

Abstract
Myelination is a complex, developmentally regulated process whereby myelin proteins and lipids are coordinately expressed by myelinating glial cells. Homozygosity mapping in nine patients with childhood onset spasticity, dystonia, cognitive dysfunction, and periventricular white matter disease revealed inactivating mutations in the FA2H gene. FA2H encodes the enzyme fatty acid 2-hydroxylase that catalyzes the 2-hydroxylation of myelin galactolipids, galactosylceramide, and its sulfated form, sulfatide. To our knowledge, this is the first identified deficiency of a lipid component of myelin and the clinical phenotype underscores the importance of the 2-hydroxylation of galactolipids for myelin maturation. In patients with autosomal-recessive unclassified leukodystrophy or complex spastic paraparesis, sequence analysis of the FA2H gene is warranted.
Main Text
Myelin is the lipid-rich organelle that surrounds axons and constitutes the white matter tracts in the nervous system.1 Myelin is composed of compact membrane layers out of which the cytoplasm has been extruded. During myelination, glial cells elaborate processes that contact axons and spirally enwrap them to form a multilamellar, compact structure. Thus, the myelin sheath is membrane dense, which reflects its role as an electrical insulator for axonal membranes. Myelination is a complex, developmentally regulated process whereby myelin proteins and lipids are coordinately expressed by myelinating glial cells.2 Myelin has a water content of ∼40% and its dry mass is characterized by a high proportion of lipid (70%–85%) and a low proportion of protein (15%–30%).1 The galactolipids galactosylceramide (GalC) and its sulfated form, sulfatide, account for approximately one-third of the lipid content of the myelin sheath.3 Using homozygosity mapping, we identified a defect in the fatty acid 2-hydroxylase (FA2H [MIM 611026]) that participates in the synthesis of the fatty acids of myelin galactolipids.
The subjects of the study were nine patients, aged 7–20 years at the time of writing, from three consanguineous Arab-Muslim families (Figure 1 and Table 1). All the patients, four males and five females, had normal early development and all presented at 4–6 years of age with gait disturbance due to lower-limb spasticity. At this age the disease did not progress in the two patients from family 3; at 10 and 12 years of age, their disease is restricted to the lower limbs with no cognitive or speech impairment. Both patients were ambulatory without external aids, although they walked with considerable difficulty. In contrast, the disease course was rapidly progressive in the patients of families 1 and 2, who already required walking aids at 7 years. When they were 7–12 years of age, their spasticity extended to the upper limbs. Dystonia was evident from a few years of onset, involving trunk, limbs, and face and interfering with articulation and swallowing. Upper-motor-neuron involvement with positive Babinski sign became evident toward 10 years of age in the patients. The patients' cognitive abilities, normal at 6 years of age with acquisition of reading and writing skills at a normal pace, deteriorated with age. At 12–14 years of age, these patients were no longer able to read or write; this deterioration was not attributed to the motor disability or the dysarthria. Cerebellar dysfunction with dysmetria and dysdiadochokinesis were additionally noted. At midadolescence, the patients lost ambulation. Patients 122 and 2761 had transient generalized seizures that resolved spontaneously. The rest of the physical examination was unremarkable and there was no clinical or biochemical indication of other organ involvement.
Figure 1.
