

Abstract
Cone-rod dystrophies are inherited dystrophies of the retina characterized by the accumulation of deposits mainly localized to the cone-rich macular region of the eye. Dystrophy can be limited to the retina or be part of a syndrome. Unlike nonsyndromic cone-rod dystrophies, syndromic cone-rod dystrophies are genetically heterogeneous with mutations in genes encoding structural, cell-adhesion, and transporter proteins. Using a genome-wide single-nucleotide polymorphism (SNP) haplotype analysis to fine map the locus and a gene-candidate approach, we identified homozygous mutations in the ancient conserved domain protein 4 gene (CNNM4) that either generate a truncated protein or occur in highly conserved regions of the protein. Given that CNNM4 is implicated in metal ion transport, cone-rod dystrophy and amelogenesis imperfecta may originate from abnormal ion homeostasis.
Main Text
Cone-rod dystrophy (CRD) can be either syndromic or nonsyndromic. Nonsyndromic CRD is due to mutations in genes implicated in photoreceptor function and development, synaptic transmission or structure, retinoid metabolism, and opsin trafficking.1 CRD also occurs in syndromes such as Bardet-Biedl (MIM 209900),2 thiamine-responsive megaloblastic anemia (Rogers syndrome [MIM 249270]),3 or spinocerebellar ataxia type 7 (MIM 164500).4 In rare instances, it has been associated with dysmorphic syndromes or metabolic dysfunctions.1 Recently, a new autosomal-recessive syndrome associating CRD and amelogenesis imperfecta (AI) has been described5 and mapped to human chromosome 2q116,7 (MIM 217080).
Using fine mapping of this chromosomal region, we report the identification of mutations in CNNM4 in three families with CRD and AI. Blood was collected from probands and affected and nonaffected first-degree family members after informed consent was obtained, and DNA was isolated from peripheral leukocytes. The affected family members had a complete eye and dental evaluation, including best-corrected Snellen visual acuity (BCVA), slit-lamp examination, funduscopy, Goldmann perimetry, electroretinography (ERG), optical coherence tomography (OCT), fundus color pictures, and autofluorescence. Affected members were carefully assessed by a dentist. The parents and unaffected siblings had a complete physical evaluation, including best-vision and fundus evaluation and dental inspection. The protocol of the study adhered to the provisions of the Declaration of Helsinki.
Family A, originating from Kosovo, is a two-generation family with no history of consanguinity that was assessed at the Jules-Gonin Eye Hospital because two of the five siblings showed poor vision (Figure 1A). The children were first seen in our hospital when they were 14 (II:1) and 7 years of age (II:4), respectively. Both patients complained of photophobia and showed pendular nystagmus. They were highly hypermetropic and had low visual acuity (II:1 right eye [RE]: 20/200, left eye [LE]: 20/100; II:4 both eyes [BE]: 20/320). Both patients' fundi showed optic disk pallor, narrow vessels, macular atrophy with pigment mottling, and peripheral deep white dot deposits mainly in the lower and nasal retina. Patient II.1, who is functionally less affected, did present peripheral bone spicules (Figure 2A) concomitant with a superior and temporal scotoma on static perimetry. Fundus autofluorescence in the same patient showed a markedly decreased macular autofluorescence due to atrophy as well as severe retinal pigment epithelium (RPE) changes in the inferior and nasal peripheral retina with levels of increased and decreased autofluorescence (Figure 2B). OCT showed decreased foveal and retinal thickness, attenuation of retinal lamination suggesting extensive loss of retinal cells, and hyperreflectivity in the choroids due to RPE and choriocapillaris atrophy (Figure 2C). The OCT device that was used is not sensitive enough for evaluating any ganglion cell loss. Full-field photopic ERGs performed according to protocols recommended by the International Society for Clinical Electrophysiology of Vision (ISCEV) were nonrecordable. Under scotopic conditions, b-wave amplitudes were markedly reduced in patient II.1 and severely reduced in patient II.4. A slightly delayed culmination time of the b-wave was observed in both patients. During the 7 year follow up of patient II.1, the remaining scotopic b-wave dropped by 40% of the lower limit for the age. At the same time, both children were followed by a dentist for AI (Figure 2D). In both children, the decidual and permanent teeth were affected. The teeth were dysplastic and yellow and brown in color, showing no enamel layer and numerous carious lesions. In addition, patient II.4 had a mandibular cyst that contained the two lower incisors and one premolar.
