

Abstract
Copy number variants (CNVs) contribute to human genetic and phenotypic diversity. However, the distribution of larger CNVs in the general population remains largely unexplored. We identify large variants in ∼2500 individuals by using Illumina SNP data, with an emphasis on “hotspots” prone to recurrent mutations. We find variants larger than 500 kb in 5%–10% of individuals and variants greater than 1 Mb in 1%–2%. In contrast to previous studies, we find limited evidence for stratification of CNVs in geographically distinct human populations. Importantly, our sample size permits a robust distinction between truly rare and polymorphic but low-frequency copy number variation. We find that a significant fraction of individual CNVs larger than 100 kb are rare and that both gene density and size are strongly anticorrelated with allele frequency. Thus, although large CNVs commonly exist in normal individuals, which suggests that size alone can not be used as a predictor of pathogenicity, such variation is generally deleterious. Considering these observations, we combine our data with published CNVs from more than 12,000 individuals contrasting control and neurological disease collections. This analysis identifies known disease loci and highlights additional CNVs (e.g., 3q29, 16p12, and 15q25.2) for further investigation. This study provides one of the first analyses of large, rare (0.1%–1%) CNVs in the general population, with insights relevant to future analyses of genetic disease.
Introduction
Copy number variants (CNVs) are insertions, deletions, and duplications of genomic sequence ranging from a kilobase to multiple megabasepairs in length and are major contributors to human genetic diversity.1–5 CNVs are known to influence both normal and disease variation,6 and there are at least two distinct, but nonexclusive, models of CNV-phenotype associations. One model involves common copy number polymorphisms (CNPs) often with multiple allelic states defined by variation in copy number and/or genomic structure. CNP genes are enriched for biological functions associated with drug response, immunity, and sensory perception, among others.7–9 Under this model, common variants that change the dosage of genes or other functional elements influence phenotypes such as HIV-1/AIDS susceptibility (MIM 609423),10 Crohn's disease (MIM 266600),11 and glomerulonephritis in systemic lupus erythematosus (MIM 152700).12
A second model involves rare CNVs that delete or duplicate typically larger genomic segments and exist in fewer allelic states (i.e., hemizygous or trisomic). These CNVs are highly penetrant and short-lived in the population, either occurring de novo or persisting for only a few generations within a pedigree. A large fraction of these variants arise by nonallelic homologous recombination (NAHR) between segmental duplications or low-copy repeats. Originally defined as genomic disorders,13 there are now dozens of clinically recognized syndromes, associated with cognitive deficits, diabetes, epilepsy, and other traits, that result from recurrent NAHR-mediated events. In some cases, variants that overlap but are distinct lead to a similar syndrome,13–17 whereas in other cases the phenotype is more variable.18–21 Additionally, recent studies of autism (MIM 209850) and schizophrenia (MIM 181500) found a bulk excess of rare CNVs in affected individuals relative to those unaffected, suggesting that some of the rare variants present in affected individuals are pathogenic.22–25 Thus, although only a limited number of rare variants have been definitively associated with disease, it is likely that a large fraction of CNV-trait associations conform to a “common disease-rare variant” hypothesis, in contrast to the “common disease-common variant” hypothesis that underpins most genome-wide association studies.
Understanding the extent to which rare CNVs influence phenotypes requires deep analyses in both disease and normal populations. Previous studies of copy-number variation in human populations have largely been restricted to hundreds of individuals and therefore unable to distinguish variants that are truly rare (<1%) from those variants that are polymorphic but at low frequency.2,26 Recent studies have begun to expand to substantially larger sample collections, but focused on analyses of specific diseases rather than the broader genomic effects of large, rare CNVs.1–4,23,25 Here, we analyze copy number variation in approximately 2500 apparently normal adult individuals by using Illumina genome-wide SNP genotype data. We find that large variants are individually rare (each found in one or a few individuals) but collectively frequent (most individuals carry one or more large CNVs) in human populations and that NAHR is a substantial mechanistic contributor to both rare and common CNVs. Analyses of size and gene content in relation to allele frequency indicate that CNVs are as a class under strong purifying selection and thus likely to be phenotypically influential. Finally, combining our data with a meta-analysis of published variants, we demonstrate the utility of our resource by suggesting candidate neurological disease loci. We describe one of the first analyses of large CNVs segregating at rare frequencies (0.1%–1%) in the general population, a framework to leverage this information in a disease study, and implications of our results for future genetic analyses.
Material and Methods
Sample Collection and SNP Genotyping
Data were obtained from three studies (Table 1): Pharmacogenomics and Risk of Cardiovascular Disease (PARC), neurologically normal individuals identified at the National Institute for Neurological Disorders and Stroke (NINDS), and the Human Genome Diversity Panel (HGDP). The PARC samples are a subset of the cohorts used in two statin trials, CAP and PRINCE,27,28 and consist of 960 middle-age (40–70 years) individuals of European descent living in the United States with moderately high levels of total cholesterol. NINDS samples were obtained from the NINDS Human Genetics Resource Center DNA and Cell Line Repository. Genotype data from NINDS were derived from two sets of neurological disease controls totaling 790 people and consist of individuals of European descent with no family history of or any first-degree relative with amyotrophic lateral sclerosis, ataxia, autism, brain aneurysm, dystonia, Parkinson disease, or schizophrenia. The HGDP consists of 1064 individuals sampled from 51 different world populations.29,30 Although a subset of the HGDP (n = 485) has been previously analyzed for CNVs,31 the analysis here was performed with independently generated genotype data and analysis tools. Our analysis of the HGDP cohort was restricted to a subset of individuals previously identified to exclude likely pairs of second-degree relatives.32 SNP genotyping data for PARC, HGDP, and NINDS were generated at the University of Washington, Stanford University, and the NINDS, respectively. PARC samples were genotyped with Illumina 317K arrays, HGDP samples were typed with Illumina 650Y arrays, and NINDS were typed with a combination of Illumina 550K and 317K with supplemental 240S SNP arrays (317K plus 240S arrays have nearly identical coverage to 550K arrays). All genotyping was performed with DNA from lymphoblastoid cell lines (LCLs), with the exception of the PRINCE subset of PARC, which used DNA from peripheral blood. Samples from any study were eliminated if they exceeded acceptable intensity noise levels or harbored obvious cell-line artifacts or mosaicism (Figure S1 available online). Intensity measurements from SNP arrays were reclustered according to the following groupings based on array platform and substudy: PARC-CAP, PARC-PRINCE, NINDS-550K, NINDS-317K+240S, and HGDP. Underlying genotyping data from these samples are being made available online (see Web Resources). This work was approved by the Human Subjects Review Committees at the University of Washington, the National Institute on Aging, and Stanford University for the PARC, NINDS, and HGDP samples, respectively.
Table 1.
