

Abstract
Despite all the research efforts made during the last few decades, most of the cases of families with breast cancer remain unexplained. Mutations in BRCA1 and BRCA2, and in other breast-cancer-susceptibility genes, account for about 25% of familial breast cancer. Linkage studies have failed to identify other breast-cancer-susceptibility genes. The selection criteria of the families, differences in the population background, or clinical and genetic heterogeneity, among other factors, might determine the power to detect the linkage signal. We have performed a SNP-based linkage scan with a total of 6000 SNP markers across the genome in 41 breast-cancer Spanish families, with an average of four breast-cancer cases per family not associated with BRCA1 or BRCA2 germline mutations. In addition, we have included three BRCA-positive families to test the power in linkage detection from a low-complexity family in which a high-penetrance mutation segregates. We have identified three regions of interest, located on 3q25, 6q24, and 21q22. The two former regions showed a suggestive linkage signal (HLOD scores 3.01 and 2.26, respectively), and the latter region showed a significant linkage signal (HLOD score 3.55). Moreover, we found that a subset of 13 families with bilateral breast cancer presented a HLOD of 3.13 on the 3q25 region. Our results suggest that several variables must be taken into account before performing a linkage study in familial breast cancer because of the high heterogeneity within non-BRCA1/2 families. Phenotypic and geographic homogeneity could be the most important factors.
Introduction
Breast cancer (BC [MIM #114480]) is the most frequent malignant tumor among women with approximately one million new cases per year around the world.1 About 5% of all BC cases are considered to be due to the segregation of a germline mutation within a family.2 The two major BC-susceptibility genes BRCA1 (MIM +113705) and BRCA2 (MIM +600185) are estimated to be involved in 20% of familial breast cancer (FBC), whereas mutations in other high-susceptibility genes (such as PTEN [MIM ∗601728], STK11 [MIM ∗602216], P53 [MIM ∗191170]) or in moderate BC-susceptibility genes (such as CHK2 [MIM +604373], PALB2 [MIM ∗610355], or BRIP1 [MIM ∗605882]) explain only about 5% of FBC.3 Thus, the majority of these families remains unexplained, and are known as non-BRCA1/2 families.
Several studies have revealed the heterogeneous nature of the non-BRCA1/2 tumors. Immunohistochemical as well as genomic alteration patterns can distinguish different classes within them,4–6 and this classification is maintained when expression analyses are performed in FBC tumors.7 Thus, this heterogeneity seems to suggest that different susceptibility genes are involved in non-BRCA1/2 FBC.
Linkage analysis is an approach commonly used in the search for genes responsible for monogenic diseases. However, during the last few years, several linkage analyses have been performed in non-BRCA1/2 families without success, via both short tandem repeat (STR or microsatellite) and SNP markers covering the whole genome.8–12 Although several candidate regions suspected to contain BC-susceptibility genes have been described in these studies, the LOD score values obtained for these regions were not significant, and the percentage of families putative linked to each region was low. In a previous study, performed in 19 families from the USA, the Netherlands, and Spain with a panel of 5000 SNP markers, we identified five regions of interest, but only one family was putative linked to each region.9 The small number of studied families, differences in the population background, and (probably the most important) the clinical and genetic heterogeneity of families might partially explain these results. In any case, the large number of candidate regions supports the genetic heterogeneity within non-BRCA1/2 families and also the likely existence of different high-penetrance genes (HPG) for breast-cancer susceptibility, each of them explaining a small number of families. In addition, some authors do not rule out polygenic or recessive models as alternative explanations,13–15 although false positive results should not be discarded.
In the present study, we aimed to minimize some of the effects on linkage analyses of the negative variables explained above. We have performed a SNP-based linkage scan by using a panel of 6000 SNP markers distributed across the genome, in a homogeneous (geographically and phenotypically) group of 41 non-BRCA1/2 families from Spain. These families presented an average of four women affected only by breast cancer, with no blood relatives affected by ovarian or male breast cancer. Moreover, we have included one BRCA1 and two BRCA2 families to evaluate the efficiency of SNP-based linkage analysis in low-complexity families in which a high-penetrance mutation segregates. We have identified three regions of interest, one of them in 21q22 region, which showed a significant linkage signal (HLOD = 3.55), and the other two in 3q25 and 6q24 regions, which showed a suggestive linkage signal (HLOD = 3.01 and 2.2, respectively). The three regions involve a total of 15 different non-BRCA1/2 families, representing 36% of the families.
