

Abstract
A current approach to mapping complex-disease-susceptibility loci in genome-wide association (GWA) studies involves leveraging the information in a reference database of dense genotype data. By modeling the patterns of linkage disequilibrium in a reference panel, genotypes not directly measured in the study samples can be imputed and tested for disease association. This imputation strategy has been successful for GWA studies in populations well represented by existing reference panels. We used genotypes at 513,008 autosomal single-nucleotide polymorphism (SNP) loci in 443 unrelated individuals from 29 worldwide populations to evaluate the “portability” of the HapMap reference panels for imputation in studies of diverse populations. When a single HapMap panel was leveraged for imputation of randomly masked genotypes, European populations had the highest imputation accuracy, followed by populations from East Asia, Central and South Asia, the Americas, Oceania, the Middle East, and Africa. For each population, we identified “optimal” mixtures of reference panels that maximized imputation accuracy, and we found that in most populations, mixtures including individuals from at least two HapMap panels produced the highest imputation accuracy. From a separate survey of additional SNPs typed in the same samples, we evaluated imputation accuracy in the scenario in which all genotypes at a given SNP position were unobserved and were imputed on the basis of data from a commercial “SNP chip,” again finding that most populations benefited from the use of combinations of two or more HapMap reference panels. Our results can serve as a guide for selecting appropriate reference panels for imputation-based GWA analysis in diverse populations.
Introduction
The recent availability of high-density single-nucleotide polymorphism (SNP) genotype databases from several human populations has facilitated the mapping of complex-disease loci in genome-wide association (GWA) studies. These databases, such as The International HapMap Project (2.5 to 4 million SNPs genome-wide1,2) and SeattleSNPs (∼7 Mb of gene-resequencing data), provide high-resolution information about allele frequencies and patterns of linkage disequilibrium (LD) among SNPs typed in the samples. They serve as “reference panels,” useful for diverse purposes in human genetics.
Information in reference panels can be leveraged in a mapping context by merging the reference genotype data with collections of data from individual GWA studies (Figure 1). Because typical GWA studies contain genotype data on, at most, a few hundred thousand to a million SNPs, a very specific missing-data pattern emerges from the union of a reference panel with a GWA data set. That is, for most SNPs, observations exist for the reference panel but not for the GWA study (Figure 1D). By modeling the pattern of LD in the reference panel and then applying the fitted model to the observed GWA study data, one can effectively impute the “missing” GWA SNP genotypes.3–9 Imputed genotypes at these SNP loci can then be used to test for association with disease, in the same way that testing occurs for SNPs that were actually genotyped in the GWA study.
Figure 1.
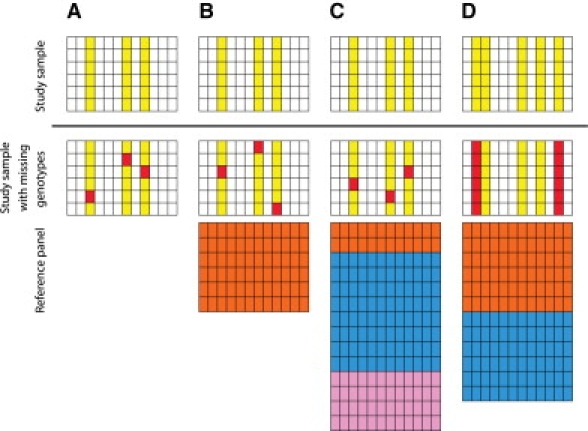
Schematic of Experimental Designs
The “Study sample” row represents data used in evaluting imputation accuracy in each design, with SNPs under consideration colored yellow. The “Study sample with missing genotypes” row represents corresponding data, with the unknown genotypes that are imputed colored in red. The “Reference panel” row represents example reference panels based on which imputation of missing genotypes or genotypes of untyped markers is performed. In a data set, each row corresponds to a haplotype and each column corresponds to a SNP position.
(A) Inference of missing genotypes, without additional reference haplotypes.
(B) Inference of missing genotypes, with a reference panel of haplotypes from a single reference sample (CEU, YRI, or CHB+JPT).
(C) Inference of missing genotypes, with a mixture reference panel, formed by the taking of a specified ratio of haplotypes from the HapMap CEU, YRI, and CHB+JPT samples.
(D) Inference of genotypes of untyped markers, with a mixture reference panel, formed by the aggregation of two or more HapMap samples.
We evaluated imputation accuracy in (A–C) for randomly masked genotypes, and in (D) for genotypes of untyped markers.
To date, most GWA studies have been conducted in populations that are well represented by the available high-density reference panels. Specifically, study samples have typically derived from populations of Northern European ancestry, for which the HapMap CEU panel—based on individuals of Northern and Western European descent, sampled in Utah—has provided additional information for imputation in association testing.10–13 However, for the purpose of genotype imputation, it is unclear how well the HapMap panels represent the patterns of genetic variation in other populations, particularly those that are more distant from the available panels, either in terms of demographic history or in terms of geographic proximity. Here, we attempt to evaluate the “portability” of these panels for imputation-based studies of diverse human populations; this work is analogous to recent assessments of the portability of informative SNPs chosen from reference panels in providing LD-based genomic coverage in diverse populations.14–18
Recently, two studies examined patterns of SNP variation in multiple human populations from around the world, providing data on samples from the Human Genome Diversity Project (HGDP) at more than 500,000 SNPs.19,20 We select one of these databases,19 and we evaluate the behavior of a missing-data-imputation algorithm in each of the sampled populations in several ways. First, using the sampled populations alone, we assess average imputation accuracy when imputing masked genotypes in the absence of a reference panel (Figure 1A). Second, we use the European American (CEU), Yoruba (YRI), and combined Chinese and Japanese (CHB+JPT) panels from the HapMap project in various combinations as reference panels, and we evaluate the properties of imputation in the sampled populations using the reference-panel data (Figures 1B and 1C). Finally, using data from a targeted high-density scan of several genomic regions on chromosome 21 in the HGDP samples,14,21 we also assess the accuracy with which genotypes of untyped markers can be imputed in these populations from the ∼500,000 typed SNPs and various combinations of HapMap reference panels (Figure 1D).
We find that when employing HapMap reference panels for imputation, genotypes from European HGDP samples are imputed with the highest accuracy, followed by samples from East Asia, Central and South Asia, the Americas, Oceania, the Middle East, and Africa. The choice of preferred HapMap reference panels for imputation in worldwide populations follows major geographic groupings. For most HGDP populations, we obtain additional gains in imputation accuracy when imputing genotypes on the basis of a mixture of available reference panels. These findings can serve as a basis for the application of imputation methods to analysis of genomic data in worldwide populations.
Material and Methods
Data
We examined a subset of 443 unrelated individuals from 29 populations in the HGDP-CEPH Human Genome Diversity Cell Line Panel, a worldwide collection of individuals from diverse locations.22 Individual genotypes obtained through the Illumina HumanHap550 SNP platform had been previously reported by Jakobsson et al.19 at 513,008 biallelic autosomal genetic markers (246 SNPs ultimately discarded by Jakobsson et al.,19 for production of their final data set of 512,762 SNPs, were included here as potentially informative for imputation).
For some analyses, we incorporated additional individuals for use as reference data in imputing missing genotypes. The reference data consisted of phased haplotypes of 210 individuals from the International HapMap Project:1,2 60 European Americans sampled from Utah, USA (abbreviated CEU), 60 Yoruba individuals from Ibadan, Nigeria (YRI), 45 Chinese individuals from Beijing, China (CHB), and 45 Japanese individuals from Tokyo, Japan (JPT). The phased HapMap data (release 21) were downloaded from the HapMap phase II data website (see Web Resources). The CHB and JPT haplotypes were combined into a single panel (CHB+JPT), and the specific origins of individual haplotypes (either CHB or JPT) were ignored. The CEU and YRI sets consisted of parents from trios; the offspring were omitted from our study but had been used in inferring haplotypes in the parents. A total of 1,958,375 autosomal markers that were polymorphic in the set of 210 HapMap individuals were used in our analyses. All except two of these SNPs (rs7008731 and rs13332778) were separately polymorphic in the CEU, YRI, and CHB+JPT panels.
