestry were used in our study. We have downloaded the genotype calls generated by the Chiamo calling algorithm; per recommendations by the Chiamo software and the WTCCC study, we only considered those genotype calls with confidence score >0.9 and treated the rest of the calls as missing genotypes. Per WTCCC guideline as specified in the “exclusion-list-snps-26_04_2007.txt” file, we have removed 30,956 SNPs from the Affymetrix Mapping 500K array from association analysis. The genomic control inflation factor14 on single-marker association analysis for this data set is 1.1. After additional quality-control procedures (removal of markers with call rate <95%, minor allele frequency <1%, or markers that are >100 kb away from any gene), a total of ∼272,000 SNPs representing 16,958 RefSeq genes were used in our pathway-based association analysis. We applied 25,000 permutation cycles and tested the association on 534 pathways and gene sets with default parameters in our software.5 The top-ranked pathway is the IL12 pathway with 20 genes (Z = 3.8, permutation p = 0.00008, Bonferroni-adjusted p = 0.043, FDR = 0.045, Table 1), whereas several additional pathways with FDR <0.25 are listed in Table S1 available online.
Table 1.
Description of the Four GWA Data Sets on Crohn Disease
| GWA Data Set | Number of Cases/Controls | Ethnicity | Array Platform | IL12/IL23 Pathway Z Score | IL12/IL23 Pathway p Value |
|---|---|---|---|---|---|
| WTCCC | 1748/1480 | European ancestry | Affymetrix Mapping 500K | 3.8 | 0.00008 |
| Ped-IBD | 647/4250 | European ancestry | Illumina HumanHap550 | 2.2 | 0.013 |
| CDCC | 1083/2507 | European ancestry | Illumina HumanHap550 | 3.3 | 0.0004 |
| CHOP-CD-AA | 40/527 | African Americans | Illumina HumanHap550 | 1.8 | 0.03 |
Examination of the 20 genes within the IL12 pathway demonstrates that a subset of these genes, including Janus kinase 2 (JAK2 [MIM 147796]), tyrosine kinase 2 (TYK2 [MIM 176941]), interleukin 12 receptor 1 (IL12RB1 [MIM 601604]), and interleukin 12 precursor (IL12B [MIM 601642]), also belong to a gene network that is typically referred to as the “IL23 pathway.”15 Given that the term “IL23 pathway” has not been annotated in the public pathway collection, we have elected to use the term “IL12/IL23 pathway” in the discussion below to be consistent with existing CD literature. Multiple plausible hits related to CD biology are contained within the IL12/IL23 pathway. Three genes, including the interleukin 12 receptor 2 (IL12RB2 [MIM 601642]) on 1p31.3, IL12B on 5q33.3, and IL12RB1 on 19p13.11, rank as the three most significant genes in the pathway based on the most significant SNP representing each gene. In cells, IL12RB1 and IL12RB2 form a high-affinity receptor for IL12, which consists of the IL12A and IL12B subunits, whereas IL12RB2 and interleukin 23 receptor (IL23R [MIM 607562]) form a receptor for IL23 signaling.16 As the first CD susceptibility gene identified by a GWA study,17 IL23R associates constitutively with JAK2 and the transcription activator STAT3 (MIM 102582).16 Interestingly, neither JAK2 nor IL12B have been implicated by any single published GWA study on CD, but they have both been recently identified as CD susceptibility genes in a meta-analysis of GWA data.2 To test their effects on pathway association, we eliminated JAK2 and IL12B from the pathway, and we still obtained significant association of the pathway with CD (Z = 3.3, permutation p = 8 × 10−5), indicating that the association results were not dependent on these known susceptibility genes. Furthermore, another two genes in the IL12/IL23 pathway, namely STAT4 (MIM 600558) and CCR5 (MIM 601373), are both in the same protein family of confirmed CD susceptibility genes (STAT3 [MIM 102582] and CCR6 [MIM 601835]).2 Therefore, variants conferring modest disease risk may not reveal themselves in multiple underpowered GWA studies, but can be readily identified by pathway-based approach in a single study.