Figure 1.
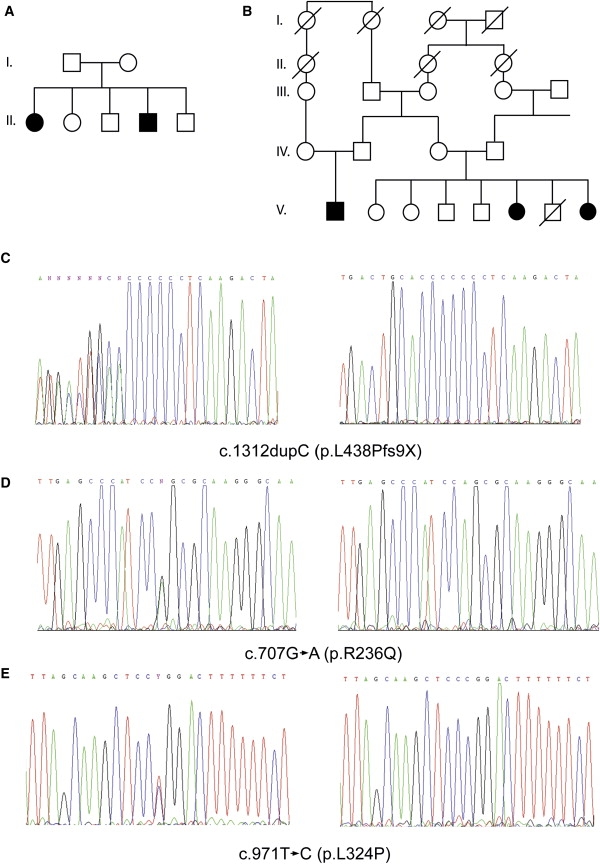
Pedigrees of Family A and B and Electropherograms
(A and B) Pedigree of family A (A) and B (B). Filled symbols represent affected individuals, and open symbols represent unaffected individuals.
(C–E) Partial electropherograms of heterozygous (left) and homozygous (right) patients from family A showing the c.1312 dupC, family B showing the c.707G→A (p.R236Q), and family C showing the c.971T→C (p.L324P) mutations in CNNM4, respectively.
Figure 2.
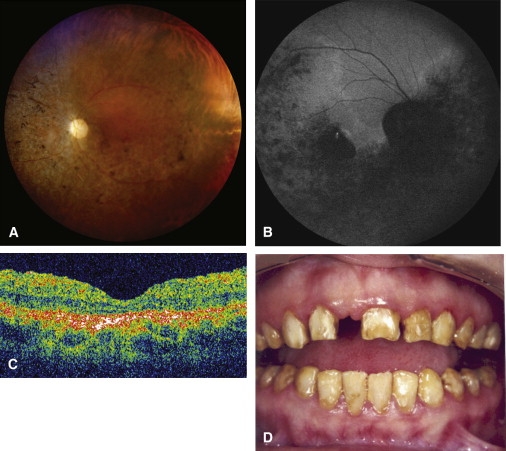
Clinical Findings in Family A
(A) Fundus photograph (patient II.1) showing optic disk pallor, narrow vessels, macular atrophy with pigment mottling, and peripheral spicules.
(B) Fundus autofluorescence (patient II.1) showing markedly decreased macular autofluorescence due to atrophy as well as severe RPE changes in the inferior and nasal peripheral retina with levels of increased and decreased autofluorescence.
(C) OCT (patient II.4) showing decreased foveal and retinal thickness, loss of well-defined retinal layers suggesting extensive loss of retinal cells, and hyperreflectivity in the choroids due to the RPE and choriocapillaris atrophy.
(D) Photograph of mouth (patient II.1) showing dysplastic teeth with yellow and brown discoloration characteristic of amelogenesis imperfecta. Upper incisor has been accidentally removed.