Summary of Data Sets
| Data Set | Platform | # Samples (before QC) | Total CNVs (HS Overlapping)a | Dels/Sample (kb) | Dups/Sample (kb) | HS Enrichmentb |
|---|---|---|---|---|---|---|
| PARCc | HumanHap300 | 936 (991) | 2664 (472) | 1.86 (179) | 0.98 (187) | 2 |
| NINDSd | HumanHap550e | 671 (790) | 4641 (932) | 5.25 (318) | 1.67 (270) | 2.2 |
| HGDPf | HumanHap650Y | 886 (941)g | 6538 (1805) | 5.3 (328) | 2.08 (288) | 3.3 |
| all | N/A | 2493 (2722) | 13843 (3209) | 4.00 (269) | 1.56 (245) | 2.5 |
HS, rearrangement hotspot; for more details, see Material and Methods or 35.
Hotspot enrichment, expressed as the ratio (# of overlapping CNVs/bp encompassed) for rearrangement hotspots versus nonhotspots.
More details regarding data set may be found at 27,28.
More details regarding data set may be found at http://ccr.coriell.org/ninds.
A subset of the data was generated as a combination of HumanHap300 plus supplemental 240S SNP Arrays.
More details regarding data set may be found at 30.
Individuals likely to be related were excluded.
CNV Discovery
All probe coordinates were mapped to the human genome assembly build 35 (hg17) by using liftOver. We used a previously developed method, based on a Hidden Markov Model (HMM), to identify homozygous deletion, heterozygous deletion, and duplication events (Figure S1).33,34 This method considers transformed LogR ratio and b-allele frequency (BAF) measurements for each probe on a per sample basis (Figure 1). Specifically, we specified a 4-state HMM that took as input the LogR intensities, transformed into standard normal measurements (Z-scores) over a chromosome, and the square root of a quantity we termed the b-deviation. The b-deviation of a probe was defined as the deviation from the expected BAF given the genotype. For homozygotes, this was defined as the minimum of BAF and 1-BAF, whereas for heterozygotes, this was defined as the absolute value of BAF −0.5. For failed genotypes or CNV probes, the b-deviation was the minimal value of these.
Figure 1.
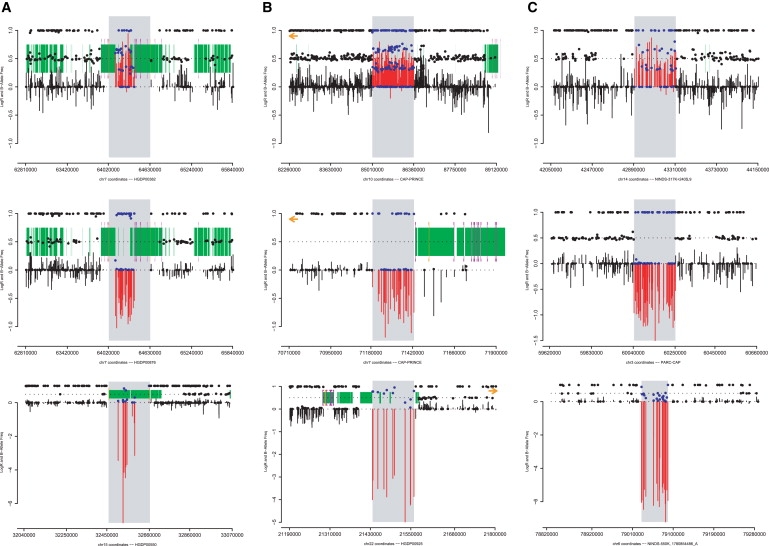
Examples of CNVs by Location and Type
Typical examples of duplications (top row), heterozygous deletions (middle row), and homozygous deletions (bottom row) as detected by using SNP arrays classified as rearrangement hotspot mediated (A), hotspot associated (B), or nonhotspot (C) (see Material and Methods for definitions). The plots show LogR ratio (vertical bars), b-allele frequency (solid points), segmental duplications in the reference assembly (green blocks), and the locations of rearrangement hotspots (purple brackets).4,35 CNVs are highlighted by gray rectangles, contrasting the LogR ratio (red) and b-allele frequency (blue) with flanking regions (black). Duplications are characterized by increased LogR ratio and heterozygous b-allele frequencies in multiple clusters, corresponding to “AAB” and “ABB” SNP genotypes, instead of a single cluster at 0.5 (“AB”). Heterozygous deletions have decreased LogR ratio and display a loss of heterozygosity. Homozygous deletions have an extremely low LogR ratio and display b-allele frequencies that fail to cluster.
The HMM analyzed each chromosome of each sample separately. HMM state assignments were merged into segments according to the following criteria: consecutive probes of the same state less than 50 kb apart were merged, and if two segments of the same state were separated by an intervening sequence of ≤5 probes and ≤10 kb, both segments and intervening sequence were called as a single variant. This yielded 460,395 HMM calls (Figure S1). Before further analysis, samples were eliminated if the hybridization did not have genome-wide LogR standard deviation ≤0.25, absolute value of the average LogR ≤0.1, and average b-deviation <0.05.
We subsequently divided putative CNVs into two categories: “small” CNVs <100 probes and <1 Mb in length and “large” with ≥100 probes or ≥1 Mb in length. All large CNVs were manually curated. Small CNVs were subject to automated filtering. Homozygous deletions were required to have ≥3 probes, median LogR Z-score ≤−4, and mean b-deviation ≥0.1 or ≥3 probes and median LogR Z-score ≤−8; heterozygous deletions were required to span ≥10 probes, have LogR Z-score ≤−1.5, and less than 10% of probes called as heterozygous; for duplications we required ≥10 probes, LogR Z-score ≥1.5, and b-deviation among heterozygote probes ≥0.075.
Rearrangement hotspots have been previously defined4,35 as regions of the genome from 50 kb to 10 Mb in size that are flanked by large segmental duplications1,3,13 of high sequence similarity (≥10 kb, ≥95% identity). These flanking duplications can result in NAHR during meiosis and therefore predispose the region to the generation of novel deletion/duplication events. Many CNVs significantly associated with human diseases map within or are bracketed by segmental duplications.14,17–20,23,25,36 Because of their significance in disease studies, rearrangement hotspots that were not identified as variant by the HMM were screened through the intensity statistic filters described above and manually inspected for false positives. This small set of calls was excluded in analyses assessing segmental duplication and hotspot enrichment to avoid ascertainment bias in the results.
All HMM CNVs and hotspot calls by intensity statistics were pooled together into a set of 18,556 variants. We manually merged calls within 1 Mb that appeared to be a result of HMM overfragmentation and discarded large calls that were possible cell-line artifacts, leaving a set of 16,751 calls. Finally, samples with >25 calls, >2 possible artifacts or false positives found during inspection of large HMM CNVs, or >2 possible artifacts during merging of HMM calls were excluded from further analysis, leaving a final set of 13,843 CNVs (Table 1). All CNV calls are listed in Table S1.