To replicate the pathway association results, we analyzed several additional GWA data sets generated on the Illumina HumanHap550 arrays with ∼550,000 SNP markers, which represent mostly different markers as the Affymetrix arrays used by WTCCC. We first examined a previously published GWA study on pediatric-onset inflammatory bowel disease (IBD) (the Ped-IBD cohort, Table 1), including 647 CD cases and 4250 control subjects of European ancestry.18 In brief, affected individuals with pediatric-onset IBD were ascertained through the Children's Hospital of Philadelphia, Children's Hospital of Wisconsin and Medical College of Wisconsin, and Cincinnati Children's Hospital Medical Center. However, because the IBD cases include subjects affected by either CD or with ulcerative colitis (UC), we only keep the 647 CD cases in our association analysis. The control group included 4250 children recruited at the Children's Hospital of Philadelphia (CHOP) of European ancestry. The Research Ethics Board of the respective hospitals and other participating centers approved the study, and written informed consent was obtained from all subjects. The genomic control inflation factor14 is calculated as 1.1, similar to the level in the WTCCC data set. With similar quality-control measures as those used on the WTCCC data set, a total of ∼373,000 SNPs representing 17,410 genes remained in the analysis. Notably, the IL12/IL23 pathway showed statistically significant association with CD (Z = 2.2, permutation p = 0.013), indicating that the pathway association results replicated with a different technical platform and marker sets.
Further replication was demonstrated in our expanded and ongoing GWA study on CD, including 1083 independent pediatric-onset CD cases and 2507 control subjects of European ancestry (the CDCC cohort, Table 1). The samples from the CDCC data set were collected from multiple centers from four geographically discrete countries, including Italy (13.3%), Scotland (15.5%), Canada (21.9%), and United States (49.3%). All patients were diagnosed prior to their nineteenth birthday and fulfilled standard CD diagnostic criteria. Phenotypic characterization was based on a modification of the Montreal classification such that the definitions of L1 and L3 were both extended to include disease within the small bowel proximal to the terminal ileum and distal to the ligament of Treitz. The control group was recruited by CHOP clinicians and nursing staff within the CHOP Health Care Network, which includes primary care clinics and outpatient practices. The control subjects did not have IBD or evidence of chronic disease on the basis of the self-reported intake questionnaire or clinician-based assessment. The research ethics board of the respective hospitals and other participating centers approved the study, and written informed consent was obtained from all subjects. We used multidimensional scaling (MDS) by PLINK19 on genotyping data to ensure the matching of ethnicity groups and to include only subjects of European ancestry. Appropriate care has been taken to ensure that none of the cases or controls overlaps with the Ped-IBD data set described above. The genomic control inflation factor14 is calculated as 1.08. After applying thorough quality-control measures, a total of ∼368,000 SNPs representing 17,404 genes remained in the analysis. The IL12/IL23 pathway still showed statistically significant association with CD (Z = 3.3, permutation p = 0.0004), further corroborating the results from the Ped-IBD cohort.
To demonstrate the diverse applicability of the pathway-based approach, we next investigated whether the association results could be replicated in a different ethnic group. Accordingly, we examined a fourth GWA study data set containing 40 African American CD cases and 527 matched-control subjects (the CHOP-CD-AA cohort, Table 1). All CD patients were identified within the CHOP through electronic examination of medical records in the EPIC clinical databases, corresponding to ∼20,000 Philadelphia children recently genotyped on the Illumina HumanHap550 platform. The controls were randomly selected from the aforementioned population sample and were genetically matched to the CD cases. The Research Ethics Board of CHOP approved the study, and written informed consent was obtained from all subjects. The genomic control inflation factor14 is calculated as 1.00. Despite the small sample size, the IL12/IL23 pathway is still significantly associated with CD (Z = 1.8, permutation p = 0.03), further confirming the potential involvement of the pathway in the pathogenesis of CD in subjects of African ancestry.
Comparison of the test statistic values for genes in the IL12/IL23 pathway between the four GWA studies demonstrates that the relative ranking of genes was quite different (Figure 1 and Table S2). Furthermore, we also examined SNPs tagging these genes in the recent Barrett et al. CD meta-analysis results2 (Table S3) and found that several previously unreported IL12/IL23 pathway genes indeed score well in the meta-analysis, albeit not reaching genome-wide significance (for example, p = 1.2 × 10−4 for rs1035127 representing IL18R1, p = 3.9 × 10−4 for rs6661505 representing JUN, p = 7.8 × 10−4 for rs374326 representing IL12RB1, p = 5.7 × 10−3 for rs12720356 representing TYK2). Despite the drastically different association results on the level of individual SNPs across studies, the pathway was consistently picked up as being associated with CD in all four GWA studies, demonstrating the power and effectiveness of pathway association approach in identifying disease-susceptibility mechanisms.