In some analyses, we used data from Conrad et al.,14 in which some of the genotypes imputed with the data of Jakobsson et al.19 were measured directly in the same HGDP samples. These analyses used an updated version of the Conrad et al.14 data from Pemberton et al.21
LD-Based Imputation
Multiple models exist for accurate imputation of missing genotypes on the basis of LD information.4–6,8,23–26 For our investigations of variation in genotype-imputation accuracy across populations, we used a recent implementation of a model related to the approach of Li and Stephens:27 the Markov Chain Haplotyping algorithm (MACH-1.0.15) of Li et al.3 (see Web Resources).
The intuition underlying this imputation approach is that collections of individuals, even those who are “unrelated,” share short stretches of DNA sequence derived identically by descent from their common ancestors. Once these stretches are identified with the use of a set of SNPs, it is possible to probabilistically predict alleles for intervening SNPs that are not measured in a given individual but are measured in other individuals. Using a hidden Markov model, the algorithm resolves a collection of unphased genotypes into imperfect mosaics of several “template” haplotypes, from which it obtains an imputation, or a “best guess,” of each unknown genotype in each individual under consideration. All of our analyses rely on these “best guess” imputations, ignoring uncertainty in the genotype estimates. Exact software settings are given in the Appendix.
Inferring Missing Genotypes without Additional Reference Individuals
To assess the impact of the proportion of missing genotypes on imputation accuracy in each population, we masked a fraction of the genotypes at random, and we then compared the estimated genotypes to the actual, masked genotypes (Figure 1A). The proportion of missing genotypes varied between 5% and 50%, with a 2.5% increment. That is, each diploid genotype was masked independently with probability equal to the specified proportion of missing genotypes. The proportion of correctly imputed alleles is reported as “imputation accuracy” throughout our analyses. For example, if the correct genotype was homozygous at a locus for a particular individual and a heterozygous genotype was imputed, then the algorithm was viewed as having produced one of two correct alleles. Similarly, if the algorithm imputed a homozygous genotype at a locus where the correct genotype was heterozygous, then we considered the algorithm to have produced one of two correct alleles. It follows that the maximum number of incorrectly imputed alleles was 2 when the unknown genotype was homozygous and 1 when the unknown genotype was heterozygous.
In each of the 29 population samples, we measured the imputation accuracy for each proportion of missing genotypes, averaging across all markers. We summarized genome-wide imputation accuracy by the weighted average of chromosome-specific imputation accuracy, using the numbers of SNPs on individual chromosomes as the weights. In our analysis of the role of the proportion of missing genotypes, an individual's missing genotypes were estimated on the basis of information strictly from other individuals in the same population sample. To obtain comparable results across populations, we restricted our analyses to a sample size of six individuals per population, the smallest sample size among the 29 populations. For each population, the six individuals were chosen randomly.
To evaluate the effect of sample size on imputation accuracy, we generated subsamples for each population and each sample size by sequentially removing individuals one at a time from the full sample. To ensure that random subsamples of individuals were used in the evaluation of imputation accuracy in each population, each of the population samples was permuted prior to the construction of subsamples. In each data set, genotypes were hidden, with a proportion of missing genotypes equal to 15%, and missing genotypes were estimated by MACH. We assessed imputation accuracy for various sample sizes for each population, and we again summarized it by the weighted average allelic-imputation accuracy across autosomes. Because imputation accuracy varies across individuals in a population, the sequence in which individuals were removed from a full population sample could conceivably influence the relationship between imputation accuracy and sample size. Therefore, to examine the importance of the particular sequence of individuals utilized in the estimation procedure, we repeated the analysis with the use of a second randomly chosen sequence of individuals in each population. Differences in imputation accuracy between the two sequences (i.e., imputation accuracies based on the first permuted sample minus corresponding values based on the second permuted sample) were negligible for most populations and sample sizes (Figures S1 and S2, available online).
Inferring Missing Genotypes with Additional Reference Individuals
Imputation Accuracy versus Panel Size
Using a single HapMap panel (either the CEU, YRI, or CHB+JPT sample) as a reference group for inferring missing genotypes (Figure 1B), we investigated the relationship between imputation accuracy and reference-panel size. For each HapMap panel, we permuted the panel and constructed random subpanels of size 10, 20, …, 120 haplotypes by sequentially adding 10 haplotypes in the order specified by our permutation. Note that each of the resulting subpanels, when viewed independently, represented a random sample of haplotypes from the appropriate HapMap panel and that a consecutive pair of haplotypes did not necessarily correspond to two haplotypes of the same individual. To obtain comparable results across HapMap panels, we considered (only in this analysis) subpanels of ≤ 120 haplotypes, despite the fact that the CHB+JPT panel had 180 haplotypes. In all populations, we utilized for imputation the same set of subpanels derived from the HapMap samples. With the use of each reference panel and its subpanels, we performed genotype imputation and evaluated the accuracy across various sizes for a given reference panel, as well as across reference panels for a given size. This analysis used the full sample from each HGDP population.
Imputation Accuracy versus Panel Composition
In addition to assessing imputation accuracy using each of the three HapMap panels in isolation, we also considered the panels combined together, and we considered other mixtures of the various panels as well (Figure 1C). To identify the mixture that produced the maximal imputation accuracy, we imputed missing genotypes in each population using mixed reference samples formed by combining individuals from the three HapMap groups. In contrast to our previous analyses, in which we considered missing genotypes on the entire autosomal genome, we imputed only unknown genotypes on one chromosome, chromosome 2, in the interest of reducing computation time. We considered a variety of mixtures, with each mixture consisting of combinations of HapMap reference haplotypes chosen according to a specified ratio.
For each ratio, we used a reference panel of maximal size, constrained by the fact that most ratios involving two or more reference panels do not permit use of all available haplotypes from the panels under consideration. The set of mixtures that we considered corresponded to the set of vectors (i1, i2, i3) of nonnegative integers with i1 + i2 + i3 = 7. For each vector, we constructed a mixture sample consisting of a1 CHB+JPT haplotypes, a2 CEU haplotypes, and a3 YRI haplotypes, so that a1, a2, and a3 were as large as possible and so that they satisfied a1: a2: a3 = i1: i2: i3. For example, the vector (i1, i2, i3) = (4, 2, 1) led to (a1, a2, a3) = (180, 90, 45).
In each population, using all individuals sampled from the population, we assessed imputation accuracy using each of 36 mixed collections of haplotypes from the three HapMap panels (corresponding to the 36 solutions to i1 + i2 + i3 = 7). For each (i1, i2, i3), within HapMap groups, haplotypes were chosen randomly among the haplotypes present, and the same randomly chosen subsets of the three HapMap panels were used as the reference panel in all HGDP populations. The random sets of haplotypes were chosen so that if h haplotypes from a HapMap population were used in one mixed collection and h′ > h haplotypes from the same HapMap population were used in another mixed collection, then it was always true that the set of h haplotypes was a subset of the set of h′ haplotypes. For (i1, i2, i3) given, the solution for the number of haplotypes, (a1, a2, a3), was obtained as described in the Appendix.