CNV Validation
We carried out validation by using array-CGH on 12 samples with a total of 98 inferred CNVs. Samples were chosen based on availability and came from the HGDP. Samples were hybridized on NimbleGen HD2 arrays with a previously characterized reference, NA15510.5 Array comparative genomic hybridization (CGH) data was normalized with qspline normalization and analyzed with the SegMNT algorithm with NimbleScan software. Given the scale of our analysis, our primary goal was to maintain high specificity to minimize the number of false-positive CNVs. However, we also assessed the extent to which CNVs may be missed in these samples in two ways. First, we considered variants inferred by CGH-segMNT from the NimbleGen array-CGH data. For this analysis, regions with known CNVs in the reference sample5 were excluded. In regions for which there was adequate (≥10) probe coverage on Illumina arrays, the fraction of CNVs inferred by NimbleGen-CGH detected via our Illumina CNVs ranges from 0.55 to 0.83, depending on the NimbleGen-CGH cutoff used (Table S3B). Using a more stringent Z-score criterion for Illumina calls increases the validation rate, but decreases the fraction of detected NimbleGen-CGH CNVs (Table S3B). Second, for regions with adequate (≥10) probe coverage, we compared the frequency of common CNVs in a previous study26,37 to those detected in our analysis. Comparing the observed number of CNVs to those expected based on frequencies in 26 we estimate a similar level of sensitivity (∼60%; data not shown).
Data Analysis
We defined rearrangement hotspots similar to previous studies4 as regions 50 kb–10 Mb in length flanked by segmental duplications >10 kb in length with >95% sequence identity. CNVs were annotated as “hotspot mediated” with respect to a hotspot if the intersection of the CNV and the predefined hotspot spans >90% of probes in the inferred CNV and >90% of probes in the hotspot. CNVs overlapping rearrangement hotspots but failing to meet these criteria were classified as “hotspot associated.” All other CNVs were classified as “nonhotspot.” CNV lengths were calculated based on the distance between the first and last array probes internal to the variant. For the purposes of calculating event frequencies, two types of CNV-region assignments were generated. When event frequency was the only parameter of interest, copy number variable regions (CNVRs) were defined by merging CNVs from different samples with any amount of overlap; this provides an upper bound on allele frequency for any given region of the genome. Alternatively, when comparing gene content, CNV length, and event frequency, CNVs from different samples were treated as allelic events only if their estimated start and end breakpoints were within 50 kb of one another. CNV gene content was determined with RefSeq gene annotation from the UCSC Genome Browser. CNV enrichment statistics were calculated based on 100 random permutations of the start coordinates of all HMM-identified CNVs, excluding those identified at hotspots based on intensity statistics alone.
Results
CNV Discovery
We analyzed Illumina genome-wide SNP array data from three genotype collections (Table 1): the Pharmacogenomics and Risk of Cardiovascular Disease project (PARC) samples, neurologically normal individuals from the National Institute for Neurological Disorder and Stroke (NINDS) Human Genetics Resource Center DNA and Cell Line Repository, and the Human Genome Diversity Panel (HGDP) samples. The individuals from PARC that we studied come from a subset of the cohorts used in two statin trials, CAP and PRINCE, and consist of 991 middle-age (40–70 years) individuals of European descent living in the United States with moderately high levels of total cholesterol.27,28 These samples were genotyped with the Illumina Human 317K SNP array. Genotype data from NINDS were derived from two sets of neurological disease controls totaling 790 people tested with the Illumina Human 550K array.38 These individuals have undergone patient interviews and were found to be free of symptoms of major neurological disease (see Material and Methods). Genotype data for the HGDP includes 1064 individuals sampled from 51 different world populations29 genotyped on the Illumina Human 650Y SNP array.30 Although a subset of the HGDP (n = 485) has been previously analyzed for CNVs,31 the analysis here was performed with independently generated genotype data and analysis tools. We excluded HGDP individuals likely to be related.32
We used a previously developed method, based on a Hidden Markov Model (HMM), to identify homozygous deletion, heterozygous deletion, and duplication events (Figure S1; Material and Methods).33,34 After quality control, we identified a total of 13,843 CNVs in 2,493 unrelated DNA samples (Table 1; Figure S2). These CNVs form 3,476 nonoverlapping CNVRs, of which 435 contain both deletions and duplications.
CNV Validation
The methodology we used has been extensively validated, and given the scope of this study, we increased the stringency of our thresholds beyond that used previously (see Material and Methods and 33). However, we also performed direct validation on 12 HGDP samples by performing comparative genomic hybridization (array-CGH) with NimbleGen HD2 oligonucleotide arrays, with a well-characterized reference sample.5 We examined 98 CNVs detected in 12 HGDP samples. By manual inspection, 64 sites were confirmed by array-CGH (Figures S3 and S4). Because of a CNV in the reference DNA sample, an additional 11 sites could not in principle be confirmed by array-CGH, but correspond to known, common CNVs (Figure S5A).2,3,39,40 Three additional sites were ambiguous, and 20 sites were not validated by array-CGH (Table S2; Figure S5B). Thus, our overall validation is 77% (Table S3A). We note that all homozygous deletions validated (12/12), and among heterozygous deletions and duplications, nonvalidated variants tend to be smaller (average of 13.2 versus 23.8 probes, p = 3.8 × 10−3, one-tailed Wilcoxon rank-sum) with less extreme Z-scores (Table S3A). At a threshold of 100 kbp, for example, ∼86% (19/22) of events validate, and all nine variants that spanned more than 30 probes validate. Additionally, given the potential for false negatives in array-CGH, this should be regarded as a conservative estimate of the true positive rate.
CNV Distribution and Segmental Duplications
We considered both the locations (Figure 2) and sizes (Figure S6) of all CNVs in the context of segmental duplications and rearrangement hotspots. As expected,2,4 we found fewer homozygous deletions than heterozygous deletions (464 versus 7737; Figure S6). At smaller sizes (∼100 kb or less), deletions are more frequent than duplications, with the opposite holding true for larger variants. The relative enrichment of deletions at smaller sizes may reflect higher de novo rates of occurrence of deletions,41 whereas their depletion at larger sizes is consistent with large deletions being more deleterious than duplications. An important caveat is that our discovery procedure emphasizes specificity over sensitivity (see Material and Methods and 33), and that power is dependent on probe counts, implying that we are underestimating the true extent of copy number variation in these genomes. Our discovery power is platform dependent and weaker for smaller variants (see below). In addition, probe coverage on SNP arrays tends to be depleted within duplicated regions of the reference assembly; for example, 0.9% of probes on the 317K SNP array are within duplications in contrast with ∼5% of the genome. However, duplications in the reference assembly are known to be enriched for copy number variation.2,4
Figure 2.