Material and Methods
Selection of Families
For this study, we selected 41 Spanish families, with an average of four BC cases per family and no mutations in either BRCA1 or BRCA2 genes. The families were recruited after informed consent by five different agencies: Spanish National Cancer Research Centre (CNIO, 8 families), Instituto Catalán de Oncología (ICO, 16 families), Hospital Santa Creu y Sant Pau (10 families), Hospital Clinico San Carlos (6 families), and Hospital de Cruces (1 family). Families were selected based on the following criteria: (1) at least three women diagnosed with breast cancer below 60 years of age, (2) no cases of ovarian cancer or male breast cancer in a blood relative, and (3) DNA samples available for genotyping from at least three women affected with breast cancer. In all families, DNA from one affected member had been screened, and mutations in either BRCA1 or BRCA2 were ruled out through different methods,16,17 including DHPLC and direct sequencing. The presence of large deletions and insertions was also analyzed by MLPA (multiplex ligation probe amplification). Finally, DNA samples from 132 members from the 41 families were available for this study. In summary, 28 families presented three or more breast-cancer cases (with or without other unrelated cancers), whereas in the other 13 families, at least one female member was affected by bilateral breast cancer. The main phenotypic features of the 41 families are shown in Table 1. This study was approved by the Ethical Committee of the Carlos III Health Institute.
Table 1.
Summary of the 41 Spanish Non-BRCA1/2 Families
| Class | Number of Families | Breast Cancer Cases | Bilateral Cases | Mean Age |
|---|---|---|---|---|
| No bilaterality | 14 | 3 | 0 | 49.13 |
| 9 | 4 | 0 | 51.12 | |
| 5 |
>4 |
0 |
51.24 |
|
| Bilaterality | 4 | 3 | 1 | 48.95 |
| 9 |
>3 |
>1 |
50.20 |
|
| Total | 41 | 156 | 15 | 49.88 |
In order to know the efficiency in linkage detection of the SNP marker panel in a set of low-complexity families, we selected 15 members (carriers and noncarriers) from one BRCA1-positive family and two BRCA2-positive families, which presented a similar structure to the non-BRCA1/2 families included in this linkage analysis (Figure 1). We considered mutation carriers as affected members (complete penetrance) to calculate the HLOD score we could expect for a single family in which a high-penetrance mutation segregates. We calculated the HLOD score for each BRCA-positive family by using different combinations of the genotyped members (see Table S1 available online), and we selected the maximum HLOD score value as representative of each original BRCA-positive family. We took as references those SNP markers flanking both genes (rs1008753-rs1385 for BRCA1 and rs390704-rs132934 for BRCA2).
Figure 1.
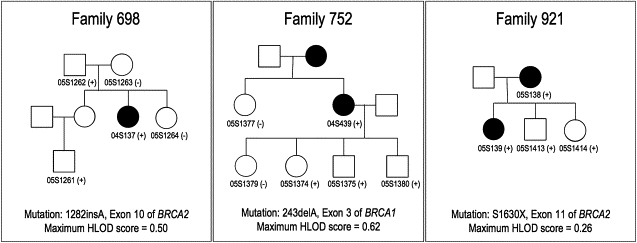
Representation of the BRCA1/2 Families
Mutation carrier individuals (+) were supposed to be affected members (complete penetrance) and their genotypes were used to obtain dominant parametric LOD scores (HLOD) for each family in either chromosome 13 or chromosome 17. Genotypes from noncarriers individuals (−) were used to observe variations in HLOD scores. When a noncarrier individual was included as an affected member (phenocopy), HLOD score dropped to even negative values in the region that harbors the mutation. When a noncarrier individual is included as a nonaffected member, the increase in HLOD score was not significant.
Markers and Genotyping
A total of 147 individuals were genotyped with Illumina BeadArray genotyping system. We genotyped 115 individuals from 36 non-BRCA1/2 families with the Illumina BeadArray linkage mapping panel version IV, which consists of 6000 genome-wide SNP markers. Another 17 individuals from 5 non-BRCA1/2 families were genotyped with Illumina BeadArray linkage mapping panel version III, which consists of a total of 5000 genome-wide SNP markers, and have been reported previously.9 The 15 individuals from the 3 BRCA-positive families were genotyped with the Illumina BeadArray linkage mapping panel version IV. We compared both linkage panels (versions III and IV) for compatibility and found no inconsistencies. However, in order to homogenize the label of the alleles to perform the whole analysis with Merlin software, we reassigned genotypes to samples genotyped with linkage panel III according to the probe designs in linkage panel IV, including missing values for those SNP markers that were not included in linkage panel III but were included in linkage panel IV. We selected linkage panel IV because it has been described that the linkage disequilibrium between SNP markers with a spacing similar to linkage panel III is limited and does not affect the linkage signal, which supports these SNP panels as reliable tools in linkage analysis.9,18
In order to confirm the familial segregation of the haplotypes obtained with the SNP markers, we selected STR markers with high heterozygosity, covering and flanking the candidate regions, with a minimal distance of 1 cM between each pair of STR markers. Four STR markers were genotyped in those families selected as putative linked to each of the candidate regions on both chromosomes 3 and 21, and another five STR markers were genotyped in those families selected as putative linked to the candidate region on chromosome 6. Genetic and physical information for all the STR markers was obtained from NCBI MapViewer and is shown in Table S2. Genotyping of the STR markers was performed with specific primers obtained from NCBI UniSTS database and the ABI 3700 DNA sequencer platform, and data analysis was carried out with Genescan software. Quality control analyses were performed with PEDSTAT and PEDCHECK programs to check family structure, and error-detection and wipe (Merlin software options) were used to evaluate genotyping reliability, as previously reported.9 We used CGHExplorer19 software to visualize the results from Merlin software.