Family B is a five-generation consanguineous family originating from Lebanon (Figure 1B). Two sisters and one cousin were affected with a disease very closely resembling that affecting patients from family A. At age 2 months, a bilateral nystagmus was noted by the parents. At age 2 years, the patients' major complaints were the presence of photophobia and difficulties seeing in darkness. The decidual and permanent teeth were yellow and brown, showing no enamel layer, with numerous carious lesions in case of the two sisters, consistent with a hypoplastic, hypomineralized AI. ERG examination of patient V.6 at 2 years according to ISCEV standards showed that the scotopic response was within the normal range, whereas the photopic response was severely attenuated. Ophthalmological examination of both affected sisters at age 12 and 6, respectively, revealed bilateral rapid nystagmus and low vision. Full-field photoscopic ERG was nonrecordable. Under scotopic conditions, ERG responses showed markedly reduced b-wave amplitudes.
The patient from family C is the only affected individual. She was first seen at the outpatient clinic at the age of 38 years. There was no history of eye disease in her family. Her parents were not known to be related. At age 6, she was legally blind (visual acuity 10/200 on both eyes) and had a very intense photoaversion that caused difficulties in moving in daylight. However, in dim light, she could move easily and was able to read. She could see only saturated colors. By age 9, she was placed in a special school for sight-disabled children. At adolescence, she lost her central vision. By age 16, she became night blind and started to have difficulties in moving in dim light. By age 20, she was severely handicapped. She had abnormal enamel from early childhood. She rapidly lost her milk teeth, and she then had severe alteration of the enamel on her adult teeth. At age 24, she had a very significant erosion of all teeth with absence of enamel, resulting in a dark yellow color, hypersensitivity, and teeth cavities. For this reason, all teeth were devitalized and capped with ceramic prostheses. At the time of presentation, her vision was limited to light perception on both eyes. Refraction was obtained only on LE: −1.25 (15°; −1.50). She had bilateral subcapsular posterior cataract. Intraocular pressure was normal at 18 mm Hg on both eyes. The fundus showed a bilateral macular atrophy and typical bone spicule-shaped pigment deposits in the midperiphery of the retina. Retinal vasculature was highly attenuated, and optic discs were pale. There was no detectable visual field at Goldmann perimetry. The ISCEV ERG showed no response. Neurological and cognitive examination was normal in all patients.
Whole-genome linkage analysis was performed on family A at the DNA array facility of the University of Lausanne with the Affymetrix human mapping 50K XbaI array according to the manufacturer's protocols. Regions of homozygosity were identified and plotted with the web site LodIRO. When present, these regions included adjacent noninformative homozygous single nucleotide polymorphism(s) (SNP). Eight homozygous regions larger than 3 Mb were observed (Figure S1 available online). One region spanned the centromere of chromosome 2 from rs2970925 to rs953320, leaving an effective homozygous region of approximately 5.4 Mb that overlapped the previously reported linked region for this disease6 (Figure S1). We therefore focused our attention on the genes in this region.
Several candidate genes were identified on the basis of homology to genes previously implicated in eye diseases, but none showed sequence variants that could be responsible for the observed phenotype. We then looked for genes coding for transcription factors and transporters expressed in eyes and teeth. A list of sequenced genes can be found in Table S1. Sequencing the coding regions of ancient conserved domain protein 4 (CNNM4 [MIM 607805]), a metal transporter, showed a homozygous one-base-pair duplication (c.1312 dupC; +1 being the A of ATG translation start) present in both affected siblings. This duplication creates a frameshift and a new putative stop codon nine residues downstream (p.L438Pfs9X) (Figure 1C). In family B, sequencing the same exon identified a homozygous c.707G→A transition inducing the modification of arginine 236 into a glutamine (p.R236Q) (Figure 1D) in all three affected individuals. In family C, a homozygous c.971T→C mutation inducing a leucine-to-proline mutation at amino acid position 324 (p.L324P) (Figure 1E) (primers and WAVE conditions for CNNM4 and mutations can be found in Table S2). All parents were heterozygous for their respective mutation. The missense mutations were located in the transmembrane region of the protein, Arginine 236 between the first and second domain and Leucine 324 just after the fourth. Arginine 236 and Leucine 324 are completely conserved among species as far as C. elegans and in human and mouse paralogs (Figure S2). These mutations were analyzed in ethnically matched control individuals and in 1248 index patients with various forms of retinal degeneration. The c.1312 dupC mutation was observed at a heterozygous state in one control individual coming from the same geographical region as family A. No mutation was observed in the screened cohort of patients.