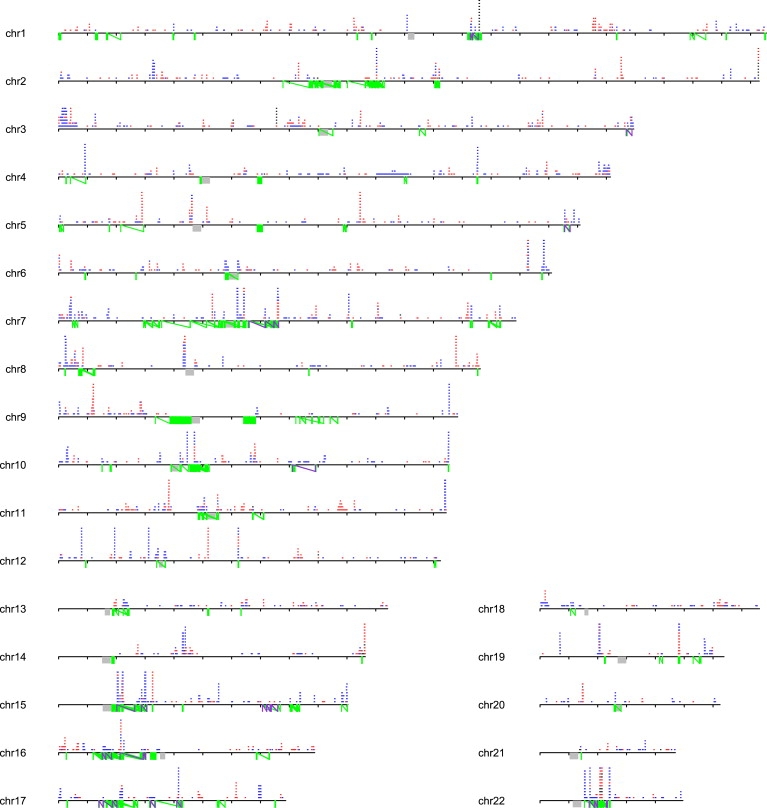
Autosomal Landscape of Large CNVs
Large CNVs are >100 kbp. Duplications (blue), deletions (red), and homozygous deletions (black) are depicted based on analysis of 2493 individuals. Chromosomes are drawn to scale (tick marks indicate 10 Mb), with the position of centromeres (gray) and predicted rearrangement hotspots (green lines connected by a diagonal) indicated. Those hotspots associated with disease are highlighted in purple. CNVs observed ten or more times for a given locus are cropped.
Rearrangement hotspots have been previously defined4,35 as regions of the genome from 50 kb to 10 Mb in size that are flanked by large (≥10 kb) duplications1,3,13 of high sequence similarity (≥95% identity). Depending on the overlap between a given CNV and the predefined genomic hotspot, we assigned CNVs as either hotspot mediated (intersection of the CNV and the predefined hotspot spans >90% of SNP probes in the CNV and >90% of SNP probes in the hotspot), hotspot associated (any CNV overlapping a hotspot that does not meet the 90% overlap criterion), or nonhotspot (Material and Methods; Table 1; Figure 2). CNVs classified as hotspot mediated are likely to have been generated through NAHR, whereas hotspot-associated CNVs occur in overlapping regions but are not necessarily NAHR events because of the discrepancies between the observed and expected breakpoints. We observe 2- to 3-fold enrichment for CNVs that are either hotspot mediated or hotspot associated relative to the number of base pairs encompassed (Table 1). Hotspot-mediated events form 32 CNVRs and have significantly higher population frequencies than hotspot-associated events (p = 7.7 × 10−6, one-tailed Wilcoxon rank-sum) and nonhotspot events (p = 2.7 × 10−9; Figure S7). We also find that 3,857 of 13,474 CNVs overlap segmental duplications, in contrast to a maximum overlap of 2,466 segments observed in 100 simulations in which CNV locations were randomly assigned to the genome (Table S4). More strikingly, we observe enrichment for pairs of related segmental duplications (>1 kb, >90% identity) near the breakpoints of CNVs, with 697 such events in the actual data versus a maximum of 42 in the randomized distributions (Table S4). The increased population frequencies of hotspot-mediated events and approximately 25-fold genomic enrichment of CNVs for flanking homologous segmental duplications are consistent with previous studies of fewer individuals.2,4,42 Importantly, here we demonstrate that NAHR is a major contributor to both common and rare copy number variation.
CNV Frequency and Burden
Within each study, we find a unimodal distribution of CNV counts, with an average of 3–7 variants and a global average of 540 kb (∼0.02% of the genome) of CNV DNA per person (Figure S2); as expected, more CNVs were identified with the higher-density array platforms because of their ability to detect smaller variants (Figure S2; Figure 3). We find that 65%–80% of individuals harbor a CNV of at least 100 kb in size, 5%–10% of individuals carry a variant at least 500 kb in length, and at least 1% of individuals harbor an event ≥1 Mb (Figure 3). Whereas at shorter lengths, the per individual CNV burden estimate is dependent on the array used (implying that we are underestimating the number of shorter CNVs; Figure S2 and 26), at larger lengths (>500 kb), differences resulting from genotyping platform largely disappear. Furthermore, PARC-CAP (DNA from cell lines) and PARC-PRINCE (blood-derived DNA) yield similar curves, suggesting that cell-line artifacts are not a major contributor to our estimates of CNV burden. Finally, comparing the two sets of neurological disease controls to either PARC or HGDP again yields no major differences. We conclude that these estimates of the impacts of large CNVs on individual human genomes are conservative but are likely to hold for the general human population.
Figure 3.
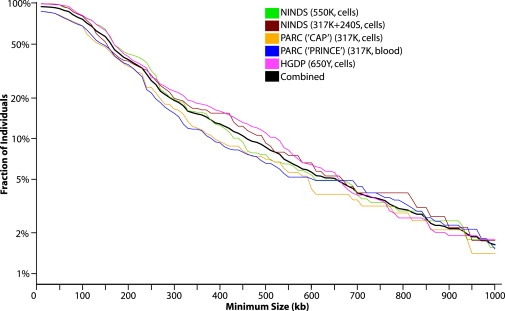
Cumulative Distributions of the Largest CNV per Individual According to Study
For 10 kb to 1 Mb in 10 kb intervals, the fraction of individuals containing one or more CNVs (y axis) of size greater or equal to a given size (x axis) is plotted according to study. Note that probe density has a significant impact at smaller CNV sizes, but that the cumulative distributions for blood-derived (PARC-PRINCE) and cell-line (PARC-CAP) DNA are similar. The average number of CNVs per individual varies by study from 3 to 7 (Figure S2).
We were also interested in the prevalence of copy number variation in human populations. The CNVs we identified collectively span ∼16% of the autosomal genome, suggesting that significant portions of the genome have the potential to vary in copy number within the normal population.2 However, polymorphic CNVRs (>1%) represent only 0.9% of the genome, whereas ∼6% of the genome is variant in CNVRs found in only one of ∼2500 individuals, indicating that the bulk (as measured by nucleotides) of the observed copy-number variation is present at ∼0.02%–1% frequency. We subsequently examined the relationship between frequency, CNV size, and gene content in greater detail (Figure 4). Because CNVRs defined by any amount of overlap could represent very different regions with little overlap (and thus dramatically affect the estimated gene content), here we calculated frequency by calling two CNVs as allelic only when the start and end coordinates of a CNV from one sample are within 50 kb of a CNV from another sample. We observe that 71% of individual CNVs (94% of CNV loci) larger than 100 kb are rare (<1%), and events >500 kb are heavily enriched for events seen in only one individual (p = 9.2 × 10−8, Pearson's chi-square; Figure S8). Furthermore, after controlling for length, rare CNVs harbor more genes than common events (p = 0.04, one-tailed Wilcoxon rank-sum), and homozygous deletions are particularly gene poor (p = 4.7 × 10−4).
Figure 4.