Application to Untyped Markers
In current GWA studies, genotypes are collected at densities on the order of ∼500,000 SNPs spread across the genome. In such a study, with the use of a reference panel, additional information can be obtained about the genotypes of SNPs not typed directly in the GWA study but measured in an external reference panel. To assess the accuracy with which the genotypes of these markers can be imputed, we used the 513,008 SNPs typed in samples from 29 populations19 in combination with the HapMap reference panels to impute genotypes of 1,445,367 SNPs. We then compared the imputed genotypes to those measured directly by Conrad et al.14 and updated by Pemberton et al.,21 which, for limited regions of the genome, consist of SNPs at higher density than those in a typical GWA study. Using this protocol, we assessed imputation accuracy at 218,345 diploid genotypes, as described below. We note that in contrast with our other analyses, in which genotypes were imputed in randomly chosen SNP positions that varied across individuals, in this analysis, for certain markers genotyped only in the reference panel, the genotypes of all individuals in the study sample were imputed. To distinguish this scenario from the “missing genotypes” scenarios of our other analyses, we refer to such markers as “untyped markers.”
Among the 2810 SNPs reported by Pemberton et al.,21 1272 were located on chromosome 21, so we restricted this analysis to chromosome 21 for convenience. Among these 1272 SNPs, 1008 had not been included in the SNP set studied by Jakobsson et al.19 Of the 1008 SNPs, 513 were genotyped in the HapMap individuals. We thus assessed imputation accuracy at these 513 SNPs by using the genotypes at 6068 SNPs from Jakobsson et al.19 and the 26,716 SNPs available on chromosome 21 in the HapMap data. Using the HapMap reference panels to impute genotypes of untyped markers in all 443 individuals studied by Jakobsson et al.,19 we measured imputation accuracy for the 513 SNPs in a set of 426 individuals. This set of 426 individuals is the intersection of the set of 927 unrelated HGDP individuals studied by Conrad et al.14 and Pemberton et al.21 with the set of 443 unrelated HGDP individuals studied by Jakobsson et al.19 The set contains at least five individuals from each of 29 populations. In total, of the 2(426)(513) = 437,076 possible alleles in which imputation accuracy could be measured, 436,690 alleles were available (that is, 386 alleles were not reported by Pemberton et al.21). As the data of Pemberton et al.21 are based on a set of individuals that overlaps with that of Jakobsson et al.,19 this experiment mimics the scenario in which a genotyping chip is used on a set of samples and imputation of additional genotypes at marker positions that were not previously typed in the same samples is of interest (Figure 1D). This scenario occurs, for instance, in meta-analyses of multiple GWA studies.28–31
In addition to reporting the proportion of alleles estimated correctly as the measure of imputation accuracy, we also calculated the square of a linear correlation coefficient between the imputed and directly measured genotypes. At each SNP for which the true genotypes were masked, we coded the possible genotypes as 0, 1, or 2, representing the possible counts of the minor allele at this SNP in the target population. Let xi denote the imputed genotype for individual i, and let denote the mean value of the imputed genotypes across individuals. Similarly, let gi and denote the analogous quantities for the true genotypes. Then, the statistic, r2, is computed as
in which n is the number of individuals in the population sample. This squared correlation coefficient was then averaged across SNPs to obtain a summary measurement for each population.
Results
Inferring Missing Genotypes without Additional Reference Individuals
Imputation accuracies for each population, as a function of the proportion of missing data, are displayed in Figure 2. Here, no reference panel has been used, and we observe a decrease in accuracy with an increasing proportion of missing data. The Pima and Colombian groups exhibited the highest imputation accuracies (>92% with 15% of genotypes missing). Across populations, the degree to which the proportion of missing genotypes affects imputation accuracy is relatively constant, as is evident in the parallel trajectories across populations in the figure. Over the range of missing-data proportions examined, we did not observe a qualitative difference in population rankings by imputation accuracy. Populations from the Americas and from Oceania had the highest imputation accuracy, followed by populations from Asia and Europe; African populations had the lowest imputation accuracy. Because the choice of the proportion of missing genotypes had relatively little influence on population rankings by imputation accuracy, especially for proportions less than ∼30%, we proceeded to subsequent analyses with a single proportion of missing genotypes equal to 15%.
Figure 2.
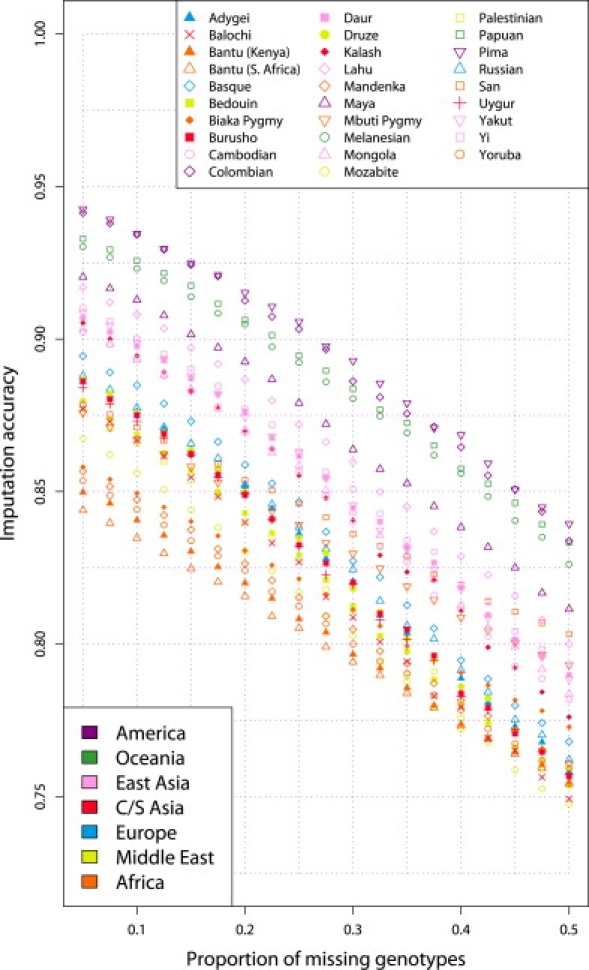
Imputation Accuracy versus Proportion of Missing Genotypes, in Each of 29 Populations
This analysis was based on samples of six individuals per population and it did not use any reference panel.
Figure 3 shows the relationship between imputation accuracy and sample size when unknown genotypes were imputed on the basis of only information from within a population sample (i.e., without a reference panel). The imputation accuracy, as measured by the proportion of alleles imputed correctly, increases as sample size increases. The pattern across populations is similar to that in Figure 2, with populations from the Americas and Oceania having the highest imputation accuracy and African populations having the lowest imputation accuracy. The boost in accuracy provided by increasing the sample size is greatest when the sample size is small.
Figure 3.
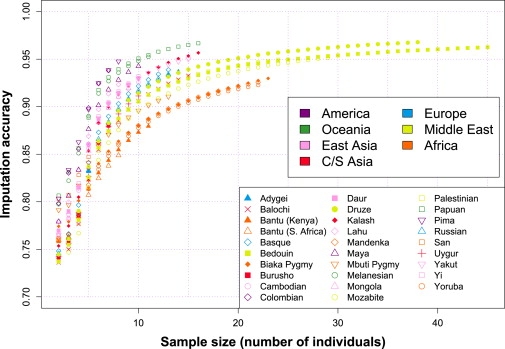
Imputation Accuracy versus Sample Size, in Each of 29 Populations
This analysis used a proportion of missing genotypes equal to 15% and did not use any reference panel.
To assess the importance of the particular sequence of individuals employed in evaluating the role of sample size, for each population sample we used an additional random ordering of individuals. Figure S1 shows the imputation accuracy as a function of sample size in the absence of a reference panel for each of two sets of permuted samples. The pointwise differences between the values in the two plots in Figure S1 are shown in Figure S2, which displays no systematic difference in imputation accuracy as a function of sample size between the two permuted samples. The maximal difference in imputation accuracy between the two permuted samples was less than 0.5% in most populations. Consequently, the impact of using a particular sequence of individuals in the evaluation of imputation accuracy appears to be minimal.
