

Abstract
Primary microcephaly (MCPH) is an autosomal-recessive congenital disorder characterized by smaller-than-normal brain size and mental retardation. MCPH is genetically heterogeneous with six known loci: MCPH1–MCPH6. We report mapping of a novel locus, MCPH7, to chromosome 1p32.3–p33 between markers D1S2797 and D1S417, corresponding to a physical distance of 8.39 Mb. Heterogeneity analysis of 24 families previously excluded from linkage to the six known MCPH loci suggested linkage of five families (20.83%) to the MCPH7 locus. In addition, four families were excluded from linkage to the MCPH7 locus as well as all of the six previously known loci, whereas the remaining 15 families could not be conclusively excluded or included. The combined maximum two-point LOD score for the linked families was 5.96 at marker D1S386 at θ = 0.0. The combined multipoint LOD score was 6.97 between markers D1S2797 and D1S417. Previously, mutations in four genes, MCPH1, CDK5RAP2, ASPM, and CENPJ, that code for centrosomal proteins have been shown to cause this disorder. Three different homozygous mutations in STIL, which codes for a pericentriolar and centrosomal protein, were identified in patients from three of the five families linked to the MCPH7 locus; all are predicted to truncate the STIL protein. Further, another recently ascertained family was homozygous for the same mutation as one of the original families. There was no evidence for a common haplotype. These results suggest that the centrosome and its associated structures are important in the control of neurogenesis in the developing human brain.
Main Text
Primary microcephaly (MCPH [MIM 251200]) is a congenital disorder in which the head circumference of a patient is greater than three standard deviations (SDs) below the age- and sex-related population mean.1 It results from hypoplasia of the cerebral cortex with a generalized reduction in the size of the entire brain.1 MCPH patients have mild to severe mental retardation without any other neurological features.1–3 It is hypothesized that the reduced size of the brain in MCPH patients is due to asymmetric divisions of neuronal progenitors in the neuroepithelium resulting in a reduced number of neurons.1,4 In most Indian families, MCPH segregates as an autosomal-recessive trait with parental consanguinity.5
MCPH is genetically heterogeneous with six known loci: MCPH1–MCPH6.1,6–11 There is no evidence for clinical differences between the MCPH families linked to different loci.12,13 So far, genes for only four of the loci, MCPH1-MCPH1 [MIM 607117], MCPH3-CDK5RAP2 [MIM 608201], MCPH5-ASPM [MIM 605481], and MCPH6-CENPJ [MIM 609279], have been identified.4,14,15 Immunofluorescence studies have shown that all of the proteins localize to the centrosome,15–17 suggesting that a centrosomal mechanism is responsible for controlling the neuron number in the developing human brain.15
In previous studies, we identified five MCPH families that were excluded from linkage to any of the known loci,3 suggesting the involvement of additional loci for MCPH. Unlinked families have also been identified in other populations.13,18 Therefore, we decided to conduct additional linkage studies to identify new MCPH loci.
We ascertained 35 (including the 5 from previous studies3) Indian families with MCPH at the Mental Retardation Clinic, National Institute of Mental Health and Neuro Sciences (NIMHANS), Bangalore. Consanguinity was observed in 25 (71.43%) families. Consanguinity was due to maternal uncle-niece marriage in 12 families and first-cousin marriage in 13 families. Four families had two affected sibships, and the remaining families had only one affected sibship. Twenty-three families had only one affected individual, ten families had two, and two families had three. Microcephaly (Figure 1) was present at birth by report in all the affected individuals, and there was no history of any neurological deficits or any malformations. The head circumferences of affected individuals at time of enrollment were 5–13 SDs below the population age- and sex-related mean. Mental retardation was present in all affected individuals and ranged from mild to severe. A normal karyotype was observed in all patients. All parents had normal intelligence and head circumferences. Informed consent for research from the patients and or their parents was obtained after the approval of the Institute's ethical committee.
Figure 1.
Photographs of a Primary Microcephaly Patient
Note reduced size of the head in this patient.