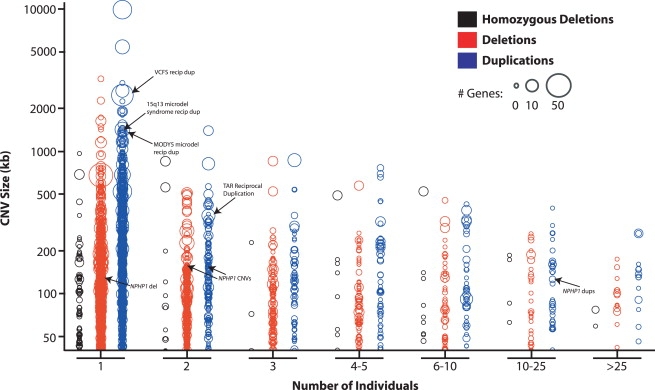
CNV Length, Gene Content, and Frequency Distributions
CNVs were plotted according to event type (color), length (y axis), frequency in the population (x axis, number of individuals from n = 2493), and number of RefSeq genes affected (circle size). To facilitate comparison across different platforms, events from different individuals were considered the same if their putative breakpoints were within 50 kb of one another. CNVs related to previously reported disease-causing variants are highlighted.
These observations are consistent with large CNVs being generally deleterious by virtue of their effects on gene dosage, consistent with previous results from smaller studies.2,43 We note that one advantage in this study is the discrimination of allele frequencies at or below 1%, including robust distinction between truly rare (<1%) and low-frequency yet polymorphic variants. Variants observed in only a handful of samples out of thousands are very unlikely to be truly polymorphic (p < 1e-9 for variants observed in 5 of 2500, for example), whereas rare and polymorphic but low-frequency CNVs are indistinguishable when analyzing dozens or hundreds of samples (e.g., HapMap). An important caveat is that incomplete sensitivity in our CNV discovery procedure (Table S3B) may nonuniformly bias our allele frequency estimates downwards; we are clearly underestimating the effects of smaller, more common CNVs, for example.26 However, our sensitivity is higher for larger events (Figure 3), and therefore would result in a bias opposite to the observed relationship between size and frequency (Figure 4).
Population Diversity
A previous study discovered CNVs within a subset of the HDGP samples and found a cumulative excess of CNVs in a few populations.31 Specifically, the Kalash, Melanesian, and Papuan populations were reported to harbor 20–30 CNVs per individual compared to a study-wide average of 7.9. We note that this study used independently generated SNP genotype data and a distinct CNV discovery algorithm31,40 from that used here. In our analysis of the same samples, we found that the Kalash, Melanesians, and Papuan individuals harbor an average of 6.4, 11.9, and 10.3 CNVs per individual, compared to a study-wide average of 7.4. Thus, although the Melanesian and Papuan harbor the highest number of CNVs on average in our analysis (Table S5), this elevation is much smaller than previously reported, and the Kalash individuals actually carry fewer CNVs than average.
Finding no evidence for population-specific undercalling in our analysis, we sought to determine if this discrepancy is due to biased overcalling in the previous analysis 31. Within the previously published SNP array and CNV annotation data, we compared standard deviation in the LogR ratio, one of the key intensity measures used to infer the presence of a CNV (Figure 1), and the number of CNVs identified. We found a strong positive correlation between average intensity noise (standard deviation in LogR) and the number of inferred CNVs within the previous study (p = 9.94 × 10−7; Figure S9A). At the sample level, there is an even stronger correlation (p ≈ 1 × 10−50; Figure S9B): samples with lower variance in LogR ratio (standard deviation < 0.18) were inferred to carry ∼5 CNVs per individual, whereas samples with intermediate variance were annotated with ∼11, and those samples with the highest variance (standard deviation > 0.25) had ∼33 CNVs per individual. We also found that intensity variance correlated strongly with the array ID (Figure S9C), and that the SNP array data used here30 showed no noise inflation in the same samples. These observations suggest that a batch artifact in the DNA handling or processing in the previous analysis31 disproportionately affected CNV annotation in particular HGDP populations. Assuming that the higher-variance samples in 31 carry similar numbers of actual CNVs as the lower-variance samples, at least 52% of the previously annotated CNVs in the higher-variance samples are false positives. These data also suggest that the CNV identification algorithm used40 has a false discovery rate that is proportionally sensitive to intensity noise. Our analysis, which normalizes signal intensity to sample noise, does not display this correlation (Figure S9D).
Normal versus Pathogenic CNVs
Previous studies have documented a statistically significant excess of rare, large CNVs in autistic and schizophrenic individuals.22,24 To search for individual loci that are risk factors for neurological disease, we compared the CNVs in our study to published data from affected individuals in nine genome-wide studies of schizophrenia, autism, and mental retardation.19,22–24,44–48 We also included CNVs identified in the control individuals from a recent large study of schizophrenia,23 and we restricted our analysis to large variants (>500 kb) to minimize platform-specific differences in detection. In total, we assembled CNVRs from 6860 affected individuals and 5674 controls (Figure S10). To rank loci with regard to potential pathogenicity, we calculated p values (Fisher's exact test) for allele frequency differences of gains, losses, and total CNVs in affected versus control samples. Because this analysis used precalled CNVs spanning a diverse set of platforms, DNA samples, study design, and CNV-identification algorithms, the resulting p values should be considered exploratory and interpreted in this context. Also, because many of the samples analyzed in this study have not been screened for neurological disease (i.e., PARC and HGDP), there are potentially a small number of affected individuals in these groups. Thus, the observation that a variant is seen in this control panel does not preclude that variant from being pathogenic. Future studies would benefit from a larger number of controls, similar to the NINDS collection, that have been excluded for neurobehavioral or neurococognitive deficits.
Most of the top-scoring loci contained CNVs from multiple studies spanning multiple diseases (Table 2). The top CNVRs are previously known pathogenic rearrangement hotspots (Table 2; Figures 5A and 5B).14,19–21,23,25,49–51 Deletions at 22q11 (MIM 192430, 188400) have been identified as pathogenic whereas the reciprocal duplications have been suggested to be benign or cause a milder phenotype.50 Correspondingly, duplications at 22q11 received a lower p value rank than the deletions (Table 2). Additionally, many of the pathogenic CNVs appear in individuals with a disease different from the disease in which pathogenicity was originally described (Table S6). This may be an artifact of merging CNVs with distinct breakpoints (e.g., Figure 5B). However, the inferred breakpoints of CNVs identified in studies of distinct diseases often overlap perfectly or nearly so (Table S6; Figure 5). Additionally, some striking examples emerge such as the presence of the 17p11.2 PMP22 microdeletion, typically associated with heredity neuropathy with liability to pressure palsies (HNPP, [MIM 162500]), among patients with schizophrenia and autism.
Table 2.