Inferring Missing Genotypes with Additional Reference Individuals
Imputation Accuracy versus Panel Size
Figure 4 shows the relationship between imputation accuracy, based on each of the three HapMap reference panels, and the size of the panels. In the first three columns, we plot the imputation accuracy from inference of missing genotypes in each population, on the basis of a single HapMap panel. In the final (rightmost) column, we plot the maximal imputation accuracy for each population, taken pointwise from the first three columns. Generally, when we used a single HapMap reference panel, higher imputation accuracies occurred in populations from the same geographic region as those of the reference panel and lower imputation accuracies occurred in African populations. With the YRI sample as the reference panel, both the highest and the lowest imputation accuracies occurred in populations from Africa (Yoruba and San, respectively).
Figure 4.

Imputation Accuracy versus Reference-Panel Size, in Each of 29 Populations, Given a Proportion of Missing Genotypes Equal to 15%
To obtain comparable results, we used the entire HapMap YRI and CEU samples but only 120 of 180 HapMap CHB+JPT reference haplotypes. The rightmost column of “maximal” imputation accuracy represents the highest accuracy achieved by one of the HapMap reference panels, taken pointwise. Populations are color-coded and symbol-coded in the same manner as in Figure 3.
We generally observed increasing imputation accuracy with increasing reference-panel size. With results averaged across all 29 populations and all three HapMap reference panels, the increase in imputation accuracy was 3.21% when the reference-panel size increased from 10 to 20 haplotypes; for subsequent additions of 10 reference haplotypes, the associated increases were 1.06%, 0.56%, 0.35%, 0.23%, 0.18%, 0.13%, 0.11%, 0.10%, 0.07%, and 0.06%. When we used the HapMap CEU or CHB+JPT sample as the reference panel, the imputation accuracy appeared to reach a plateau as the reference-panel size approached 120 haplotypes. However, we did not observe as clear a plateau when using the HapMap YRI sample as the reference panel, particularly for the Yoruba HGDP sample.
When we considered the maximal imputation accuracy attained by use of a single HapMap reference panel of 120 haplotypes, European populations generally had the highest accuracy, followed by populations from East Asia, Central and South Asia, the Americas, the Middle East, Oceania, and Africa (Figure 4). The maximal imputation accuracies of populations within a geographic region displayed more variation in Africa and the Middle East than in other geographic regions. For example, when using 120 haplotypes from the reference panel, we found that African and Middle Eastern populations had a wider range of maximal imputation accuracies (9.8% for African populations and 2.8% for Middle Eastern populations) than, for instance, the Central and South Asian populations (<1% between the highest and lowest accuracies).
Figure 5 summarizes with a bar plot the maximal imputation accuracy achieved by one of the HapMap reference panels, each 120 haplotypes in size, for each population. The colors of the bars indicate which HapMap panel was utilized for producing the maximal imputation accuracy. In African populations, we obtained the maximal imputation accuracy by using the HapMap YRI sample as the reference panel. Populations from Europe, Central and South Asia, and the Middle East, as well as the Maya population from the Americas, attained their maximal imputation accuracies with the HapMap CEU panel, whereas populations from East Asia and Oceania, as well as the Pima and Colombian populations from the Americas, achieved their maximal accuracies with the HapMap CHB+JPT reference panel.
Figure 5.
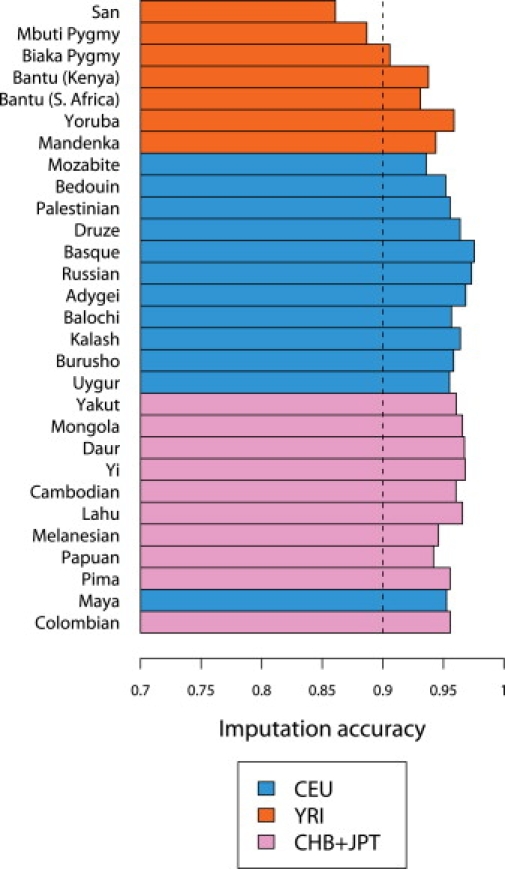
The Maximal Imputation Accuracy Achieved by One of the Three HapMap Reference Panels, in Each of 29 Populations, Given a Proportion of Missing Genotypes Equal to 15%
This plot corresponds to the imputation accuracy obtained with a reference-panel size of 120 haplotypes, shown in the rightmost column (MAX) of Figure 4. For convenience in interpreting the figure, the vertical dashed line indicates 90% imputation accuracy.
Imputation Accuracy versus Panel Composition
For each population, Figure 6 displays the imputation accuracy on the basis of mixtures of HapMap reference panels, indicating with a darkened circle the mixture of HapMap samples that produced the maximal imputation accuracy. The vertices of a triangle in Figure 6 represent imputation accuracies based solely on a single HapMap group, and the interior points represent imputation accuracies achieved by the use of mixtures of HapMap reference haplotypes (see Material and Methods). The colors correspond to the nine quantiles of the observed imputation accuracies across all mixtures and all populations, with darker colors representing higher imputation accuracies. Each point in a triangle is colored according to the imputation accuracy produced by the panel mixture corresponding to the point.
Figure 6.

Imputation Accuracy in Each of 29 Populations Achieved by Utilizing Mixtures of HapMap Samples Chosen According to Specified Ratios
Each triangle represents imputation accuracy, for a given population, based on various mixtures of HapMap reference panels. The vertices of a triangle represent imputation accuracy based on single HapMap groups, whereas the edges and interior points represent imputation accuracy attained by the use of mixtures of HapMap reference panels. Darker colors indicate higher imputation accuracy; a darkened circle indicates the maximal imputation accuracy for a population. The spacing of the cutoffs for the various colors was set so that across all 29 populations, each color would be used equally often. The set of mixtures corresponded to the set of vectors (i1, i2, i3) of nonnegative integers, with i1 + i2 + i3 = 7. For each vector, we used as the reference panel the largest possible mixture sample that consisted of a1, a2, and a3 HapMap CHB+JPT, CEU, and YRI individuals, respectively, and that satisfied a1:a2:a3 = i1:i2:i3. Corresponding numbers of HapMap haplotypes in the mixtures, (a1, a2, a3), are shown in the larger triangle. Imputation accuracy was evaluated with the use of only chromosome 2, with a proportion of missing genotypes equal to 15%.