Immunohistochemical localization of CNNM4 was evaluated in retina of 2-month-old (2M) C57Bl/J6 mice with a rabbit polyclonal antibody raised against amino acids 21–200 of human CNNM4 obtained from Santa Cruz Biotechnology. At 2M, a strong signal was observed in the different parts of the eye (Figure 3). In the cornea, CNNM4 localization was mainly observed in the epithelium surrounding the nuclei, in the keratocytes, and in the endoderm. In the retina, CNNM4 localization was concentrated in the ganglion cell layers, the inner (IPLs) and outer plexiform layers (OPLs), and the inner and outer photoreceptor segments (Figure 3), where it was mainly localized to the cytoplasm compartment, surrounding the nucleus. IPLs and OPLs consist of connecting fibrils and dendrites from ganglion, bipolar, amacrine, Muller, and horizontal cells and rod and cone photoreceptors. The nuclei of the inner and outer cell layers as well as the RPE also showed CNNM4 staining, but at a lower level. This staining is consistent with the observation that CNNM4 was localized in dendrites and soma of cultured neurons.8
Figure 3.
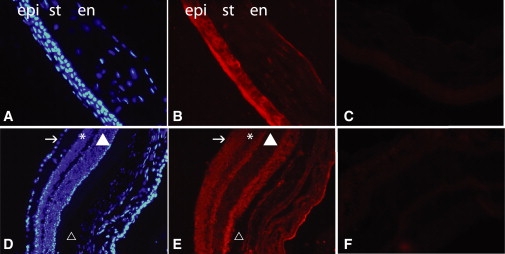
CNNM4 Immunostaining of Mouse Cornea and Retina
CNNM4 immunostaining of cornea (A–C) and retina (D–F) of 2-month-old mouse retina. In the cornea (B), CNNM4 is mainly localized in the epithelium (epi) surrounding the nuclei, in the keratocyte present in the stroma (st), and in the endothelium (en). (A) shows DAPI staining of the same slide, and (C) shows negative control without primary antibody. In the retina (E), immunostaining is mainly localized in the ganglion cell layer (indicated by the arrow), the inner and outer plexiform layers, and the outer segments of the photoreceptors. (D) shows DAPI staining of the same slide, and (F) shows negative control of without primary antibody. Symbols are used as follows: arrow, ganglion cell layer; asterisk, inner cell layer; filled triangle, outer cell layer; triangle, retinal pigment epithelium. The rabbit polyclonal antibody raised against amino acids 21–200 of human CNNM4 was used in a 1:200 dilution on 12 μm cryosections of eyes of 2-month-old C57Bl/6 mice. Secondary antibodies conjugated to Alexa Fluor 594 (Molecular Probes, Invitrogen) were diluted at 1:1000. Sections were stained with DAPI for visualizing nuclei before mounting in Cityfluor (Cityfluor Ltd.).
AI describes a group of inherited disorders primarily affecting the formation of enamel, a tissue with low protein and high mineral content. During tooth formation, ameloblasts secrete the four major matrix proteins and proteases: amelogenin, ameloblastin, enamelin, and enamelysin. Although this part of tooth development is relatively well characterized, not much is known about the maturation of enamel, during which the protein content is slowly being reduced and the mineral content is increased in order to produce enamel, the strongest tissue of the body.9,10 Because CNNM4 is implicated in ion transport,8,11 possibly of magnesium, the hypoplastic and hypomineralized form of AI present in these families could result from aberrant mineralization of enamel. We therefore investigated the localization of CNNM4 in teeth of postnatal day 2 (P2) mice by immunohistology. Staining was observed ubiquitously in the tooth and was strongest in the cell body of the ameloblasts (Figure 4). Transcription of Cnnm4 in the various parts of the eye and teeth was confirmed by reverse transcriptase-polymerase chain reaction. In addition, we evaluated by quantitative PCR the relative levels of expression between the four members of the CNNM family in brain and eye. Notably, Cnnm4 was at its highest expression levels in the retina, whereas Cnnm1, Cnnm2, and Cnnm3 mRNA expression was markedly higher in brain than in retina (Figure S3).