Genetic Parameters
The analysis of all families combined was performed by assuming that all families were of the same genetic and homogeneous background, as we demonstrated in a previous stratification study in the Spanish population.20 For SNP analysis, we calculated the allele frequencies considering all the genotyped individuals (ALL frequencies). For microsatellite analysis, we genotyped an additional sample of 95 unrelated healthy Spanish individuals to obtain better allele frequency estimates. A genetic map with meioses derived from CEPH pedigrees was constructed by Illumina21 and used for SNP analysis. For microsatellite data, we used the STR genetic map constructed by deCODE Genetics Inc.22
Statistical Analysis
We estimated LOD score values via Merlin software23,24 in two different ways: (1) multipoint and singlepoint nonparametric linkage analyses (NPL); and (2) multipoint and singlepoint parametric linkage analyses (HLOD) were estimated assuming a dominant model based on previous publications.25 In the model, the susceptibility allele is supposed to have a population frequency of 0.003, and risks were modeled in 7 age categories and implemented in 14 liability classes, with separate classes for unaffected and affected individuals.26 Multipoint and singlepoint analyses for the whole family set and for each family individually (with the perFamily option in Merlin software) were performed with ALL frequencies. We considered candidate linkage regions those regions that showed NPL score with associated p value < 0.01 and HLOD score higher than 2.20 (suggestive linkage). Additional fine-mapping microsatellite data was also analyzed with Merlin software, and parametric and nonparametric analyses were performed.
In order to evaluate the robustness of our linkage results, we used Merlin's simulation option to estimate empirically the probability that the observed results could be obtained by chance. Ten thousand genome-wide replicates were analyzed under the null hypothesis of no linkage to breast cancer, and the number of regions with a HLOD score over the specified threshold was determined in each genome-wide replicate.
Results
Linkage Analysis
We have performed a linkage analysis in 41 non-BRCA1/2 families with a panel of 6000 SNP markers across the genome. The results of multipoint nonparametric (NPL) and parametric (HLOD) analyses for all chromosomes are shown in Figure 2. Candidate regions were determined as those regions that showed NPL score with associated p value < 0.01 and HLOD score higher than 2.2. According to these criteria, we selected three regions on three different chromosomes as regions that might contain breast-cancer-susceptibility genes: 3q25 region with a maximum NPL score of 2.46 (p value = 0.007) and maximum HLOD score of 3.01 (alpha = 0.51); 6q24 region with a maximum NPL score of 2.65 (p value = 0.004) and maximum HLOD score of 2.26 (alpha = 0.501); and 21q22 region with a maximum NPL score of 4.37 (p value = 0.00001) and maximum HLOD score of 3.55 (alpha = 0.751). The size of the regions was about 10 Mb for the regions on both chromosomes 3 and 6, and about 2 Mb for the region on chromosome 21. All the information of the candidate regions is summarized in Table 2.
Figure 2.
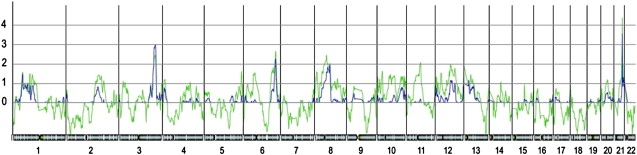
Representation of LOD Scores for All the Autosomal Chromosomes
The y axis represents LOD score values and x axis represents the autosomal chromosomes. The green line represents the nonparametric LOD score (NPL), whereas the blue line represents dominant parametric LOD score (HLOD) score for the 22 autosomal chromosomes. Vertical lines represent the chromosomal boundaries.
Table 2.