With only a few exceptions, the panel mixture that led to the maximal imputation accuracy for a particular population had as its primary component the same HapMap reference panel that individually produced the maximal imputation accuracy shown in Figure 5. Specifically, the YRI panel was the primary component of the mixture for all African populations, the CEU panel was the primary component for all European populations, and the CHB+JPT panel was the primary component for populations from East Asia, Oceania, and the Americas. However, populations from the Middle East and Central or South Asia did not display such homogeneous patterns for the major contributing HapMap panel in the optimal mixture. In two Middle Eastern groups, Mozabite and Bedouin, the HapMap YRI and CEU samples contributed equally to their optimal mixtures of reference haplotypes, whereas in the other two Middle Eastern groups, Palestinian and Druze, the CEU sample alone served as the major contributing HapMap reference panel. For populations from Central or South Asia, the major contributing HapMap panels were the CEU sample in the Balochi group and the CHB+JPT sample in the Kalash and Uygur groups; the optimal mixture for the Burusho group contained equal contributions from the HapMap CEU and CHB+JPT samples.
Compared with imputation accuracy obtained with only a single HapMap reference group (Figures 4 and 5), in 23 of 29 populations, the major contributing HapMap sample in the mixtures that produced the maximal imputation accuracies corresponded to the single highest-accuracy panel in the analysis of individual HapMap panels. In the Kalash, Uygur, and Maya populations, the major contributing HapMap samples differed from the samples that produced the highest imputation accuracy when we evaluated HapMap panels separately; the Mozabite, Bedouin, and Burusho populations each had two HapMap panels contributing the same number of reference haplotypes in the optimal mixtures.
When we considered imputation accuracy across populations on the basis of the 36 mixtures of reference panels, European and East Asian populations had generally higher imputation accuracies that fell within the top quantiles. With the exception of the Yoruba population, African populations had substantially lower imputation accuracies that fell mostly within the bottom quantiles. The highest imputation accuracy across all points in Figure 6 was 97.83%, in the Basque population (based on a mixture consisting of 48 CHB+JPT haplotypes, all 120 CEU haplotypes, and no YRI haplotypes). The lowest imputation accuracy among all points tested—the minimum value across all 29 × 36 choices of a population sample and a reference panel—was 78.20%, in the San population (based on the entire CHB+JPT panel of 180 haplotypes). Whereas the use of mixed reference panels resulted in increased imputation accuracy in all populations, the choice of all 210 HapMap individuals as the reference panel did not yield the highest imputation accuracy in any of the 29 populations. However, this choice generally produced imputation accuracy similar to that of the optimal mixture; across populations, the mean difference between imputation accuracy based on the optimal mixture and that based on the full HapMap sample was 0.0059. This value was less than the mean difference between imputation accuracy based on the optimal mixture and that based on the optimal vertex (0.0079).
Application to Untyped Markers
Figure 7 and Table S1 present imputation accuracy for inference of unknown genotypes in the untyped chromosome 21 markers of Jakobsson et al.,19 based on individual HapMap panels and on mixtures of two or three HapMap panels. As indicated by the bar plot in Figure 7, five of seven combinations of HapMap panels produced the highest imputation accuracy in at least one population. The two combinations that did not serve as the optimal reference panel in any of the populations were the HapMap CEU sample and the combination of the YRI and CHB+JPT samples. With the exception of five groups (San, Mbuti Pigmy, Yoruba, Mandenka, and Lahu), most of the populations that we examined benefited from use of a combination of two or more HapMap samples as the reference panel for imputation of genotypes at untyped markers on chromosome 21. The highest maximal imputation accuracy was 96.05%, occurring in a European population, Adygei, and the lowest maximal imputation accuracy was 89.12%, occurring in an African population, San.
Figure 7.
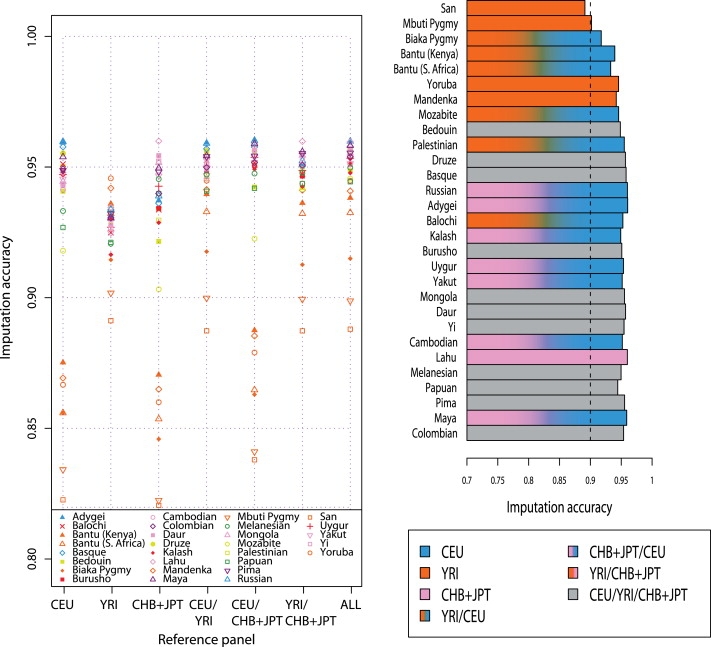
Imputation Accuracy for Inference of Genotypes of Untyped Markers, Based on One, Two, or All Three HapMap Reference Panels
The plot on the left shows imputation accuracy based on each of seven choices. The bar plot on the right represents the maximal imputation accuracy among the seven choices, and it is colored according to the choice of optimal reference panel. For convenience in interpreting the figure, the vertical dashed line indicates 90% imputation accuracy. Each HapMap panel was used with its original size.
In this setting, where mixtures of HapMap panels are coarser than those displayed in Figure 6, for 11 of 29 populations, the imputation accuracy was the highest when we constructed the reference panel from all available HapMap individuals. Seven of these 11 groups represent populations of Eurasia, with some degree of dissimilarity from the HapMap groups in northern and western Europe and in China and Japan; the other four are from Oceania and the Americas.
We obtained comparable results for the choice of reference panel when, in place of imputation accuracy, we considered the squared correlation of imputed and measured genotypes, r2, as a measure of the performance of the genotype-imputation procedure (Figure 8 and Table S2). Unlike in Figure 7, however, populations from the Americas had the highest values of r2. Across populations, the highest maximal r2, 0.9618, occurred in the Pima population and the lowest maximal r2, 0.7397, occurred in the Mbuti Pygmy population. Among the seven combinations of the HapMap panels, the CHB+JPT sample was the only panel that did not serve as the optimal panel for any of the populations. In 25 of 29 populations, the use of two or three HapMap samples produced the maximal r2 between the imputed genotypes and those directly measured by Conrad et al.14 A single HapMap panel (YRI) produced the highest r2 in San, Yoruba, and Mandenka populations; another individual panel (CEU) produced the highest r2 in the Russian population. When we used all available HapMap individuals as the reference panel, we obtained the maximal r2 in nine populations, eight of which were among the 11 populations for which imputation accuracies were the highest when the full HapMap set was used (Figure 7).
Figure 8.