Figure 4.
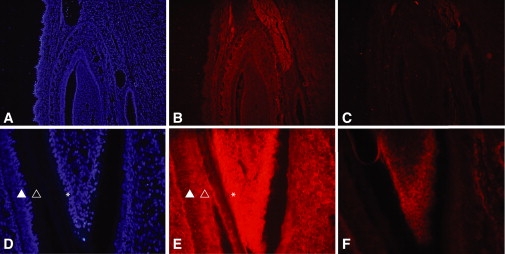
CNNM4 Immunostaining of Mouse Lower Incisor
Immunostaining of a lower incisor of a 2-day-old mouse. CNNM4 is localized in all tissues of the mandibula, including muscle and connective tissue (B). DAPI staining of the same slide is shown in (A), and negative control without primary antibody is shown in (C). At higher magnification (400×), CNNM4 is observed in the ameloblasts (indicated by the filled triangle) and their cell body (indicated by the open triangle) as well as in the odontoblasts (indicated by the asterisk) (E). (D) shows DAPI staining of the same slide, and (F) shows negative control without secondary antibody.
CNNM4 is characterized by an “ancient conserved domain” that is evolutionarily conserved in species ranging from bacteria to zebrafish to mammals.11 We therefore evaluated in zebrafish the effect of a morpholino (MO)-based knockdown expression of cnnm4 that potentially mimicked the loss-of-function mutation observed in family A. From 1 to 3 days postfertilization (dpf), transient tachycardia was observed in most of the embryos treated with MO. This tachycardia was not observed anymore at 5 dpf. At that stage, the eyes of the morphants showed a reduction of approximately 35% of the number of ganglion cells on toluidine blue-stained sections. This reduction was rescued by approximately 50% with injection of human CNNM4 mRNA (Figure 5). Zebrafish do not have buccal teeth, but instead have two sets of pharyngeal teeth. Tooth initiation occurs approximately 36 to 48 hr postfertilization (hpf), and the first two germs (I1 and I2) become mineralized by 3 and 5 dpf, respectively,12 at a time when morpholinos reach their limit of action. Zebrafish teeth were therefore not examined.
Figure 5.
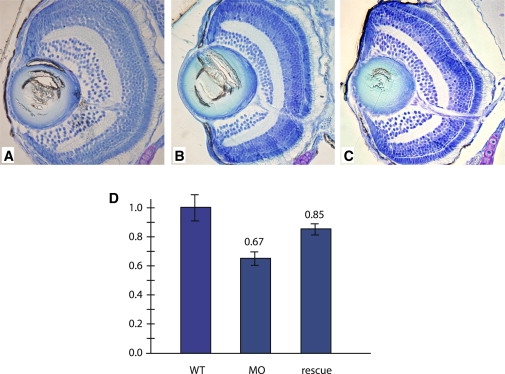
Toluidine Blue-Staining Sections of a Zebrafish Eye
Toluidine blue-stained sections of 5 dpf zebrafish embryos.
(A) Wild-type embryos.
(B) Morphant embryos.
(C) Rescued embryos.
(D) Ganglion cell number was quantified in the indicated zebrafish eye and then compared to wild-type set to 1 (n = 12 eyes in each category). The following abbreviations were used: WT, wild-type; MO, morpholino-treated embryos; rescue, morpholino-treated embryos injected with CNNM4 mRNA. Morpholinos were designed for targeting the exon 2-intron 2 splice site. One hundred micromolar solutions of either cnnm4 morpholino (MO: ATTTT GCCAC TGTCC ACTCA CTGTA) or control-morpholino (ct-MO: ATaTT cCCAC TcTCC ACTgA CTcTA) were injected into the eggs at the 1–2 cell stage. In rescue experiments, the human full-length CNNM4 cDNA was subcloned in pCDNA3.1− and transcribed from the T7 promoter. Forty pg of mRNA were injected in eggs together with the MO. Embryos were evaluated at 5 dpf, and the number of ganglion cell layers (GCLs) was counted on toluidine blue-stained sections containing the optic nerve. The number of GCLs from WT embryos was set to 1, and the counts in the morphants and rescued embryos were compared. Bars represent standard errors.