Maximum LOD Scores in the Three Candidate Regions
|
From |
To |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Region | SNP | Mb | SNP | Mb | Families | NPL (Max) | p Value | HLOD (Max) |
| 3 | q25.33-q26.2 | rs1472578 | 160.29 | rs1920122 | 170.98 | 6(6) | 2.46 | 0.007 | 3.01 |
| 6 | q24.3-q25.1 | rs612928 | 144.64 | rs1407491 | 153.03 | 7(5) | 2.65 | 0.004 | 2.26 |
| 21 | q22.13 | rs1012959 | 36.98 | rs2836301 | 38.59 | 5(5) | 4.37 | 0.00001 | 3.55 |
Nonparametric (NPL) and parametric (HLOD) LOD scores obtained from the whole set of 41 families for the three candidate regions. The number of putative linked families is shown before and after fine-mapping strategy. In bold and italics is shown the significant HLOD value for chromosome 21.
We wanted to know the power that the SNP panel has to detect the linkage signal in low-complexity families; furthermore, we included three families with known mutations in either BRCA1 or BRCA2 genes in this study (Figure 1). Therefore, we performed parametric linkage analysis in a set of different combinations of the genotyped members for each family, and we calculated the HLOD score for every single combination (see Table S1). We observed that the maximum HLOD score from BRCA-positive families data varied from 0.26 to 0.62, for a single family (Figure 1). HLOD score was calculated for each of the 41 non-BRCA1/2 families, and, in order to avoid a high rate of false-positive results, we determined those families that showed HLOD score ∼0.50 as putative linked families to each candidate region. We observed that even in a family in which a high-penetrance mutation segregates, the HLOD score depended on the genotyped members used to calculate it (genotypical dependence, see Table S1). We also observed that the HLOD score decreased even to negative values when a noncarrier individual was labeled as affected (phenocopy effect, see Table S1).
We selected six families for chromosome 3, seven families for chromosome 6, and five families for chromosome 21 as putative linked families, with HLOD scores ranging from 0.46 to 1.28. Two families showed a similar linkage signal in both chromosomes 3 and 6, and another family showed a similar linkage signal in both chromosomes 3 and 21. Finally, these 15 different families were included for the next step, the fine-mapping strategy via STR markers, and the clinical features of these families are shown in Table 3.
Table 3.
Clinical Features of the Linked Families for Each Candidate Region
| Chromosome | Region | Family ID | Breast Cases | Bilateral Cases | Mean Age | Related Cancers |
|---|---|---|---|---|---|---|
| 3 | 3q25 | 3 | 4 | 1 | 47.4 | |
| 5 | 7 | 1 | 50.3 | lymphoma | ||
| 10 | 8 | 1 | 52.8 | leukemia | ||
| 21 | 3 | 0 | 50.3 | colon | ||
| 27 | 4 | 0 | 47.5 | gastric | ||
|
24 |
6 |
1 |
59.5 |
lymphoma |
||
| 6 | 6q24 | 2 | 4 | 0 | 38.2 | thyroid |
| 6 | 5 | 1 | 51.1 | bladder | ||
| 31 | 3 | 0 | 43.1 | CNS/CRCa | ||
| 33 | 5 | 0 | 50.8 | |||
| 35 |
4 |
0 |
49.2 |
|||
| 21 | 21q22 | 8 | 3 | 2 | 46.7 | colon |
| 9 | 4 | 0 | 40.7 | |||
| 18 | 4 | 1 | 46.1 | |||
| 41 | 4 | 0 | 51.5 | |||
| 24 | 6 | 1 | 59.5 | lymphoma |
Family 24 was confirmed as linked to both chromosome 3 and 21 candidate regions.
CNS, central nervous system tumor; CRC, colorectal cancer.
Simulation Study
In order to examine the false positive rates in our data, we generated 10,000 random genome-wide scan replicates of the data via Merlin software, and we calculated how many genome-wide scans with a maximum HLOD ≥ 3.60 could be expected by chance. We calculated the number of replicates with a maximum HLOD score higher than or equal to 3.60, because this is the threshold for significant linkage estimated for STR markers.27 We observed that only 155 replicate scans showed HLOD ≥ 3.60, giving an empirical p value of 0.015. The empirical threshold for significance was HLOD > 3.10 (p value = 0.05).