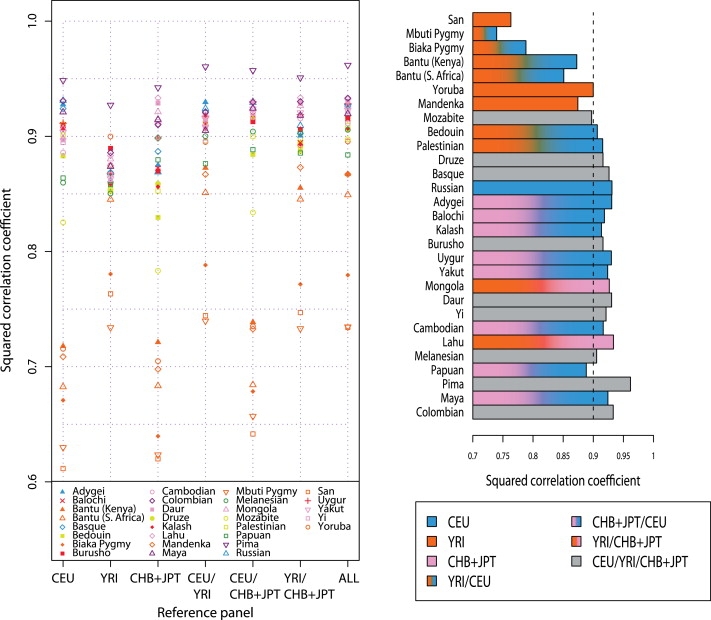
Squared Correlation Coefficient, r2, between the Genotypes Imputed from the Data of Jakobsson et al.19 and Those Directly Measured in the Data of Pemberton et al.,21 Based on One, Two, or All Three HapMap Reference Panels
The plot on the left shows r2 based on each of seven choices. The bar plot on the right represents the maximal r2 among the seven choices and is colored according to the choice of optimal reference panel. For convenience in interpreting the figure, the vertical dashed line indicates a squared correlation coefficient of 0.9. Each HapMap panel was used with its original size.
Discussion
Until now, nearly all imputation-based GWA studies have been performed in populations of European descent. As genotyping costs decrease, it is likely that such studies will begin to include individuals from an increasing diversity of populations. As a result of the success of recent studies that have leveraged external reference samples for imputation of unmeasured genotypes and of the potential that we have demonstrated for accurate genotype imputation in diverse populations, it is likely that the imputation approach can be successfully applied to GWA studies in which the sampled individuals are more distantly related to the samples that make up available reference panels. This investigation can therefore serve as an initial resource for the design and analysis of imputation-based GWA studies in these diverse populations.
We characterized the levels of LD in 29 HGDP populations using the practical metric of imputation accuracy, the ability to estimate missing genotypes on the basis of patterns of LD. Although our evaluations of imputation accuracy on the basis of the HGDP samples alone (without the use of a reference database) are somewhat constrained by the small sample sizes, we obtained relative imputation accuracies among the HGDP populations that reflect previously observed levels of LD. For example, these imputation-accuracy comparisons correspond closely to the pairwise LD calculations described by Jakobsson et al.19 Indeed, the Spearman correlation coefficient of population rankings by imputation accuracy at 15% missing data (Figure 2) and population rankings by the pairwise LD statistic, r2, for markers at 10 kb distance (Figure S4 of Jakobsson et al.19) was 0.9680 (Tables 1 and 2).
Table 1.
Statistics Compared across Imputation Scenarios
| Scenario Number | Figure Displaying Scenario Results | Type of Statistic | Description of Imputation Scenario |
|---|---|---|---|
| 1 | 2 | Imputation accuracy | 15% randomly missing genotypes; imputation without reference panels |
| 2 | 5 | Imputation accuracy | 15% randomly missing genotypes; imputation with the optimal single HapMap reference panel (among 3 choices) |
| 3 | 6 | Imputation accuracy | 15% randomly missing genotypes; imputation with the optimal mixture HapMap reference panel (among 36 choices) |
| 4 | 7 | Imputation accuracy | Untyped markers; imputation with the optimal combination of HapMap reference panels (among 7 choices) |
| 5 | 8 | Squared correlation coefficient between imputed and measured genotypes | Untyped markers; imputation with the optimal combination of HapMap reference panels (among 7 choices) |
| 6 | S4 in Jakobsson et al.19 | Linkage disequilibrium statistic, r2, at 10 kb | N/A |
Table 2.
Spearman and Pearson Correlation Coefficients between Measures of Imputation Accuracy in Various Scenarios
| Scenario Number | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 0.3910 | 0.5499 | 0.5217 | 0.6177 | 0.9680 | |
| 2 | 0.3008 | 0.8852 | 0.8035 | 0.7453 | 0.4263 | |
| 3 | 0.3755 | 0.9760 | 0.8744 | 0.8980 | 0.5795 | |
| 4 | 0.3601 | 0.9699 | 0.9856 | 0.9034 | 0.5542 | |
| 5 | 0.4405 | 0.9301 | 0.9653 | 0.9683 | 0.6507 | |
| 6 | 0.9677 | 0.4225 | 0.5100 | 0.4971 | 0.5732 |
For each scenario in Table 1, we obtained a list of values of the appropriate statistic for the 29 populations, and the correlation coefficients between pairs among these lists are shown in this table. An entry in the table represents the correlation coefficient between lists for the scenarios in the appropriate row and column. The Spearman and Pearson correlation coefficients are shown in the upper and lower triangular areas on either side of the blank cells, respectively.
Our assessments of which reference panels are most appropriate for imputation in different populations are reminiscent of evaluations of tag SNP portability in the same populations.14,15,21 When considering the three HapMap samples separately, for nearly all populations, we obtained the maximal imputation accuracies in the data of Conrad et al.14 and Pemberton et al.21 by using the same HapMap groups that produced the highest proportion of variation tagged (PVT) as reported by these studies. The only exception was the Mozabite population, in which the CEU panel achieved the highest imputation accuracy and the YRI panel achieved the highest PVT. Nonetheless, these results were compatible, because both optimal mixtures of HapMap samples in Mozabites—the one that produced the highest imputation accuracy and the one that produced the highest PVT21—contained equal proportions of the HapMap CEU and YRI panels.
More generally, we observed a notable consistency in the PVT and imputation-accuracy results for mixture reference panels. In 24 of 29 populations, the major contributing HapMap group in the optimal mixture for the purpose of genotype imputation (Figure 6) corresponded to the major group in the optimal mixture for the purpose of tag SNP selection.21 In the Burusho population, the optimal mixture for imputation contained equal numbers of HapMap CEU and CHB+JPT components, whereas the CEU panel alone served as the major contributing HapMap group in the optimal mixture for tag SNP selection.21 In the other four populations (Uygur and Kalash from Central and South Asia and Colombian and Maya from the Americas), the major contributing HapMap group was the HapMap CHB+JPT panel in the optimal mixture for imputation and the CEU panel in the optimal mixture for selection of tag SNPs.
Caution needs to be exercised in comparing imputation-accuracy results from our study with tag SNP results from Conrad et al.14 and Pemberton et al.21 In our evaluation of the effect of panel size on imputation accuracy with the use of individual HapMap panels (Figure 3), we adjusted for differences in panel size by studying HapMap samples of equal size (120 haplotypes), whereas in assessing the potential of mixture panels for use in inferring unknown genotypes (Figure 6), we utilized up to 180 haplotypes from the CHB+JPT reference group to allow for the use of all available HapMap samples. Pemberton et al.,21 on the other hand, used subsets of the CHB+JPT panel of 120 haplotypes throughout their mixture analyses. Our decision to utilize the HapMap CHB+JPT panel in its entirety could in part explain the increased utility of the CHB+JPT panel in the optimal mixtures for the five aforementioned Central and South Asian and American populations.
Although LD levels predicted imputation accuracy extremely well when we imputed genotypes without reference panels, with reference panels, LD levels were less predictive of imputation accuracy (e.g., Tables 1 and 2, Spearman correlation coefficient of 0.5795 between the maximal imputation accuracy in Figure 6 and the pairwise LD statistic, r2, at 10 kb). African populations, whose levels of LD were generally quite similar,19 varied considerably in imputation accuracy, with the highest values occurring in the lower-LD Yoruba population and the lowest values occurring in the higher-LD Mbuti Pygmy and San populations. Instead of being highest for populations from the Americas and Oceania, who exhibit the highest LD levels, imputation accuracy was highest in most analyses for European and East Asian populations that are closely related to populations from the reference panels. When the squared correlation coefficient between imputed and measured genotypes was used as the measure of imputation performance, however, the rankings of populations matched the pattern expected on the basis of LD levels somewhat more closely (Tables 1 and 2).