CNNM proteins are also known under the name of ACDP1 to −4 and cyclin M1 to −4. The 775 amino acid CNNM4 protein has four transmembrane domains, a sequence motif present in cyclin box, a cyclic nucleotide-monophosphate (cNMP)-binding domain, two cystathionine-beta-synthase (CBS) domains, and a DUF21 domain.11 CBS domains are small intracellular modules, usually found in two or four copies, and their function is still debated.13 Proteins with CBS domains have been implicated as metabolic sensors (Cystathionine beta-synthase) and also implicated in ATP binding (AMP-activated protein kinase), nucleotide biosynthesis (inosine-5′-mono-phosphate dehydrogenase, IMPDH), and intracellular trafficking and protein-protein interactions (CLC family).13 Interestingly, mutations in IMPDH1 (OMIM: 146690) and in the gamma-2 regulatory subunit of AMP-activated protein kinase (MIM 602643) cause retinitis pigmentosa (RP10 [MIM 180105])14 and familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome (MIM 600858),15 respectively.
Although the exact function of CNNM4 is still unknown, several members of this family have been investigated. In bacteria, CorC, formed by the highly conserved core protein region of all CNNM, is involved in magnesium and cobalt efflux,11 whereas in Saccharamyces cerevisiae, Mam3p, an ortholog of CNNM4, is implicated in manganese toxicity.16 CNNM4 is expressed in many tissues, and recent studies on spinal cord dorsal horn neurons have shown that it is located on or close to the plasma membrane, where it interacts with cytochrome oxydase 11 (COX11 [MIM 603648]) to regulate metal ion homeostasis.8 Metal ions are indispensable to many physiological processes, in particular to the visual cycle, in which, among other functions, magnesium is critical for adjusting the Ca2+ sensitivity of the membrane guanylate cyclase 2D (GUCY2D [MIM 600179]), an enzyme that produces cGMP in photoreceptor cells when stimulated by guanylyl cyclase-activating protein-1 (GUCA1A [MIM 600364]). Under physiological conditions, activation of photoreceptor GUCY2D is caused by Mg2+ and Ca2+ exchange in the EF-hands of GCAP1. Mutations in GUCY2D and GUCA1A have been associated with CRD (MIM 601777)17 and with congenital amaurosis, a severe form of CRD (MIM 204000),18 and cone dystrophy (MIM 600364), respectively.19 Magnesium is also essential in enamel formation. It is found at high concentrations in enamel fluid surrounding the forming enamel crystals, where it can compete with Ca ions for adsorption onto these crystals.20
This study identifies mutations in the CNNM4 gene that result in the autosomal-recessive CRD with AI. The nature of the mutations suggests a loss-of-function mechanism and paves the way to a new therapeutic approach by viral-mediated gene replacement. It also highlights a new gene involved in the maturation of amelogenin, a gene that will be of some importance in understanding the development of caries.
The experiments performed in animals adhered to the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmology and vision research and were approved by the Veterinary Service of the State of Valais (Switzerland).