Fine Mapping Strategy
In order to confirm or rule out the haplotypical segregation in our candidate families, we performed the additional genotyping of a panel of high-heterozygosity STR markers in the three candidate regions (Table S2). We analyzed a total of 13 STR markers (four STR markers in both chromosomes 3 and 21 and five STR markers in chromosome 6) in those families putative linked to each candidate region. We calculated the HLOD score by using Merlin software, for both SNP and STR markers (Table 4). The HLOD score of the candidate regions in both chromosomes 3 and 21 was maintained or even increased (confirming the haplotypical segregation of both SNP and STR markers in all the families putative linked to these regions), whereas the impact of fine-mapping in the candidate region on chromosome 6 was evident as the HLOD score fell from 4.93 (SNP markers) to 3.40 (SNP+STR markers). The decrease in the HLOD score on this region may be explained by the fact that there was no segregation of STR markers in the two families putative linked to both chromosomes 3 and 6, even though the haplotypical segregation with both SNP and STR markers was confirmed in chromosome 3. When these two families were removed from the data set in chromosome 6, the HLOD score increased from 3.33 (SNP markers) to 3.39 (SNP+STR markers). Only the haplotypical segregation of family 24 was confirmed in both chromosomes 3 and 21 after microsatellite data analysis. Figure S1 represents how STR markers allowed us to confirm (Figure S1A) or to rule out (Figure S1B) the haplotypical segregation in two candidate families identified by SNP markers as putative linked to chromosome 6. These results support the fine-mapping strategy as a useful haplotype validation tool in linkage studies. Thus, we corroborated the three chromosomal regions to be candidates to harbor breast-cancer-susceptibility genes, although no informative recombination events were found and we were not able to narrow down the size of the regions.
Table 4.
Maximum HLOD Scores in the Three Candidate Regions via SNP and STR Markers
|
From |
To |
||||||
|---|---|---|---|---|---|---|---|
| Chromosome | Region | SNP | Mb | SNP | Mb | HLOD SNPs | HLOD SNPs + FM STRs |
| 3 | q25.33-q26.2 | rs1472578 | 160.29 | rs905129 | 172.43 | 5.33 | 5.46 |
| 6 | q24.3-q25.1 | rs612928 | 144.64 | rs1407491 | 153.03 | 4.93/3.33 | 3.40/3.39 |
| 21 | q22.11-q22.13 | rs762173 | 32.75 | rs2836301 | 38.59 | 3.57 | 3.68 |
Parametric (HLOD) LOD scores obtained from the candidates families for the three candidate regions before and after fine-mapping. In chromosome 6, HLOD score is shown for the seven initial putative linked families and the five final putative linked families. The three regions were confirmed after fine-mapping.
Subgroup Analysis
We observed that the regions on both chromosomes 21 and 3, which showed the highest HLOD scores, were associated with a higher number of affected cases per family (4.7 cases versus 3.8 cases, p value < 0.05) and a higher percentage of bilaterality (14.9% versus 6.9%, p value < 0.04). Moreover, we observed that families linked to chromosome 6 had an earlier age of onset on average, although not significant (46.6 years versus 50.37 years, p value = 0.20) (see Table 3). According to these data, we have calculated the HLOD score for those families that presented cases affected by bilaterality (13 families), for those that presented more than 4 cases (23 families), and for those families with a mean age less than or equal to 50 years (21 families). The most interesting result was that the subset of families with bilaterality showed a HLOD = 3.13 for region on chromosome 3, but no linkage signal to either chromosome 21 or chromosome 6 (Table S3).
Discussion
In the present linkage study performed across 41 Spanish non-BRCA1/2 families, significant linkage signal was observed for one candidate region on chromosome 21 (HLOD score 3.55), with 5 (12.2%) families linked, and suggestive linkage signal was observed for candidate regions on both chromosomes 3 and 6 (HLOD score = 3.01 and 2.26, respectively) with 6 (14.6%) and 5 (12.2%) families putative linked, respectively. These results suggest the presence of several putative HPG for breast cancer and each one could explain a low percentage of multiple-case families.
To date, numerous linkage studies have been performed on families with breast cancer,8–12 identifying different candidate regions, which showed no significant or suggestive LOD scores. In addition, and probably due to the small number of families in some cases, geographic differences, or clinical and genetic heterogeneity of the families in others, none of the candidate regions have been confirmed in a new series of non-BRCA1/2 families. Similarly, our group performed a previous SNP-based linkage study, which included 19 families from the USA, the Netherlands, and Spain.9 We identified five candidate regions on five different chromosomes, but only one family was linked to each region. These results suggested that heterogeneity among families from different geographic areas could mask linkage signal, especially when the number of families is small.
Because of this fact, we decided to perform a new SNP-based linkage analysis including a more homogeneous (phenotypically and geographically) set of families of 41 Spanish non-BRCA1/2 breast-cancer families from a nonstratified population,20 with an average of 4 females affected only by breast cancer (Table 1). None of the regions described previously were validated in the present study and, vice versa, none of the three regions described in the present study were found in previous studies. Furthermore, all these data might suggest population specificity, although we cannot discard the effect of randomness.