All 35 families were typed for three markers from each of the six known loci,3 and multipoint LOD scores were calculated with Mapmaker/Homoz; 24 of the 35 families could be excluded from linkage to these loci (data not shown).19,20 Next, three affected individuals from a single excluded consanguineous pedigree, IIS-17 (Figure S1 available online), were selected and genotyped with 222 markers from the CHLC/Weber Human Screening Set 10 (Research Genetics). Homozygosity for a single marker (D1S2134) in all three individuals was found, and 14 additional markers (Table S1) from the region were genotyped; a segregating haplotype was visually identified. Affected individual V-2 was homozygous for all 15 markers. However, heterozygosities in affected individual V-3 at D1S2797 and in affected individual V-1 at D1S417 delineated a minimum candidate region of 4.14 cM (physical length = 8.39 Mb) for a novel MCPH locus, MCPH7, at chromosome 1p32.3–p33 (Figure S1).
All 24 unlinked families were then genotyped for four markers (D1S2797, D1S2134, D1S197, and D1S386) in the region. Two-point LOD scores were calculated with the MLINK program from LINKAGE Package version 5.120 under the assumption of an autosomal-recessive mode of inheritance and a disease-allele frequency of 1/300.1 Population-specific allele frequencies were calculated by genotyping 30 unrelated normal Indian individuals. Multipoint linkage analysis was carried out with Genehunter.20 HOMOG was utilized for assessing the evidence for linkage and heterogeneity with the multipoint results.20 Evidence for both linkage and heterogeneity (p = 0.001, alpha = 0.25) was found. Five families (IIS-17, IIS-28, IIS-3, IIS-35, and IIS-40) had a greater than 70% conditional probability of linkage to this novel locus (Figures S1 and S2). The next highest conditional probability of linkage was 0.3. In addition, four of the families could be excluded from linkage to this region, suggesting the existence of at least one more MCPH locus. The remaining families were either too small and/or uninformative for establishing or ruling out linkage.
The five linked families were then genotyped for the additional 11 markers in the region (Table S1). The combined maximum two-point LOD score observed for the 15 markers in the five linked families was 5.96 for D1S386 at θ = 0.0. A combined multipoint analysis gave a maximum LOD score of 6.97 for the region flanked by D1S2797 and D1S417 (Figure S3). The candidate region delineated by family IIS-17 (between D1S2797 and D1S417) was not refined by any of the other families.
The MCPH7 candidate region harbors 77 genes, including 59 genes expressed in the brain (University of California, Santa Cruz [UCSC] Genome Bioinformatics site). One of these genes, diencephalon/mesencephalon homeobox 1 (DMBX1 [MIM 607410]) was found to be interesting and was screened for mutations in families IIS-17 and IIS-28, but no mutations were identified (data not shown). Because four previously identified MCPH genes are expressed in the centrosome, we used bioinformatic analysis to look for genes expressed in the centrosome or involved in spindle pole body function among the 59 brain-expressing genes. Two such genes, MOBKL2C and STIL (also known as SIL [MIM 181590]), lie between markers D1S2797 and D1S2874 in a physical distance of 980 Kb (Figure 2). We first screened the entire coding region of STIL for mutations in our MCPH7-linked families by using intronic primers (Table S2).
Figure 2.
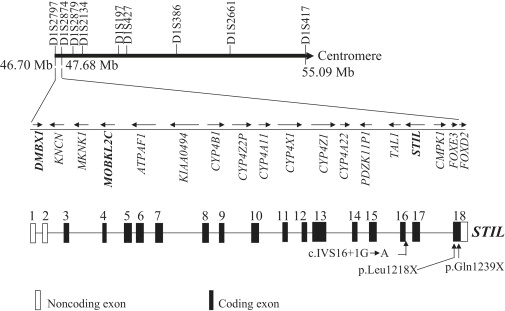
The Candidate Region of the MCPH7 Locus between Markers D1S2797 and D1S417
The known genes between markers D1S2797 and D1S2874 are shown. The arrows depict the direction of transcription of the genes. The genomic structure of STIL is also shown below. Three mutations identified during this study are marked in STIL.
DNA sequence analysis of STIL in family IIS-17 revealed a homozygous nonsense mutation (c.3715C→T/p.Gln1239X) in exon 18 in all three patients, resulting in the introduction of a premature stop codon; both parents were heterozygous for this mutation (Figure 3A). This mutation was not present in 202 population-matched control chromosomes (Table S3).