Loci Enriched for CNVs in Autism, Mental Retardation, and Schizophrenia Identified by Disease Meta-Analysis
| Chr | Start | Stop | Length | Note | NAHRa | Total CNVs | Type | Disease CNVs | Control CNVs | Diseases and Studies | CNV p Value | Locus p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr15 | 27,015,263 | 30,650,000 | 3,634,737 | Prader-Willi/15q13 | yes | 19 | loss | 19 | 0 | schizophrenia23 | 1.08E-05 | |
| chr15 | 18,376,200 | 30,756,771 | 12,380,571 | 58 | gain | 45 | 13 | autism,19,22,45–47 mental retardation,44 schizophrenia,23 controls,23 this study | 2.69E-04 | 1.54E-07 | ||
| chr22 | 17,014,900 | 19,993,127 | 2,978,227 | VCFS | yes | 31 | loss | 31 | 0 | autism,19,45,47 mental retardation,44 schizophrenia23,48 | 7.93E-09 | |
| chr22 | 17,200,000 | 21,546,762 | 4,346,762 | 14 | gain | 9 | 5 | autism,19,45–47 schizophrenia,23 controls,23 this study | 0.330 | 9.53E-07 | ||
| chr1 | 142,540,000 | 146,059,433 | 3,519,433 | 1q21 | yes | 27 | loss | 24 | 3 | autism,19 schizophrenia,23 controls23 | 1.67E-04 | |
| chr1 | 142,800,580 | 146,009,436 | 3,208,856 | 15 | gain | 12 | 3 | autism,19,45 mental retardation,44 schizophrenia,23 controls23 | 0.041 | 2.16E-05 | ||
| chr22 | 45,144,027 | 49,509,153 | 4,365,126 | Terminal 22 del syndrome | no | 4 | loss | 4 | 0 | autism22,45,47 | 0.090 | |
| chr22 | 47,572,875 | 48,323,417 | 750,542 | 6 | gain | 5 | 1 | autism,19,46,47 schizophrenia,23 controls23 | 0.160 | 0.022 | ||
| chr16 | 29,470,951 | 30,252,473 | 781,522 | 16p11.2 | yes | 11 | loss | 8 | 3 | autism,19,22,46,47 controls23 | 0.186 | |
| chr16 | 29,474,810 | 30,235,818 | 761,008 | 7 | gain | 6 | 1 | autism,19,47 schizophrenia,23,24 this study | 0.100 | 0.039 | ||
| chr17 | 14,000,000 | 15,421,835 | 1,421,835 | CMT1A/HNPP | yes | 7 | loss | 6 | 1 | autism,19,46 schizophrenia,23 controls23 | 0.100 | |
| chr17 | 12,650,000 | 15,540,000 | 2,890,000 | 5 | gain | 4 | 1 | autism,45 mental retardation,44 schizophrenia,23 controls23 | 0.252 | 0.041 | ||
| chr16 | 60,141,700 | 61,581,600 | 1,439,900 | 16q21, CDH8 | no | 4 | loss | 4 | 0 | autism45 | 0.090 | |
| chr16 | 60,552,237 | 61,294,685 | 742,448 | 1 | gain | 1 | 0 | schizophrenia23 | 0.547 | 0.049 | ||
| chr11 | 78,120,000 | 85,610,000 | 7,490,000 | 11q14.1 | no | 3 | loss | 3 | 0 | mental retardation,44 schizophrenia23 | 0.164 | |
| chr11 | 84,304,683 | 85,042,205 | 737,522 | 1 | gain | 1 | 0 | schizophrenia23 | 0.547 | 0.090 | ||
| chr2 | 185,118,087 | 185,909,729 | 791,642 | 2q32.1 | no | 1 | loss | 1 | 0 | schizophrenia23 | 0.547 | |
| chr2 | 184,270,000 | 186,892,000 | 2,622,000 | 3 | gain | 3 | 0 | autism45 | 0.164 | 0.090 | ||
| chr15 | 82,573,421 | 83,631,697 | 1,058,276 | 15q25 | yes | 4 | loss | 4 | 0 | autism,46,47 schizophrenia23 | 0.090 | |
| chr15 | na | na | na | 0 | gain | 0 | 0 | none | 1 | 0.090 | ||
| chr9 | 140575 | 1175526 | 1,034,951 | 9p24 | no | 1 | loss | 1 | 0 | schizophrenia,23 | 0.547 | |
| chr9 | 206456 | 1599250 | 1,392,794 | 3 | gain | 3 | 0 | autism,45 schizophrenia,23 | 0.164 | 0.090 | ||
| chr3 | 197,179,156 | 198,842,299 | 1,663,143 | 3q29 | yes | 3 | loss | 3 | 0 | schizophrenia23,24 | 0.164 | |
| chr3 | 198,325,925 | 199,384,429 | 1,058,504 | 2 | gain | 1 | 1 | schizophrenia,23 controls23 | 1 | 0.252 | ||
| chr16 | 21,693,739 | 22,611,363 | 917,624 | 16p12 | yes | 5b | loss | 5b | 0 | autism,47 schizophrenia23 | 0.049 | |
| chr16 | 21,441,805 | 22,688,093 | 1,246,288 | 5 | gain | 2 | 3 | autism,47 schizophrenia,23 controls23 | 1 | 0.261 | ||
| chr16 | 80,722,684 | 82,227,917 | 1,505,233 | 16q23.3, CDH13 | no | 2 | loss | 0 | 2 | controls,23 this study | 1 | |
| chr16 | 80,737,839 | 82,208,451 | 1,470,612 | 4 | gain | 4 | 0 | autism,46 schizophrenia23,24 | 0.090 | 0.436 |
Indicates if there are large segmental duplications near breakpoints in hg17.
A deletion of ∼480 kb in size in a schizophrenic sample is included in this count.
Figure 5.
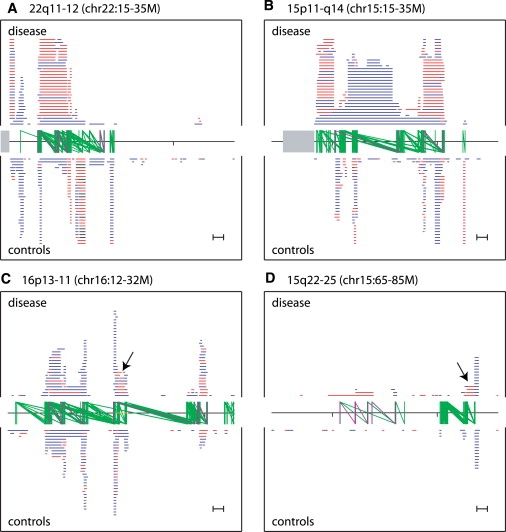
Comparison of CNVs >100 kb in Affected versus Unaffected Individuals at Four Selected Loci Scoring Highly for Potential Pathogenicity
Duplications, deletions, and homozygous deletions are plotted blue, red, and black, respectively, in human reference assembly coordinates (x axis in each plot). Tick marks are spaced 10 Mb apart, centromeres are indicated in gray, and hotspots are shown as two green vertical lines connected by a green diagonal. Scale in bottom right indicates 1 Mb. Rearrangement hotspots that have been associated with disease are highlighted in purple. Plotting is cropped after 30 overlapping CNVs at a given locus.
(A and B) Known disease loci.
(A) 22q11-12. Disease hotspots (left to right): VCFS, critical region; VCFS, distal region, Distal 22q11 deletion syndrome (MIM 611867).55
(B) 15q11-q14. Disease hotspots: Prader-Willi/Angelman Syndrome BP1-BP3 (MIM 176270, 105830), and 15q13.3 (MIM 612001).20
(C and D) Candidate disease loci.