In order to know the HLOD score to be expected from a low-complexity family in which a HPG segregates, we included three families with known mutations in either BRCA1 or BRCA2 genes and similar familial structure to our non-BRCA1/2 families. We found that these families presented a maximum HLOD score between 0.26 and 0.62 (Figure 1), suggesting that a high amount of information could be missed if we applied the standard thresholds of 1.5, established in high-complexity families.11 Thus, we decided to select those families that showed a HLOD score around 0.50 as families putative linked to each region, resulting in a total of 15 different families being selected for the three regions (Table 2). We corroborated the linkage signal in these regions by using a panel of STR markers, which covered and flanked the candidate regions. We also ruled out the haplotypical segregation of two families putative linked to chromosome 6 with microsatellite data, although these families segregated in both chromosomes 3 and 6 with SNP markers and were confirmed in chromosome 3. Finally, we considered 6 families as putative linked to 3q25 region, 5 families to 6q24 region, and 5 families to 21q22 region (Table 4).
We also analyzed whether the thresholds estimated when genome-wide scans were based on STR markers27 were reliable or not for genome-wide scans based on high-density maps of low-heterozygosity markers. Furthermore, we generated 10,000 random genome-wide scan replicates of the data and we observed that the probability of finding HLOD scores greater than 3.60 was significant (empirical p value = 0.015). This result supports the use of the established thresholds for SNP markers as well as for STR markers.
Our results highlight different factors that may affect linkage studies in FBC. Probably the most important points for consideration is the genetic heterogeneity among non-BRCA1/2 families and also the genetic heterogeneity among human populations. The regions described during the past few years as genomic areas potentially harboring HPG for breast cancer and those found in the present report are represented in Table 5. Only those regions associated with either a single big family or a small group of families that presented HLOD scores ≥1.5 are included in this table. These two groups probably represent the scenario in which non-BRCA1/2 families could be represented, e.g., various susceptibility genes and each of them explaining a low number of families. In addition, an important characteristic that in some cases may be observed is the linkage signal of a single family to two or three different regions. In these cases, the probability that two HPGs are segregating together through different generations is very low. An example of this fact is a family we previously studied, which was linked to both chromosomes 11 and 14 (see Table 5, FAM153). We calculated the probability that the four studied members share two loci by chance in 1/540,000 (unpublished data). Furthermore, we can discard neither that only one, or even none, of these regions contain a causal gene, nor that two moderate-penetrance genes interact among them or with other low-penetrance genes.
Table 5.
Summary of the Studies Performed in Non-BRCA1/2 Families during the Last Years
| Study | Center | Family | Chr. | Region | LOD score |
|---|---|---|---|---|---|
| [11] | Australia | 699003 | 2 | 2p21 | 1.67 |
| IARC | 2191 | 4 | 4p14-q12 | 1.84a | |
| 20 | 20q13.1 | 1.80 | |||
| MAYO151 | 3 | 3p14 | 1.52 | ||
| 11 | 11p13 | 1.59 | |||
| Netherlands | RUL153 | 11 | 11q14 | 1.67b | |
| UK | EUR60a | 15 | 15q14 | 1.50 | |
| EUR60b | 4 | 4q13.1-q13.2 | 1.91 | ||
| 22 |
22q13.2 |
2.62 |
|||
| [10] |
Finland |
2 |
2q32 |
1.61 |
|
| [8] | Sweden | Family 14 | 10 | 10q23 | 1.66 |
| 19 | 19q13 | 1.52 | |||
| 17 |
17p13 |
1.51 |
|||
| [9] | CNIO | FAM3395 | 2 | 2p22.3 | 1.92 |
| FAM2191 | 4 | 4p14q12 | 1.8a | ||
| FAM153 | 11 | 11q13.5q14.3 | 2.2b | ||
| 14 |
14q21.1q21.3 |
2.2 |
|||
| Present study | Spain | 6 families | 3 | 3q25-33 | 3.01 |
| 5 families | 6 | 6q24 | 2.26 | ||
| 5 families | 21 | 21q22.13 | 3.55 |
In order to make possible a complete comparison between the studies, chromosomal regions for the study reported in [11] have been estimated from genetic distances associated with STR markers presented by the authors. LOD scores were performed with the same dominant model adapted by authors of [26] in all the studies.
Same family studied.
Same family studied.
Although we cannot quantify the percentage of non-BRCA1/2 families that could be explained by the candidate regions in Table 5, it is likely to be less than 0.10 per region. Data from the study conducted by the BCLC11 estimated 0.18 as the proportion of families that would be explained by those genes included in the regions on both chromosomes 2 and 4, and 0.06 as the proportion of families that would be explained by those genes included in the region on chromosome 22. In our study, we estimated in 0.36 (15/41 families) the proportion of families that might be explained by genes located on the three regions: 0.12 (5/41 families) for both chromosomes 6 and 21, and 0.14 (6/41 families) for chromosome 3. Although these results can be biased by possible false positive cases, they could support the idea that there are many HPGs involved in familial breast cancer, probably associated with subsets of homogeneous populations or with common phenotypic features. In this way, it is important to highlight the association between the 2p21 region and the group of families with more than four breast cancer cases (LOD = 2.4) pointed out by the BCLC.11 In our study, we have found a similar result with a subset of families with bilateral breast cancer. This group presented a unique linkage signal on chromosome 3q25 (HLOD = 3.13), which represents an interesting association for further studies.