Figure 3.
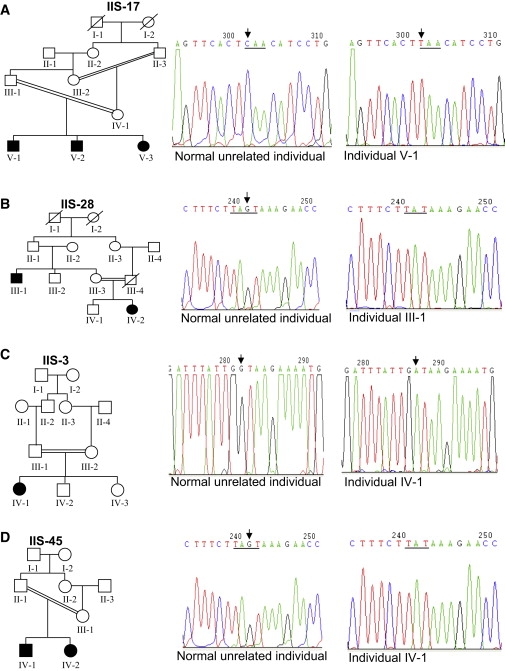
DNA Sequence Analysis of STIL for Mutations in Individuals from Families IIS-17, IIS-28, IIS-3, and IIS-45
(A) Note a homozygous C→T change in affected individual V-1 from family IIS-17. Both parents III-1 and IV-1 are heterozygous for this change. Two other affected individuals, V-2 and V-3, are homozygous for this change. This change converts the CAA codon to a premature stop codon TAA. The CAA and TAA codons are underlined in chromatograms of the normal unrelated individual and the affected individual V-1. Arrows mark the position of the nucleotide change.
(B) Note the deletion of the nucleotide residue G in affected individual III-1 from family IIS-28. The deleted nucleotide residue G is marked by an arrow in the chromatogram of the normal unrelated individual. The other affected individual, IV-2, is also homozygous for this deletion, and the rest of the individuals, II-1, II-2, III-2, III-3, and IV-1, are heterozygous for this deletion. The region around the deleted nucleotide residue G is underlined in chromatograms.
(C) Note a homozygous G→A change affecting the donor splice site in the affected individual IV-1 from family IIS-3. Both parents (III-1 and III-2) and normal siblings (IV-2 and IV-3) are heterozygous for this change. Arrows mark the position of nucleotide residues involved in G→A change in chromatograms.
(D) Note the deletion of the nucleotide residue G in affected individual IV-1 from family IIS-45. The deleted nucleotide residue G is marked by an arrow in the chromatogram of the normal unrelated individual. The other affected individual, IV-2, is also homozygous for this deletion, and both parents II-1 and III-1 are heterozygous for this deletion. The region around the deleted nucleotide residue G is underlined in chromatograms.
DNA sequence analysis in family IIS-28 revealed a homozygous 1 base pair (bp) deletion (c.3655 delG) in exon 18 in both patients (III-1 and IV-2), and this mutation segregated in this family (Figure 3B). This mutation was not present in 206 control chromosomes (Table S3) and is predicted to truncate the STIL protein (p.Leu1218X). DNA sequence analysis in family IIS-3 revealed a donor splice-site mutation (c.IVS16+1G→A) in a homozygous state in an affected individual, IV-1 (Figure 3C); this mutation segregated in this family and was not present in 208 control chromosomes (Table S3). This donor splice site was conserved across species (Figure S4A). The investigation of the effect of this mutation on splicing by the Splice Site Prediction by Neural Network program showed that the donor-motif probability is reduced from 0.99 to 0.0, suggesting inactivation of the donor splice site. Mutations affecting donor splice sites are described in a number of human genetic disorders,21 including in ASPM in a MCPH patient.22 Mutations at the conserved donor splice site, leading to the disease phenotype, can result in several types of abnormal mRNA involving exon skipping, a combination of exon skipping and use of cryptic sites, or inclusion of intronic sequences in the mature mRNA.21 The most likely result, the inclusion of intron 16 sequences in the mRNA, will introduce a new TAG stop codon from nucleotide positions +43 to +45 in intron 16, resulting in a truncated STIL protein (Figure S4B).