Acknowledgments
This work was supported in part by grant 32-111948 from the Swiss National Science Foundation. We thank the families for their participation and Tatiana Favez, Céline Agosti, Carole Herkenne, Nathalie Voirol, Nicolas Lonfat, Keith Harshman, Otto Hagenbuchle, and Yann Leuba for technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org
LodIRO, http://www.lodiro.ch
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Hamel C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams N.A., Awadein A., Toma H.S. The retinal ciliopathies. Ophthalmic Genet. 2007;28:113–125. doi: 10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- 3.Fleming J.C., Tartaglini E., Steinkamp M.P., Schorderet D.F., Cohen N., Neufeld E.J. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat. Genet. 1999;22:305–308. doi: 10.1038/10379. [DOI] [PubMed] [Google Scholar]
- 4.Aleman T.S., Cideciyan A.V., Volpe N.J., Stevanin G., Brice A., Jacobson S.G. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp. Eye Res. 2002;74:737–745. doi: 10.1006/exer.2002.1169. [DOI] [PubMed] [Google Scholar]
- 5.Jalili I.K., Smith N.J. A progressive cone-rod dystrophy and amelogenesis imperfecta: A new syndrome. J. Med. Genet. 1988;25:738–740. doi: 10.1136/jmg.25.11.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downey L.M., Keen T.J., Jalili I.K., McHale J., Aldred M.J., Robertson S.P., Mighell A., Fayle S., Wissinger B., Inglehearn C.F. Identification of a locus on chromosome 2q11 at which recessive amelogenesis imperfecta and cone-rod dystrophy cosegregate. Eur. J. Hum. Genet. 2002;10:865–869. doi: 10.1038/sj.ejhg.5200884. [DOI] [PubMed] [Google Scholar]
- 7.Michaelides M., Bloch-Zupan A., Holder G.E., Hunt D.M., Moore A.T. An autosomal recessive cone-rod dystrophy associated with amelogenesis imperfecta. J. Med. Genet. 2004;41:468–473. doi: 10.1136/jmg.2003.015792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo D., Ling J., Wang M.H., She J.X., Gu J., Wang C.Y. Physical interaction and functional coupling between ACDP4 and the intracellular ion chaperone COX11, an implication of the role of ACDP4 in essential metal ion transport and homeostasis. Mol. Pain. 2005;1:15. doi: 10.1186/1744-8069-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford P.J., Aldred M., Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J. Rare Dis. 2007;2:17. doi: 10.1186/1750-1172-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J.C., Chun Y.H., Al H.T., Simmer J.P. Enamel formation and amelogenesis imperfecta. Cells Tissues Organs. 2007;186:78–85. doi: 10.1159/000102683. [DOI] [PubMed] [Google Scholar]
- 11.Wang C.Y., Shi J.D., Yang P., Kumar P.G., Li Q.Z., Run Q.G., Su Y.C., Scott H.S., Kao K.J., She J.X. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP) Gene. 2003;306:37–44. doi: 10.1016/s0378-1119(02)01210-6. [DOI] [PubMed] [Google Scholar]
- 12.Huysseune A., Van der Heyden C., Sire J.Y. Early development of the zebrafish (Danio rerio) pharyngeal dentition (Teleostei, Cyprinidae) Anat. Embryol. (Berl.) 1998;198:289–305. doi: 10.1007/s004290050185. [DOI] [PubMed] [Google Scholar]
- 13.Ignoul S., Eggermont J. CBS domains: Structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005;289:C1369–C1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 14.Bowne S.J., Sullivan L.S., Blanton S.H., Cepko C.L., Blackshaw S., Birch D.G., Hughbanks-Wheaton D., Heckenlively J.R., Daiger S.P. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gollob M.H., Seger J.J., Gollob T.N., Tapscott T., Gonzales O., Bachinski L., Roberts R. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 2001;104:3030–3033. doi: 10.1161/hc5001.102111. [DOI] [PubMed] [Google Scholar]
- 16.Yang M., Jensen L.T., Gardner A.J., Culotta V.C. Manganese toxicity and Saccharomyces cerevisiae Mam3p, a member of the ACDP (ancient conserved domain protein) family. Biochem. J. 2005;386:479–487. doi: 10.1042/BJ20041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelsell R.E., Gregory-Evans K., Payne A.M., Perrault I., Kaplan J., Yang R.B., Garbers D.L., Bird A.C., Moore A.T., Hunt D.M. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 1998;7:1179–1184. doi: 10.1093/hmg/7.7.1179. [DOI] [PubMed] [Google Scholar]
- 18.Perrault I., Rozet J.M., Calvas P., Gerber S., Camuzat A., Dollfus H., Chatelin S., Souied E., Ghazi I., Leowski C. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 19.Payne A.M., Downes S.M., Bessant D.A., Taylor R., Holder G.E., Warren M.J., Bird A.C., Bhattacharya S.S. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 20.Aoba T. Recent observations on enamel crystal formation during mammalian amelogenesis. Anat. Rec. 1996;245:208–218. doi: 10.1002/(SICI)1097-0185(199606)245:2<208::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.