The accuracy with which genotypes can be imputed with the use of a reference panel is a function of multiple factors, including the similarity of haplotypes in the study sample and reference panel, as well as the allele frequencies and levels of LD in the study sample. For most populations in which imputation accuracy was high, the high value might have been expected on the basis of at least one of these factors. For the Basque population, who had the highest imputation accuracy in some analyses, a lower imputation accuracy might have been expected because of the status of the population as a linguistic isolate. However, previous analyses of the same samples have found this population to be genetically similar to other European populations, with similar levels of LD,19,20 so that a similar imputation accuracy for Basques and other European populations is not surprising. Another factor that could have contributed to high imputation accuracy in Basques and other Europeans is the possibility that European reference haplotypes might have been estimated more accurately than East Asian reference haplotypes, as a result of the availability of offspring in trios. Finally, the properties of the markers studied in the HapMap reference samples might influence imputation accuracy; many of the markers used were probably chosen for being informative about LD in Europeans, potentially leading to increased imputation accuracy in European populations.
Here, we have not extensively examined the ability of LD-based algorithms to impute genotypes at SNPs of specific allele frequencies. Our data do, however, permit a preliminary investigation of the effect of allele frequency on imputation accuracy in different populations. For each population, Figure 9 compares imputation accuracy for untyped markers with MAF greater than 0.2 and untyped markers with MAF ≤ 0.2. In all 29 populations, the genotypes of markers in the lower-MAF category were imputed with fewer errors. African populations showed a high variability in the difference in imputation accuracy between lower-MAF and higher-MAF markers (Figure S3), with a difference as high as 8.2% in the San population. In most non-African populations, genotypes of higher-MAF markers were imputed almost as accurately as were those of lower-MAF markers—most notably in the Mozabite population, for whom the difference in imputation accuracies was only 0.3%. These observations are due, in part, to the distributions of allele frequencies at the imputed SNPs; populations whose MAF > 0.2 and MAF ≤ 0.2 markers had a larger difference in mean MAF (Table S3) tended to display larger differences in imputation accuracy between the two SNP sets. A larger reference-panel size will be of some help in increasing the potential for accurate imputation; the extent to which rare alleles are satisfactorily imputed will be more easily tested in projects that include larger reference sample sizes and, consequently, that include rarer alleles.
Figure 9.
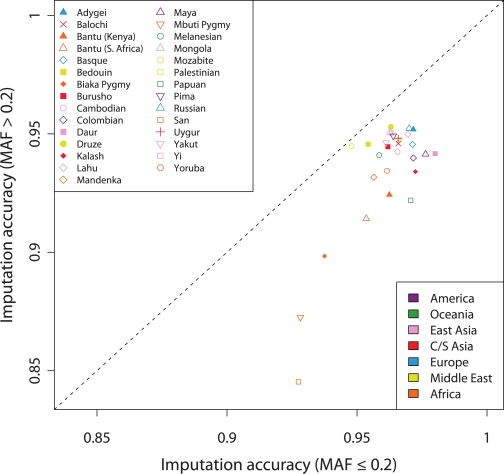
Imputation Accuracy for Genotypes at Untyped Markers in the Jakobsson et al.19 Data with Minor-Allele Frequency > 0.2 versus Imputation Accuracy for Genotypes at Untyped Markers with Minor-Allele Frequency ≤ 0.2
For a given population, we separated markers into two categories on the basis of their MAF in the population, on average placing 220 markers into the lower-MAF category and 293 into the higher-MAF category. Using the imputed genotypes described in Figures 7 and 8 for each of the seven reference-panel choices, we determined the imputation accuracy, separately restricting our attention to low-MAF markers and high-MAF markers. For each population, the highest of these seven numbers for the high-MAF markers is plotted on the y axis and the highest of these seven numbers for the low-MAF markers is plotted on the x axis (in some cases, the underlying optimal reference panel differed for the high-MAF and low-MAF markers). The diagonal dashed line indicates identical imputation accuracy for the two MAF categories. The difference between the imputation accuracy of the low-MAF markers and that of the high-MAF markers is plotted in Figure S3.
An examination of reference-panel size could assist in characterizing the way in which imputation accuracy changes for alleles in different frequency categories as reference panels are enlarged; we note, however, that our analysis of imputation accuracy and reference-panel size is restricted to the marker sets directly measured in the genome scan itself, whereas in practice, the accuracies of all imputed SNPs would be of interest. Because they were included on a commercial SNP chip, the SNPs available for testing are tag SNPs that have a somewhat regular spacing. If alleles at a tag SNP are masked, then the distance from that SNP to the nearest tag SNPs used in imputation might be greater than the corresponding distance for a randomly chosen SNP. Additionally, tag SNPs tend to have higher allele frequencies, at least for the populations in which the SNPs were discovered and the populations for which the chips were designed. Conclusions about the value of larger reference panels should be interpreted in this light and might potentially benefit from results obtained in simulations.32
In evaluating genome-wide imputation accuracies, results from rare SNPs are hidden by the large number of testable genotypes at SNPs with more frequent minor alleles. Furthermore, assessment of imputation accuracy of heterozygous genotypes at rare SNPs is obscured by the imputation-accuracy statistic that we use here. For instance, a procedure that always imputes the major allele will, on average, achieve 99.9% accuracy at a SNP with MAF of 1/1000. However, this high level of accuracy can hide a high error rate for individuals with the rare allele. As detection of rare alleles and their interactions becomes more feasible in association studies, it will be of interest to more carefully assess the accuracy with which rare alleles can be imputed.
We note that whereas our investigations that did not rely on a reference panel were affected by the sizes of the HGDP samples, our imputation-accuracy evaluations that utilized reference panels were not strongly dependent on sample size. This result is due to the manner in which we conducted our investigations, which was motivated by current strategies for imputation-based mapping in GWA studies. Specifically, conditional on the reference haplotypes, we analyzed the study samples independently rather than including other study individuals when imputing genotypes of each particular study individual. Therefore, average imputation accuracies reported here are unbiased estimates of what would be obtained from study of the entire population, provided that the individuals chosen were sampled randomly from the population.
Because of the conditional independence of study individuals during the analysis (given the reference haplotypes), the scheme that we used to evaluate optimal mixtures (e.g., Figure 6) also mimicked the current setting for analyses of GWA data, in which the information for imputing a single unobserved genotype comes entirely from the reference panel. Although for this particular investigation we did not force all genotypes to be unobserved at specified loci, instead masking individual genotypes completely at random, our imputation-accuracy results obtained with the use of randomly masked genotypes (Figures 4–6) were similar to those obtained with completely untyped markers (Figures 7 and 8). Results from our detailed investigation of optimal mixtures might therefore serve as a basis for methods that appropriately weigh reference samples from the various panels while utilizing all available information.
An alternative approach to evaluating optimal reference-panel composition, which we did not pursue, is to identify the mixture that produced the maximal imputation accuracy among mixtures of a fixed panel size, in order to more thoroughly evaluate the maximal imputation accuracy as a function of reference-panel size. This approach is constrained by the difference in the HapMap reference-panel sample sizes, so it cannot consider a mixture sample larger than 120 haplotypes (60 individuals), the smallest HapMap reference-panel size. Thus, taking into consideration the effect of reference-panel size on imputation accuracy (Figure 4), our use of the largest mixed sample permitted by a given ratio is motivated by the goal of imputing based on as many reference individuals as possible, given currently available databases. Although the optimal mixtures shown in Figure 6 for the 29 populations were not composed of all 420 haplotypes (from 210 unrelated HapMap individuals), the difference between the maximal accuracy and that obtained with the use of all haplotypes was relatively small in many cases, and for such populations, the collection of all haplotypes would form a convenient reference.