A new family, IIS-45, has been ascertained after our initial mapping studies of the 24 families and the identification of STIL mutations in MCPH7-linked families IIS-17, IIS-28, and IIS-3 as described above. Haplotype analysis of this family with three markers from each of the six known MCPH1–MCPH6 loci3 and four markers (D1S2797, D1S2874, D1S197, and D1S386) from the novel MCPH7 locus suggested localization of this family to the MCPH7 locus (Figure S5). DNA sequence analysis found that it has the same 1 bp deletion (c.3655 delG) as seen in IIS-28 (Figure 3D). Haplotype analysis suggested that mutations were not from a single founder (Figure S5).
We were not able to find mutations in families IIS-35 and IIS-40. It is possible that mutations in these families lie in the regulatory region or in intronic regions not covered by the primers used for sequencing or that the families have mutations in the other candidate gene of this region. It is also possible that these families do not actually map to this locus.
Two types of mutations were identified in our families (Table S4): nonsense and deletion mutations leading to a premature stop codon and a mutation affecting a donor splice site. Two families were homozygous for the same deletion mutation. It is interesting to note that these three mutations in STIL and almost all the mutations in previously reported MCPH genes are protein-truncating mutations,3,4,14,15,18,22 suggesting that the loss of function of these genes leads to MCPH.
STIL (SCL/TAL1 interrupting locus) is necessary for proper mitotic spindle organization in zebrafish and human cells and localizes to the mitotic spindle poles only during metaphase.23 In HeLa cells, STIL localizes to the pericentriolar region, but it does not completely colocalize with the gamma-tubulin of the centrosome.23 Cells undergoing anaphase lacked STIL localization to the spindle poles, and virtually no STIL expression was seen in interphase cells.23 Interestingly, all four other MCPH proteins (MCPH1, CDK5RAP2, ASPM, and CENPJ) localize to the centrosome at the interphase, metaphase, and anaphase stages of the cell cycle.15–17 STIL is an early-response gene that is ubiquitously expressed in proliferating cells and during early embryonic development.24,25 STIL consists of 18 exons (Figure 2). The human and the mouse STIL show an overall identity of 73% at the amino acid level.26 Reverse transcriptase-polymerase chain reaction analysis showed that STIL is expressed in human fetal tissues, including the fetal brain from a spontaneous abortion at 16 weeks of gestation (Figure S6). The development of human cerebral cortex starts with proliferation of neuronal progenitors present in the walls of cerebral ventricles. A major addition of neurons in cortex occurs at approximately 4 to 5 fetal months.27 Most neurons reach their destination in the cortex by the 24th week of gestation, guided by radial glia.28 Therefore, the expression of STIL in the brain of a 16-week-old human fetus supports its role in neuronal cell proliferation. Further, the RNA in situ hybridization in mouse embryos at embryonic day 14.5 has shown that STIL is expressed in the developing cerebral cortex29 like other MCPH genes.4,14,15
Pfaff et al.23 have identified a homozygous lethal zebrafish mutant, cassiopeia (csp), as a mitotic mutant. csp mutant embryos have an increased mitotic index, have highly disorganized mitotic spindles, and often lack one or both centrosomes.23 The csp phenotype was caused by loss-of-function mutations in zebrafish stil.23 The mitotic spindle defects in csp mutants are similar to those in Drosophila abnormal spindle (asp) mutant.30 It is interesting to note that mutations in ASPM, the human ortholog of Drosophila melanogaster asp, is a major cause of MCPH.3,18,22 The similarity in the mitotic spindle defects between zebrafish stil mutants and Drosophila asp mutants supports the role of Stil in mitotic spindle pole organization.