Figure 1.
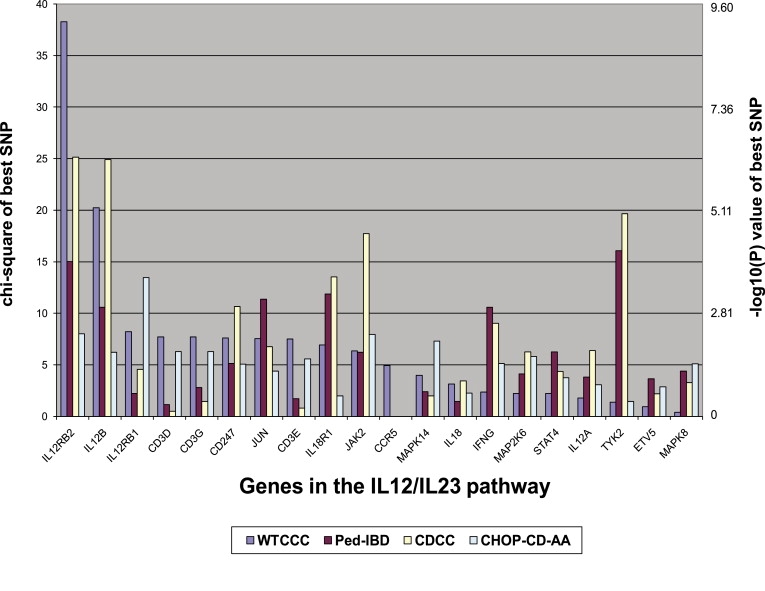
The Chi-Square Values and Negative Logarithm of p Values of the Most Significant SNPs for Each of the 20 Genes in the IL12/IL23 Pathway in Four GWA Data Sets
The CCR5 gene is not represented by SNPs in the Illumina arrays. The relative ranking of the genes are different between the four GWA studies; however, as a group, they collectively show moderately high association signals in each of the GWA studies.
Although our study represents a successful example of a pathway-based approach in relation with GWA studies, several caveats need to be addressed. First, the success of such an analysis depends on the accurate annotation of each pathway as well as the comprehensiveness of the pathway collection, with respect to the disease of interest. For example, previous GWA studies have implicated autophagy-related molecules in the pathogenesis of CD, on the basis of association signals at the immunity-related GTPase M protein (IRGM [MIM 608212]) locus20 and the autophagy-related 16-like 1 gene (ATG16L1 [MIM 610767]) locus.21 Given that this pathway is not well characterized (in fact, IRGM is not even annotated as a RefSeq gene), we constructed a hypothetical pathway containing all genes with “autophagy” in their RefSeq description as well as IRGM (15 genes in the Affymetrix array, 17 genes in the Illumina array). Weak evidence of association was identified for the autophagy pathway, with p value of 0.002, 0.05, 0.24, and 0.29 for the four data sets in Table 1, respectively. Similarly, the nucleotide-binding oligomerization domain 2 (NOD2 [MIM 605956]) gene is a well-known CD susceptibility gene,22,23 but it was not annotated in any pathway used in our study, so our approach cannot account for the effect of this gene on CD susceptibility. These two examples demonstrate that deficiencies in pathway collection could lead to loss of power to detect genuine disease-susceptibility mechanisms.
Another potential limitation of the pathway association approach is that the method cannot readily distinguish the scenario in which each of the susceptibility genes confers moderate risk to disease versus the scenario in which major effect genes in a pathway play dominant roles. For some diseases, such as type 1 diabetes, major effect loci (HLA loci) exist that may easily mask the contribution from other loci with small effect sizes. To demonstrate this, we have analyzed the WTCCC data set on type 1 diabetes and found that essentially any pathway containing the HLA genes generates highly significant p values, even if HLA genes are the only genes with evidence of association to the disease in the pathway. Thus, appropriate adjustments may be required to reflect the contribution of minor effect loci in certain disease areas.