Appendix
Software Settings
The MACH-implemented options that we used included mle, mldetails, interimInterval, rounds, errorRate, compact, greedy, autoFlip, and mask. The first two options generate SNP-specific information (e.g., marker name, allele labels, minor-allele frequency [MAF], etc.), as well as genotype-level maximum-likelihood estimates of genotypes, allele dosage, confidence scores, and posterior probabilities for the three possible genotypes; “interimInterval” outputs intermediate imputation results; “rounds” specifies the number of runs for the Markov sampler (set to 20); “errorRate” provides to the algorithm an omnibus measure reflecting a combination of genotyping error, gene conversion, recurrent mutation, and assay inconsistencies between multiple platforms or laboratories (set to 10−3); “compact” reduces memory requirements at the cost of computational time; “greedy” treats the reference panel (not the combination of study and reference samples) as the only source of reference haplotypes; “autoFlip” switches the alleles at a given locus in the study samples to the complementary alleles when it is discovered that the reference panel uses the complements of the alleles used for the study sample. The “mask” option, used throughout our analyses except in application to untyped markers, specifies the proportion of genotype data to be randomly masked for evaluation of imputation accuracy.
Obtaining Mixtures of HapMap Reference Panels
Here, we solve for the numbers of haplotypes, (a1, a2, a3), that maximize the total number of haplotypes present when a ratio of integers, i1:i2:i3, is specified for the relative numbers of haplotypes in three groups.
Suppose that positive integers k and n are given, that ij is an integer in [0, k] for each j from 1 to n, and that . Suppose also that for each j from 1 to n, a positive integer Aj is given and that aj is an integer in [0, Aj]. We aim to find a = (a1, a2, …, an) such that is as large as possible and such that a1:a2:…:an = i1:i2:…:in.
Without loss of generality, suppose that i1 ≥ i2 ≥ … ≥ in. Because a1:a2:…:an = i1:i2:…:in, a1ij/i1 must be an integer for each j. Because
in which gcd represents the greatest common divisor, for each j, a1 must be a multiple of i1/ gcd(i1, ij), given that ij/ gcd(i1, ij) and i1/ gcd(i1, ij) are relatively prime. It follows that a1 is a multiple of lcm(i1/ gcd(i1, i2),…,i1/ gcd(i1, in)), in which lcm represents the least common multiple. Considering that aj = a1ij/i1 and aj ≤ Aj for each j, a1 ≤ min (A1, A2i1/i2,…,Ani1/in). As a result, the solution for a1 in the vector a that maximizes is
| (1) |
The other components of a are obtained with the use of aj = a1ij/i1.
In our analysis, k = 7, n = 3, and (A1, A2, A3) = (180, 120, 120). For each (i1, i2, i3) with i1 + i2 + i3 = 7, we obtain (a1, a2, a3) with the use of Equation 1. We chose k = 7 because this is the smallest value that permits use of the full HapMap. With k = 7, use of the full HapMap corresponds to the point (i1, i2, i3) = (3, 2, 2).
Supplemental Data
Supplemental Data include three figures and three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
HapMap phase II data, http://ftp.hapmap.org/phasing/2006-07_phaseII/phased/
MACH software, http://www.sph.umich.edu/csg/abecasis/mach/
Seattle SNPs Variation Discovery Resource, http://pga.gs.washington.edu
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01 GM081441, R01 HL090564, and U01 HL084729), the Burroughs Wellcome Fund, and the Alfred P. Sloan Foundation and by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services (Z01AG000949-02).
References
- 1.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Ding J., Abecasis G.R. Mach 1.0: Rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. 2006;79:S2290. [Google Scholar]
- 4.Nicolae D.L. Testing untyped alleles (TUNA) - applications to genome-wide association studies. Genet. Epidemiol. 2006;30:718–727. doi: 10.1002/gepi.20182. [DOI] [PubMed] [Google Scholar]
- 5.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 6.Servin B., Stephens M. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z., Schaid D.J. Methods to impute missing genotypes for population data. Hum. Genet. 2007;122:495–504. doi: 10.1007/s00439-007-0427-y. [DOI] [PubMed] [Google Scholar]
- 8.Browning S.R. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum. Genet. 2008;124:439–450. doi: 10.1007/s00439-008-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan Y., Stephens M. Practical issues in imputation-based association mapping. PLoS Genet. 2008;4:e1000279. doi: 10.1371/journal.pgen.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiner A.P., Barber M.J., Guan Y., Ridker P.M., Lange L.A., Chasman D.I., Walston J.D., Cooper G.M., Jenny N.S., Rieder M.J. Polymorphisms of the HNF1A gene encoding hepatocyte nuclear factor-1α are associated with C-reactive protein. Am. J. Hum. Genet. 2008;82:1193–1201. doi: 10.1016/j.ajhg.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willer C.J., Sanna S., Jackson A.U., Scuteri A., Bonnycastle L.L., Clarke R., Heath S.C., Timpson N.J., Najjar S.S., Stringham H.M. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conrad D.F., Jakobsson M., Coop G., Wen X., Wall J.D., Rosenberg N.A., Pritchard J.K. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 15.González-Neira A., Ke X., Lao O., Calafell F., Navarro A., Comas D., Cann H., Bumpstead S., Ghori J., Hunt S. The portability of tagSNPs across populations: a worldwide survey. Genome Res. 2006;16:323–330. doi: 10.1101/gr.4138406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu S., Pakstis A.J., Li H., Speed W.C., Kidd J.R., Kidd K.K. Significant variation in haplotype block structure but conservation in tagSNP patterns among global populations. Eur. J. Hum. Genet. 2007;15:302–312. doi: 10.1038/sj.ejhg.5201751. [DOI] [PubMed] [Google Scholar]
- 17.Gu C.C., Yu K., Ketkar S., Templeton A.R., Rao D.C. On transferability of genome-wide tagSNPs. Genet. Epidemiol. 2008;32:89–97. doi: 10.1002/gepi.20269. [DOI] [PubMed] [Google Scholar]
- 18.Xing J., Witherspoon D.J., Watkins W.S., Zhang Y., Tolpinrud W., Jorde L.B. HapMap tagSNP transferability in multiple populations: general guidelines. Genomics. 2008;92:41–51. doi: 10.1016/j.ygeno.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype, and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 20.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 21.Pemberton T.J., Jakobsson M., Conrad D.F., Coop G., Wall J.D., Pritchard J.K., Patel P.I., Rosenberg N.A. Using population mixtures to optimize the utility of genomic databases: linkage disequilibrium and association study design in India. Ann. Hum. Genet. 2008;72:535–546. doi: 10.1111/j.1469-1809.2008.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cann H.M., de Toma C., Cazes L., Legrand M.F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 23.Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin D.Y., Hu Y., Huang B.E. Simple and efficient analysis of disease association with missing genotype data. Am. J. Hum. Genet. 2008;82:444–452. doi: 10.1016/j.ajhg.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li N., Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lettre G., Jackson A.U., Gieger C., Schumacher F.R., Berndt S.I., Sanna S., Eyheramendy S., Voight B.F., Butler J.L., Guiducci C. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loos R.J.F., Lindgren C.M., Li S., Wheeler E., Zhao J.H., Prokopenko I., Inouye M., Freathy R.M., Attwood A.P., Beckmann J.S. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 2008;40:768–775. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeggini E., Scott L.J., Saxena R., Voight B.F., Diabetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;5:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pei Y.F., Li J., Zhang L., Papasian C.J., Deng H.W. Analyses and comparison of accuracy of different genotype imputation methods. PLoS ONE. 2008;3:e3551. doi: 10.1371/journal.pone.0003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.