Stil (Sil) mutations have been studied in the mouse. Sil−/− (null) mice die after embryonic day 10.5 with midline and left-right asymmetry defects, whereas Sil+/− (heterozygous) mice were normal.25 The null mice showed the arrest of neural tube closure and lack of midline separation at the anterior end of the cranial folds, leading to holoprosencephaly-like defects.25 This suggests that Sil has a role in embryonic brain development. Karkera et al.26 screened this gene for mutations in 83 familial holoprosencephaly probands, but they did not find any mutations. The histological examination of Sil−/− embryos showed significantly fewer cells in the neural fold than normal, which was found to be due to apoptosis rather than the lack of a proliferation defect.25 Therefore, the reduced number of neurons in the cerebral cortex of MCPH patients appears to be due to apoptosis of neurons rather than reduced neuron production as suggested earlier.12
In summary, we have mapped a novel MCPH locus, MCPH7, in 5/24 Indian families unlinked to known MCPH1–MCPH6 loci, and we provide evidence for STIL as the causative gene. Three different mutations in STIL were identified in four families. The present data will be helpful in identifying the extent of the involvement of this locus in MCPH families unlinked to the other six known MCPH loci from other populations and lead to better diagnosis and management of MCPH patients in the future.
Acknowledgments
This work was financially supported by the Sir Dorabji Tata Center for Tropical Diseases, Bangalore. We thank L. Krishna, M. Markandaya, Sabina Rao, and M.N. Nagashri for their help in patient recruitment and human fetal tissue samples.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
The Berkley Drosophila Genome Project Splice Site Prediction by Neural Network program, http://www.fruitfly.org/seq_tools/splice.html
Marshfield linkage maps for genetic distances, http://research.marshfieldclinic.org/genetics/
Mouse Gene Expression Database, http://www.informatics.jax.org/expression.shtml
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Bioinformatics site, http://genome.ucsc.edu/
References
- 1.Jackson A.P., McHale D.P., Campbell D.A., Jafri H., Rashid Y., Mannan J., Karbani G., Corry P., Levene M.L., Mueller R.F. Primary autosomal microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 1998;63:541–546. doi: 10.1086/301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woods C.G., Bond J., Enard W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar A., Blanton S.H., Babu M., Markandaya M., Girimaji S.C. Genetic analysis of primary microcephaly in Indian families: Novel ASPM mutations. Clin. Genet. 2004;66:341–348. doi: 10.1111/j.1399-0004.2004.00304.x. [DOI] [PubMed] [Google Scholar]
- 4.Bond J., Roberts E., Mochida G.H., Hampshire D.J., Scott S., Askham J.M., Springell K., Mahadevan M., Crow Y.J., Markham A.F. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 5.Cowie V. The genetics and sub-classification of microcephaly. J. Ment. Defic. Res. 1960;4:42–47. doi: 10.1111/j.1365-2788.1960.tb00751.x. [DOI] [PubMed] [Google Scholar]
- 6.Roberts E., Jackson A.P., Carradice A.C., Deeble V.J., Mannan J., Rashid Y., McHae D.P., Markham A.F., Lench N.J., Woods C.G. The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur. J. Hum. Genet. 1999;7:815–820. doi: 10.1038/sj.ejhg.5200385. [DOI] [PubMed] [Google Scholar]
- 7.Moynihan L., Jackson A.P., Roberts E., Karbani G., Lewis I., Corry P., Turner G., Mueller R.F., Lench N.J., Woods C.G. A third locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am. J. Hum. Genet. 2000;66:724–777. doi: 10.1086/302777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamieson C.R., Govaerts C., Abramowicz M.J. Primary autosomal recessive microcephaly: Homozygosity mapping of MCPH4 to chromosome 15. Am. J. Hum. Genet. 1999;65:1465–1469. doi: 10.1086/302640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pattison L., Crow Y.J., Deeble V.J., Jackson A.P., Jafri H., Rashid Y., Roberts E., Woods C.G. A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am. J. Hum. Genet. 2000;67:1578–1580. doi: 10.1086/316910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamieson C.R., Fryns J.P., Jacobs J., Matthijs G., Abramowicz M.J. Primary autosomal recessive microcephaly: MCPH5 maps to 1q25–1q32. Am. J. Hum. Genet. 