(C) 16p11-13. An inversion-containing region found in 7/8 analyzed HapMap samples42 has been colored orange along the x axis. Disease hotspots from left to right: 16p13 deletion syndrome distal and proximal regions,56 16p11.2-p12.2 deletion syndrome,15 and 16p11 region associated with autism.19,49
(D) 15q22-25. Disease hotspots from left to right: 15q24 deletion syndrome BP0-BP1, BP1-BP2, and BP2-BP3.17
We find other loci that are of lower rank, but which are suggestive of being pathogenic. Based on observing deletions in four affected individuals, a recent study23 suggested 16p12 (chr16:21.7-22.6M) as a candidate risk locus for schizophrenia. Our meta-analysis lends support to this hypothesis, but also suggests that the locus may be more broadly related to neurological disease, by highlighting a published deletion present in an autistic individual47 and none in an additional 2493 controls (this study). Of interest, but unknown relevance, is the observation that 7 of 8 HapMap samples analyzed by fosmid-end sequence pair mapping have a sequence-validated ∼1.1 Mb inversion event overlapping this locus.5,42 Manual inspection also suggests 3q29 (chr3: 196.9-198.9; Figure 5C) deletions as causative for schizophrenia. Although we find no control samples carrying this deletion, published data from two independent studies of schizophrenia include deletions in three affected individuals (Table 2).24 This is additionally supported by previous reports of a 3q29 microdeletion syndrome (MIM 609425) with clinical features that include mental retardation and other neurologic abnormalities.36 Finally, deletions of 15q25 (chr15: 82.5-83.6M; Figure 5D) are present in four affected individuals (two autism, two schizophrenia) identified in three independent studies; interestingly, an adjacent but nonoverlapping deletion within 15q25 has been reported in a child with mental retardation.52 In each of the above regions, there are large, highly identical duplications near the breakpoints suggesting that there are recurrent mutational events mediated by NAHR (Table 2; Figure 5D).
Discussion
Our results highlight the biological significance of rare copy number variation. We find that the majority of people harbor CNVs larger than 100 kb, in line with previous studies,23,26,38 and we robustly estimate that at least 1% of individuals carry variants greater than 1 Mb. The latter is well within the range considered pathogenic by some array-based studies.44 With CNVRs defined by any overlap as an upper bound on CNV frequency, ∼61% of observed CNVs (98% of CNVRs) larger than 100 kb are present at frequencies less than 1%. All events larger than 1 Mb were observed in only one or two normal individuals, and we also observe that rare variants are comparatively gene enriched. Thus, although large variants are commonly seen in human populations, these variants are generally deleterious in relation to both their size and gene content. This conclusion is consistent with results from previous analyses restricted to smaller sample sizes,2 and with recent experiments conducted on high-density arrays showing that common CNVs tend to be very small (<10 kb).26
Within the HGDP, we observe significantly less population-specific variation in total CNV content than previously reported. Our analysis suggests that an artifact in sample handling and data analysis contributed significantly to the previously reported excess of population-stratified variants.31 Deeper population screens to assess the distribution of large and rare CNVs in the human population are clearly warranted, because although such variants may segregate within specific populations because of genetic drift, others may contribute disproportionately to disease susceptibility or alternatively be adaptive within those populations.
In this analysis, we find a 25-fold enrichment for CNVs between pairs of homologous segmental duplications (Table S4). This effect is most striking for the largest CNVs (Figures 2 and 5) and replicates earlier surveys that implicate NAHR as a substantial contributor to the spectrum of copy number variation in human populations.2–4 We also demonstrated that predicted NAHR-mediated events occur more frequently across both rare and polymorphic CNVs (Figure S7). These results have relevance to existing genome-wide association studies for several reasons. First, because of recurrence, NAHR-driven mutations are less likely to be effectively “tagged” via linkage disequilibrium with neighboring SNPs, even when appearing at polymorphic frequencies. Second, SNP arrays cannot directly detect many known variants and particularly lack probe coverage (and therefore detection power) in and around duplicated sequences.33 Thus, assessment of variation at both hotspots and their breakpoints is currently incomplete, and the actual contribution of NAHR is underestimated here42 and in other SNP-based studies of copy number variation. Third, despite this bias, we found many CNVs that affect the breakpoints of rearrangement hotspots (Figures 2 and 5). These polymorphisms alter the number of duplicated copies at NAHR breakpoints and therefore may change the likelihood of a future mutation. Such a mechanism may explain how diseases caused by dominant, rare, sporadic CNVs could exhibit signatures of heritability: variants affecting potential NAHR breakpoints may be commonly segregating risk factors (one generation removed) even if the pathogenic CNV is itself not heritable (or only briefly so). Pathogenic microdeletions at 17q21.3, which in all known cases originate from a parental chromosome bearing a large inversion with a duplication architecture distinct from the reference assembly, is one clear example of this phenomenon.16,53 Collectively, these observations imply that variation within hotspots and their breakpoints is understudied and potentially critical to the genetic basis for human disease.
Although it is becoming clear that rare CNVs in general are important to common traits, a major challenge remains to identify individual variants that are pathogenic. One solution to this challenge will be in the accumulation of very large sample sets. We conducted a meta-analysis of more than 12,500 samples from 11 collections, including several neurological disease studies and two large control sets. Although a subset of our control set had not been screened for neurologic disease, this analysis clearly identifies known pathogenic loci, including the rearrangement hotspots at 22q11.2, 15q13.3, and the recently described 1q21.31.21,23,25 In addition, it provides further support for deletions at 16p12 as causative for neurological disease, indicates that deletions at 3q29 may be associated with schizophrenia in addition to mental retardation, and identifies loci that to our knowledge have not been previously reported and may be worthy of follow-up, in particular hotspot-mediated deletions at 15q25.2. We also note that seemingly diverse diseases (autism, mental retardation, and schizophrenia) are often associated with the same CNVR; although in some cases this may result from breakpoint-resolution artifacts, individuals diagnosed with one disease often carry CNVs associated with a distinct disorder (Table S6; Figure 5). These observations reinforce the conclusions from recent studies showing that similar CNVs are pathogenic in patients affected by distinct (and often multiple) neurological diseases.19,21,50,54 Imperfect diagnoses or individuals with several distinct diseases may account for this observation. However, an alternative explanation is that these loci are more general risk factors with the particular manifestation sensitive to genetic modifier or environmental effects. In any case, expanded collections of reliable, high-resolution CNV maps in both healthy and disease individuals will be critical for better characterizing the biological impact of rare CNVs in human populations.
Supplemental Data
Supplemental Data include ten figures and six tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Eichler lab structural variation data, http://hgsv.washington.edu
HGDP SNP genotyping data from Li et al.30, http://hagsc.org/
HGDP SNP genotyping data analyzed in Jakobsson et al.31, http://neurogenetics.nia.nih.gov/paperdata/public/
NimbleScan software, http://www.nimblegen.com/products/software/index.html
NINDS Human Genetics Resource Center DNA and Cell Line Repository, http://ccr.coriell.org/NINDS
NINDS sample genotype data are available from dbGAP, http://www.ncbi.nlm.nih.gov/gap
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PARC genotype data are available from pharmGKB, http://www.pharmgkb.org/
UCSC Human Genome Browser (liftOver utility and RefSeq genes annotation), http://genome.ucsc.edu/
Acknowledgments
We would like to thank A. Singleton for sharing genotype data, generated with support from the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services (Z01-AG000932-01). A.I. is supported by the National Human Genome Research Institute Training Grant (T32 HG00035). G.M.C. is supported by a Merck, Jane Coffin Childs Fellowship P.M.R. has received research support relevant to the content of this manuscript from the National Heart, Lung, and Blood Institute, the National Cancer Institute, the Donald W. Reynolds Foundation, Roche Diagnostics, and Amgen, Inc. D.I.C. acknowledges support from the Donald W. Reynolds Foundation and the National Institutes of Health. The PARC project is supported by the National Heart, Lung, and Blood Institute (HL01069757). E.E.E. acknowledges the support of the National Institutes of Health (HD043569, HG004120) and is an investigator of the Howard Hughes Medical Institute. The authors have no conflicts of interest to declare.