We also caution that the assignment of test statistic for a SNP to its closest gene may not always be correct, given that the culprit gene could be farther away from the risk SNP than the closest gene. For example, in our data, the SNP rs7546245 was assigned to represent its closest gene IL12RB2 (22 kb away); however, the SNP is only 25 kb away from IL23R, a well-known CD susceptibility gene. In fact, rs7546245 is in moderate linkage disequilibrium (r2 = 0.48) with rs11805303 (the most significant SNP for IL23R reported by WTCCC13), suggesting that it may indeed tag a variant within IL23R. Similar examples in GWA studies have been previously reported: the most significant SNP for lactase persistence is not located within the biochemically verified gene LCT (MIM 603202) but within introns of a nearby gene;24,25 similarly, the most significant SNP for human height near the Indian hedgehog homolog (IHH [MIM 600726]) locus is located within a nearby gene rather than IHH, which is known to be the true causal gene on the basis of monogenic syndrome caused by mutation in the gene as well as knockout mouse phenotype.26 Altogether, these issues need to be considered when performing and interpreting pathway-based association tests on GWA studies.
Our study has significant implications with respect to CD biology and may broaden the options for therapy. Treatment with a monoclonal antibody against IL12 in patients with active CD has been shown to induce clinical responses and remissions.27,28 Molecular evidence implicating the role for key members of the pathway, such as STAT429 and MAP kinase,30,31 is also accumulating. Although GWA studies have been proven useful to identify common disease-susceptibility variants in specific human populations, biologically important genes do not necessarily contain common variants with high odds ratio. Therefore, by identifying the ensemble of biologically relevant genes, pathway-based approaches can help formulate hypotheses. As such, other members in the IL12/IL23 pathway may also serve as functional candidates for detailed molecular studies or as promising therapeutic targets. Furthermore, some additional pathways may not reach stringent criteria for significance but are still worth examining. For example, the list in Table S1 contains two T cell receptor signaling pathways from different pathway sources, confirming the involvement of cellular immunity in CD pathogenesis.32,33 In fact, PTPN2 (MIM 176887) and PTPN22 (MIM 600716), two T cell protein tyrosine phosphatases, have now been firmly established as CD susceptibility genes.2 In addition, ATP-binding cassette (ABC) transporters rank as the fourth most significant pathway, and ABCB1 (MIM 171050) in the pathway has been previously associated with inflammatory bowel diseases.34,35
Our study also has significant implications regarding efficient analysis of GWA data. The successful identification and replication of a CD susceptibility pathway demonstrates the power and applicability of the pathway-based approach in the analysis of GWA data. By switching the focus from single SNPs to gene pathways and gene networks, we can potentially aggregate information from multiple susceptibility loci and eliminate the notoriously inherent noise in GWA data. In conclusion, in order to take the full advantage of the information carried in high-density GWA data sets, we need to take steps that go beyond the conventional approaches of assessing only top SNP hits.
Acknowledgments
We would like to thank all participating subjects and families. This research was supported by the Children's Hospital of Philadelphia, the Primary Children's Medical Center Foundation, DK069513, M01-RR00064, M01 RR002172-26, and C06-RR11234 from the National Center for Research Resources. We also thank the Wellcome Trust Case Control Consortium for making the Affymetrix GeneChip Mapping 500K data sets publicly available. All genome-wide genotyping by the Illumina arrays was funded by an Institute Development Award from the Children's Hospital of Philadelphia.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioCarta database, http://www.biocarta.com/
Chiamo software, http://www.stats.ox.ac.uk/∼marchini/software/gwas/chiamo.html
Gene Ontology database, http://www.geneontology.org
KEGG database, http://www.genome.ad.jp/kegg/pathway.html
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim
Pathway association software, http://www.openbioinformatics.org/gengen/
UCSC Genome Browser, http://www.genome.ucsc.edu
WTCCC, http://www.wtccc.org.uk
References