2000;67:1575–1577. doi: 10.1086/316909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leal G.F., Roberts E., Silva E.O., Costa S.M., Hampshire D.J., Woods C.G. A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J. Med. Genet. 2003;40:540–542. doi: 10.1136/jmg.40.7.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mochida G.H., Walsh C.A. Molecular genetics of human microcephaly. Curr. Opin. Neurol. 2001;14:151–156. doi: 10.1097/00019052-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Roberts E., Hampshire D.J., Pattison L., Springell K., Jafri H., Corry P., Mannon J., Rashid Y., Crow Y., Bond J., Woods C.G. Autosomal recessive primary microcephaly: An analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 2002;39:718–721. doi: 10.1136/jmg.39.10.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson A.P., Eastwood H., Bell S.M., Adu J., Toomes C., Carr I.M., Roberts E., Hampshire D.J., Crow Y.J., Mighell A.J. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bond J., Roberts E., Springell K., Lizarraga S., Scott S., Higgins J., Hampshire D.J., Morrison E.E., Leal G.F., Silva E.O. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 16.Zhong X., Liu L., Zhao A., Pfeifer G.P., Xu X. The abnormal spindle-like, microcephaly-associated (ASPM) gene encodes a centrosomal protein. Cell Cycle. 2005;4:1227–1229. doi: 10.4161/cc.4.9.2029. [DOI] [PubMed] [Google Scholar]
- 17.Zhong X., Pfeifer G.P., Xu X. Microcephalin encodes a centrosomal protein. Cell Cycle. 2006;5:457–458. doi: 10.4161/cc.5.4.2481. [DOI] [PubMed] [Google Scholar]
- 18.Gul A., Hassan M.J., Mahmood S., Chen W., Rahmani S., Naseer M.I., Dellefave L., Muhammad N., Rafiq M.A., Ansar M. Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: Novel sequence variants in ASPM gene. Neurogenetics. 2006;7:105–110. doi: 10.1007/s10048-006-0042-4. [DOI] [PubMed] [Google Scholar]
- 19.Lander E.S., Bodstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 20.Lathrop G.M., Lalouel J.M., Juliet C., Ott J. Strategies for multilocus linkage analysis in humans. Proc. Natl. Acad. Sci. USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krawczak M., Reiss J., Cooper D.N. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: Causes and consequences. Hum. Genet. 1992;90:41–45. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 22.Bond J., Scott S., Hampshire D.J., Springell K., Corry P., Abramowicz M.J., Mochida G.H., Hennekam R.C., Maher E.R., Fryns J.P. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am. J. Hum. Genet. 2003;73:1170–1177. doi: 10.1086/379085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaff K.L., Straub C.T., Chiang K., Bear D.M., Zhou Y., Zon L.I. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol. Cell. Biol. 2007;27:5887–5897. doi: 10.1128/MCB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izraeli S., Colaizzo-Anas T., Bertness V.L., Mani K., Aplan P.D., Kirsch I.R. Expression of the SIL gene is correlated with growth induction and cellular proliferation. Cell Growth Differ. 1997;8:1171–1179. [PubMed] [Google Scholar]
- 25.Izraeli S., Lowe L.A., Bertness V.L., Good D.J., Dorward D.W., Kirsch I.R., Kuehn M.R. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature. 1999;399:691–694. doi: 10.1038/21429. [DOI] [PubMed] [Google Scholar]
- 26.Karkera J.D., Izraeli S., Roessler E., Dutra A., Kirsch I., Muenke M. The genomic structure, chromosomal localization, and analysis of SIL as a candidate gene for holoprosencephaly. Cytogenet. Genome Res. 2002;97:62–67. doi: 10.1159/000064057. [DOI] [PubMed] [Google Scholar]
- 27.Jones K.L. Fifth Edition. W.B. Saunders Company; Philadelphia: 1997. Smith's Recognizable Patterns of Human Malformation. [Google Scholar]
- 28.Gardiner M. Molecular genetics of neonatal epilepsies: Recent advances. Journal of the Arab Neonatology Forum. 2006;3:17–23. [Google Scholar]
- 29.Smith C.M., Finger J.H., Hayamizu T.F., McCright I.J., Eppig J.T., Kadin J.A., Richardson J.E., Ringwald M. The mouse Gene Expression Database (GXD): 2007 update. Nucleic Acids Res. 2007;35(Database issue):D618–D623. doi: 10.1093/nar/gkl1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Carmo Avides M., Glover D.M. Abnormal spindle protein, Asp, and the integrity of mitotic centrosomal microtubule organizing centers. Science. 1999;283:1733–1735. doi: 10.1126/science.283.5408.1733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.