References
- 1.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Maner S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 4.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 6.Feuk L., Carson A.R., Scherer S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 7.Cooper G.M., Nickerson D.A., Eichler E.E. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet. 2007;39:S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 8.Trask B.J., Massa H., Brand-Arpon V., Chan K., Friedman C., Nguyen O.T., Eichler E., van den Engh G., Rouquier S., Shizuya H., Giorgi D. Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum. Mol. Genet. 1998;7:2007–2020. doi: 10.1093/hmg/7.13.2007. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen D.Q., Webber C., Ponting C.P. Bias of selection on human copy-number variants. PLoS Genet. 2006;2:e20. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez E., Kulkarni H., Bolivar H., Mangano A., Sanchez R., Catano G., Nibbs R.J., Freedman B.I., Quinones M.P., Bamshad M.J. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 11.Fellermann K., Stange D.E., Schaeffeler E., Schmalzl H., Wehkamp J., Bevins C.L., Reinisch W., Teml A., Schwab M., Lichter P. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 13.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 14.Christian S.L., Fantes J.A., Mewborn S.K., Huang B., Ledbetter D.H. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13) Hum. Mol. Genet. 1999;8:1025–1037. doi: 10.1093/hmg/8.6.1025. [DOI] [PubMed] [Google Scholar]
- 15.Ballif B.C., Hornor S.A., Jenkins E., Madan-Khetarpal S., Surti U., Jackson K.E., Asamoah A., Brock P.L., Gowans G.C., Conway R.L. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 16.Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays. Hum. Genet. 2008;124:1–17. doi: 10.1007/s00439-008-0513-9. [DOI] [PubMed] [Google Scholar]
- 17.Sharp A.J., Selzer R.R., Veltman J.A., Gimelli S., Gimelli G., Striano P., Coppola A., Regan R., Price S.M., Knoers N.V. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet. 2007;16:567–572. doi: 10.1093/hmg/ddm016. [DOI] [PubMed] [Google Scholar]
- 18.Mefford H.C., Clauin S., Sharp A.J., Moller R.S., Ullmann R., Kapur R., Pinkel D., Cooper G.M., Ventura M., Ropers H.H. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 20.Sharp A.J., Mefford H.C., Li K., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., De Gregori M., Ciccone R. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 25.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 27.Albert M.A., Danielson E., Rifai N., Ridker P.M. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70. doi: 10.1001/jama.286.1.64. [DOI] [PubMed] [Google Scholar]
- 28.Simon J.A., Lin F., Hulley S.B., Blanche P.J., Waters D., Shiboski S., Rotter J.I., Nickerson D.A., Yang H., Saad M., Krauss R.M. Phenotypic predictors of response to simvastatin therapy among African-Americans and Caucasians: the Cholesterol and Pharmacogenetics (CAP) Study. Am. J. Cardiol. 2006;97:843–850. doi: 10.1016/j.amjcard.2005.09.134. [DOI] [PubMed] [Google Scholar]
- 29.Cann H.M., de Toma C., Cazes L., Legrand M.F., Morel V., Piouffre L., Bodmer J., Bonne-Tamir B., Cambon-Thomsen A., Chen Z. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 30.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 31.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg N.A. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann. Hum. Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 33.Cooper G.M., Zerr T., Kidd J.M., Eichler E.E., Nickerson D.A. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat. Genet. 2008;40:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Day N., Hemmaplardh A., Thurman R.E., Stamatoyannopoulos J.A., Noble W.S. Unsupervised segmentation of continuous genomic data. Bioinformatics. 2007;23:1424–1426. doi: 10.1093/bioinformatics/btm096. [DOI] [PubMed] [Google Scholar]
- 35.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 36.Willatt L., Cox J., Barber J., Cabanas E.D., Collins A., Donnai D., FitzPatrick D.R., Maher E., Martin H., Parnau J. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon-Sanchez J., Scholz S., Fung H.C., Matarin M., Hernandez D., Gibbs J.R., Britton A., de Vrieze F.W., Peckham E., Gwinn-Hardy K. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- 39.McCarroll S.A., Hadnott T.N., Perry G.H., Sabeti P.C., Zody M.C., Barrett J.C., Dallaire S., Gabriel S.B., Lee C., Daly M.J., Altshuler D.M. Common deletion polymorphisms in the human genome. Nat. Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 40.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conrad D.F., Andrews T.D., Carter N.P., Hurles M.E., Pritchard J.K. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 44.de Vries B.B., Pfundt R., Leisink M., Koolen D.A., Vissers L.E., Janssen I.M., Reijmersdal S., Nillesen W.M., Huys E.H., Leeuw N. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Autism Genome Project Consortium Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christian S.L., Brune C.W., Sudi J., Kumar R.A., Liu S., Karamohamed S., Badner J.A., Matsui S., Conroy J., McQuaid D. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry. 2008;63:1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 49.Kumar R.A., KaraMohamed S., Sudi J., Conrad D.F., Brune C., Badner J.A., Gilliam T.C., Nowak N.J., Cook E.H., Dobyns W.B., Christian S.L. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 50.McDermid H.E., Morrow B.E. Genomic disorders on 22q11. Am. J. Hum. Genet. 2002;70:1077–1088. doi: 10.1086/340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lupski J.R., Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagenstaller J., Spranger S., Lorenz-Depiereux B., Kazmierczak B., Nathrath M., Wahl D., Heye B., Glaser D., Liebscher V., Meitinger T., Strom T.M. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zody M.C., Jiang Z., Fung H.C., Antonacci F., Hillier L.W., Cardone M.F., Graves T.A., Kidd J.M., Cheng Z., Abouelleil A. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet. 2008 doi: 10.1038/ng.193. in press. Published online August 10, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Antshel K.M., Aneja A., Strunge L., Peebles J., Fremont W.P., Stallone K., Abdulsabur N., Higgins A.M., Shprintzen R.J., Kates W.R. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J. Autism Dev. Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 55.Ben-Shachar S., Ou Z., Shaw C.A., Belmont J.W., Patel M.S., Hummel M., Amato S., Tartaglia N., Berg J., Sutton V.R. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am. J. Hum. Genet. 2008;82:214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hannes F.D., Sharp A.J., Mefford H.C., de Ravel T., Ruivenkamp C.A., Breuning M.H., Fryns J.P., Devriendt K., Van Buggenhout G., Vogels A. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J. Med. Genet. 2008 doi: 10.1136/jmg.2007.055202. in press. Published online June 11, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.