- 1.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 2.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeggini E., Scott L.J., Saxena R., Voight B.F., Marchini J.L., Hu T., de Bakker P.I., Abecasis G.R., Almgren P., Andersen G. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baum A.E., Hamshere M., Green E., Cichon S., Rietschel M., Noethen M.M., Craddock N., McMahon F.J. Meta-analysis of two genome-wide association studies of bipolar disorder reveals important points of agreement. Mol. Psychiatry. 2008;13:466–467. doi: 10.1038/mp.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K., Li M., Bucan M. Pathway-based approaches for analysis of genome-wide association studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casci T. Association studies: The best of the rest. Nat. Rev. Genet. 2007;8:907. [Google Scholar]
- 7.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollander M., Wolfe D.A. Wiley; New York: 1999. Nonparametric Statistical Methods. [Google Scholar]
- 9.Reiner A., Yekutieli D., Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 10.Karolchik D., Kuhn R.M., Baertsch R., Barber G.P., Clawson H., Diekhans M., Giardine B., Harte R.A., Hinrichs A.S., Hsu F. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 15.Abraham C., Cho J.H. IL-23 and Autoimmunity: New Insights into the Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Med. 2009;60:97–110. doi: 10.1146/annurev.med.60.051407.123757. [DOI] [PubMed] [Google Scholar]
- 16.Parham C., Chirica M., Timans J., Vaisberg E., Travis M., Cheung J., Pflanz S., Zhang R., Singh K.P., Vega F. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 17.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kugathasan S., Baldassano R.N., Bradfield J.P., Sleiman P.M., Imielinski M., Guthery S.L., Cucchiara S., Kim C.E., Frackelton E.C., Annaiah K. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat. Genet. 2008;40:1211–1215. doi: 10.1038/ng.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkes M., Barrett J.C., Prescott N.J., Tremelling M., Anderson C.A., Fisher S.A., Roberts R.G., Nimmo E.R., Cummings F.R., Soars D. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampe J., Franke A., Rosenstiel P., Till A., Teuber M., Huse K., Albrecht M., Mayr G., De La Vega F.M., Briggs J. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 22.Hugot J.P., Chamaillard M., Zouali H., Lesage S., Cezard J.P., Belaiche J., Almer S., Tysk C., O'Morain C.A., Gassull M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 23.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 24.Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enattah N.S., Sahi T., Savilahti E., Terwilliger J.D., Peltonen L., Jarvela I. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 26.Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mannon P.J., Fuss I.J., Mayer L., Elson C.O., Sandborn W.J., Present D., Dolin B., Goodman N., Groden C., Hornung R.L. Anti-interleukin-12 antibody for active Crohn's disease. N. Engl. J. Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 28.Sandborn W.J., Feagan B.G., Fedorak R.N., Scherl E., Fleisher M.R., Katz S., Johanns J., Blank M., Rutgeerts P. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–1141. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Pang Y.H., Zheng C.Q., Yang X.Z., Zhang W.J. Increased expression and activation of IL-12-induced Stat4 signaling in the mucosa of ulcerative colitis patients. Cell. Immunol. 2007;248:115–120. doi: 10.1016/j.cellimm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Schindler J.F., Monahan J.B., Smith W.G. p38 pathway kinases as anti-inflammatory drug targets. J. Dent. Res. 2007;86:800–811. doi: 10.1177/154405910708600902. [DOI] [PubMed] [Google Scholar]
- 31.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta. 2005;1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Mitsuhashi M., Targan S.R. Ex vivo simulation of IgG Fc and T-cell receptor functions: An application to inflammatory bowel disease. Inflamm. Bowel Dis. 2008;14:1061–1067. doi: 10.1002/ibd.20428. [DOI] [PubMed] [Google Scholar]
- 33.Romagnani P., Annunziato F., Baccari M.C., Parronchi P. T cells and cytokines in Crohn's disease. Curr. Opin. Immunol. 1997;9:793–799. doi: 10.1016/s0952-7915(97)80180-x. [DOI] [PubMed] [Google Scholar]
- 34.Ardizzone S., Maconi G., Bianchi V., Russo A., Colombo E., Cassinotti A., Penati C., Tenchini M.L., Bianchi Porro G. Multidrug resistance 1 gene polymorphism and susceptibility to inflammatory bowel disease. Inflamm. Bowel Dis. 2007;13:516–523. doi: 10.1002/ibd.20108. [DOI] [PubMed] [Google Scholar]
- 35.Onnie C.M., Fisher S.A., Pattni R., Sanderson J., Forbes A., Lewis C.M., Mathew C.G. Associations of allelic variants of the multidrug resistance gene (ABCB1 or MDR1) and inflammatory bowel disease and their effects on disease behavior: A case-control and meta-analysis study. Inflamm. Bowel Dis. 2006;12:263–271. doi: 10.1097/01.MIB.0000209791.98866.ba. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.