In summary, we consider that, in order to achieve greater power, future linkage studies should contemplate the possibility of studying sets of families with more homogeneous features, phenotypical and/or geographical, instead of large studies with heterogeneous sets of families. In addition, we think that it is important to include low-complexity pedigrees of BRCA1/2 families as an internal control, in order to consider the new LOD score thresholds that can be expected instead of the classical thresholds based on high-complexity BRCA1/2 pedigrees. This strategy could probably permit the identification of putative candidate regions associated with homogeneous groups of families.
Supplemental Data
Supplemental Data include one figure and three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI MapViewer database, http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid=9606
NCBI UniSTS database, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank Emilio Gonzalez and Charo Alonso from the CeGen Unit at the Spanish National Cancer Research Centre and Victoria Fernandez for their technical support. We also thank Ana Osorio, Roger Milne, and Lorenzo Melchor from the Human Genetics Group for their scientific support and advice. We want to thank Emily Hodges from Cold Spring Harbor Laboratory for the English grammatical review and her useful corrections. JMRR grant sponsor was Ministerio de Educacion y Ciencia, FPU AP-2005-1720. This study has been funded by projects FIS-04-2240, FIS 05/0864, and PI-081120 from the Fondo de Investigaciones Sanitarias, RD06/0020/1060 and RD06/0020/1051 from Carlos III Health Institute, 2005SGR00018 from Catalan Health Institute and Autonomous Government of Catalonia, and by the Asociación Española Contra el Cáncer and the Centre of Biomedical Research on Rare Disease (CIBERER). The authors declare no conflict of interest.
References
- 1.Parkin D.M., Bray F., Ferlay J., Pisani P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Nathanson K.L., Wooster R., Weber B.L. Breast cancer genetics: what we know and what we need. Nat. Med. 2001;7:552–556. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- 3.Stratton M.R., Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 4.Honrado E., Osorio A., Palacios J., Milne R.L., Sanchez L., Diez O., Cazorla A., Syrjakoski K., Huntsman D., Heikkila P. Immunohistochemical expression of DNA repair proteins in familial breast cancer differentiate BRCA2-associated tumors. J. Clin. Oncol. 2005;23:7503–7511. doi: 10.1200/JCO.2005.01.3698. [DOI] [PubMed] [Google Scholar]
- 5.Melchor L., Honrado E., Garcia M.J., Alvarez S., Palacios J., Osorio A., Nathanson K.L., Benitez J. Distinct genomic aberration patterns are found in familial breast cancer associated with different immunohistochemical subtypes. Oncogene. 2008;27:3165–3175. doi: 10.1038/sj.onc.1210975. [DOI] [PubMed] [Google Scholar]
- 6.Oldenburg R.A., Kroeze-Jansema K., Meijers-Heijboer H., van Asperen C.J., Hoogerbrugge N., van Leeuwen I., Vasen H.F., Cleton-Jansen A.M., Kraan J., Houwing-Duistermaat J.J. Characterization of familial non-BRCA1/2 breast tumors by loss of heterozygosity and immunophenotyping. Clin. Cancer Res. 2006;12:1693–1700. doi: 10.1158/1078-0432.CCR-05-2230. [DOI] [PubMed] [Google Scholar]
- 7.Hedenfalk I., Ringner M., Ben-Dor A., Yakhini Z., Chen Y., Chebil G., Ach R., Loman N., Olsson H., Meltzer P. Molecular classification of familial non-BRCA1/BRCA2 breast cancer. Proc. Natl. Acad. Sci. USA. 2003;100:2532–2537. doi: 10.1073/pnas.0533805100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergman A., Karlsson P., Berggren J., Martinsson T., Bjorck K., Nilsson S., Wahlstrom J., Wallgren A., Nordling M. Genome-wide linkage scan for breast cancer susceptibility loci in Swedish hereditary non-BRCA1/2 families: suggestive linkage to 10q23.32-q25.3. Genes Chromosomes Cancer. 2007;46:302–309. doi: 10.1002/gcc.20405. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Neira A., Rosa-Rosa J.M., Osorio A., Gonzalez E., Southey M., Sinilnikova O., Lynch H., Oldenburg R.A., van Asperen C.J., Hoogerbrugge N. Genomewide high-density SNP linkage analysis of non-BRCA1/2 breast cancer families identifies various candidate regions and has greater power than microsatellite studies. BMC Genomics. 2007;8:299. doi: 10.1186/1471-2164-8-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huusko P., Juo S.H., Gillanders E., Sarantaus L., Kainu T., Vahteristo P., Allinen M., Jones M., Rapakko K., Eerola H. Genome-wide scanning for linkage in Finnish breast cancer families. Eur. J. Hum. Genet. 2004;12:98–104. doi: 10.1038/sj.ejhg.5201091. [DOI] [PubMed] [Google Scholar]
- 11.Smith P., McGuffog L., Easton D.F., Mann G.J., Pupo G.M., Newman B., Chenevix-Trench G., Szabo C., Southey M., Renard H. A genome wide linkage search for breast cancer susceptibility genes. Genes Chromosomes Cancer. 2006;45:646–655. doi: 10.1002/gcc.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson D., Szabo C.I., Mangion J., Oldenburg R.A., Odefrey F., Seal S., Barfoot R., Kroeze-Jansema K., Teare D., Rahman N. Evaluation of linkage of breast cancer to the putative BRCA3 locus on chromosome 13q21 in 128 multiple case families from the Breast Cancer Linkage Consortium. Proc. Natl. Acad. Sci. USA. 2002;99:827–831. doi: 10.1073/pnas.012584499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antoniou A.C., Easton D.F. Polygenic inheritance of breast cancer: implications for design of association studies. Genet. Epidemiol. 2003;25:190–202. doi: 10.1002/gepi.10261. [DOI] [PubMed] [Google Scholar]
- 14.Cui J., Antoniou A.C., Dite G.S., Southey M.C., Venter D.J., Easton D.F., Giles G.G., McCredie M.R., Hopper J.L. After BRCA1 and BRCA2-what next? Multifactorial segregation analyses of three-generation, population-based Australian families affected by female breast cancer. Am. J. Hum. Genet. 2001;68:420–431. doi: 10.1086/318187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pharoah P.D., Antoniou A.C., Easton D.F., Ponder B.A. Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 16.Diez O., Osorio A., Duran M., Martinez-Ferrandis J.I., de la Hoya M., Salazar R., Vega A., Campos B., Rodriguez-Lopez R., Velasco E. Analysis of BRCA1 and BRCA2 genes in Spanish breast/ovarian cancer patients: a high proportion of mutations unique to Spain and evidence of founder effects. Hum. Mutat. 2003;22:301–312. doi: 10.1002/humu.10260. [DOI] [PubMed] [Google Scholar]
- 17.Osorio A., Barroso A., Martinez B., Cebrian A., San Roman J.M., Lobo F., Robledo M., Benitez J. Molecular analysis of the BRCA1 and BRCA2 genes in 32 breast and/or ovarian cancer Spanish families. Br. J. Cancer. 2000;82:1266–1270. doi: 10.1054/bjoc.1999.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray S.S. Evaluation of linkage disequilibrium and its effect on non-parametric multipoint linkage analysis using two high density single-nucleotide polymorphism mapping panels. BMC Genet. 2005;6(Suppl 1):S85. doi: 10.1186/1471-2156-6-S1-S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lingjaerde O.C., Baumbusch L.O., Liestol K., Glad I.K., Borresen-Dale A.L. CGH-Explorer: a program for analysis of array-CGH data. Bioinformatics. 2005;21:821–822. doi: 10.1093/bioinformatics/bti113. [DOI] [PubMed] [Google Scholar]
- 20.Milne R.L., Ribas G., Gonzalez-Neira A., Fagerholm R., Salas A., Gonzalez E., Dopazo J., Nevanlinna H., Robledo M., Benitez J. ERCC4 associated with breast cancer risk: a two-stage case-control study using high-throughput genotyping. Cancer Res. 2006;66:9420–9427. doi: 10.1158/0008-5472.CAN-06-1418. [DOI] [PubMed] [Google Scholar]
- 21.Murray S.S., Oliphant A., Shen R., McBride C., Steeke R.J., Shannon S.G., Rubano T., Kermani B.G., Fan J.B., Chee M.S. A highly informative SNP linkage panel for human genetic studies. Nat. Methods. 2004;1:113–117. doi: 10.1038/nmeth712. [DOI] [PubMed] [Google Scholar]
- 22.Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 23.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 24.Kruglyak L. Nonparametric linkage tests are model free. Am. J. Hum. Genet. 1997;61:254–255. doi: 10.1016/S0002-9297(07)64305-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claus E.B., Risch N., Thompson W.D. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am. J. Hum. Genet. 1991;48:232–242. [PMC free article] [PubMed] [Google Scholar]
- 26.Easton D.F., Bishop D.T., Ford D., Crockford G.P. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- 27.Lander E., Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat. Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.