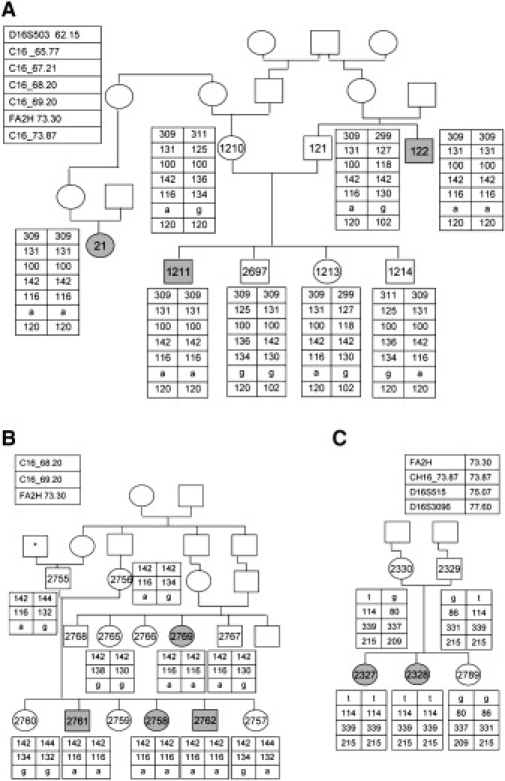
The Families' Pedigree and Haplotype along the Critical Region on Chromosome 16
(A) Family 1 pedigree and their haplotype along the critical region on chromosome 16. Patients' symbols are filled. Numbered symbols represent individuals whose DNA samples were available for analysis. The polymorphic microsatellite markers and their chromosomal locations (in Mb) are given in the upper-left panel. C16 _65.77 stands for hg18_chr16:65767359-65767624, C16_67.21 stands for hg18_chr16:67214180-67214437, C16_68.20 stands for hg18_ chr16:68218628-68218887, C16_69.20 stands for hg18_chr16:69227890-69228133, and C16_73.87 stands for hg18_chr16:73875745-73875997.
(B) Family 2 pedigree. The asterisk represents an individual whose family name is identical to that of family 1.
(C) Family 3 pedigree. The polymorphic microsatellite markers and their location are given at the upper-left and upper-right panel, for family 2 and family 3, respectively.
Table 1.
Clinical and Radiological Findings in the Patients
| Family-Patient | 1-122 | 1-1211 | 1-21 | 2-2761 | 2-2758 | 2-2769 | 2-2762 | 3-2327 | 3-2328 |
|---|---|---|---|---|---|---|---|---|---|
| Current age | 20 | 18 | 14 | 17 | 11 | 8 | 7 | 12 | 10 |
| Age at onset | 4.5 | 6 | 5 | 4.5 | 4.5 | 5 | 4 | 4 | 6 |
| Initial symptom | walking difficulty | foot inversion | foot inversion | walking difficulty | walking difficulty | foot inversion | walking difficulty | walking difficulty | walking difficulty |
| Spasticity | + | + | + | + | + | + | + | + | + |
| Dystonia | + | + | + | + | + | + | + | − | − |
| Cerebellar signs | + | + | − | + | − | + | − | − | − |
| Transfer to special education | 14 | 11 | 12 | 14 | RC | RC | RC | RC | RC |
| Wheelchair-bound | 14 | 13 | 14 | 12 | 11 | walking | walking | walking | walking |
| Dysarthria onset | 6 | 7 | 6 | 7 | 5 | 7 | 5 | − | − |
| Seizures onset | 15 | − | − | 12 | − | − | − | − | − |
| Initial brain MRI findings | PPRT | PPRT, TCC, CA | PPRT, TCC, CA | PPRT, TCC, CA | PPRT | PPRT | PPRT | not done | normal |
All numbers refer to age in years, RC, Regular Class; clinical signs are as follows: present (+) or not present (−). PPRT, prolonged periventricular relaxation time; TCC, thin corpus callosum; and CA, cerebellar atrophy.
Laboratory investigations including analysis of plasma lactate, creatine kinase, ammonia, amino acids, very-long-chain fatty acids, and urinary organic acids were all normal. In muscle mitochondria, the activities of the five mitochondrial respiratory-chain complexes were within the control range, and the pathological and histochemical studies were normal. The brain stem auditory evoked potentials of patient 1211 at 18 years of age were normal, but pattern visual evoked potentials were delayed with a latency of 130 ms (Rt) and 123 ms (Lt) (normal up to 110 ms), suggesting a demyelinating process; electromyography and nerve conduction velocity were normal. Biopsy of sural nerve of patient 1211 at age 17 years revealed slight decrease of myelinated nerve fibers, but the ratio of large to small fibers and the number of nonmyelinated fibers were normal. The myelin thickness was proportional to the axonal diameter and few fibers exhibited Wallerian axonal degeneration. Myelin compaction was intact (Figure 2) as evidenced by a space of 15 nanometers between the major dense lines4. Brain MRI revealed a uniform pattern among patients from families 1 and 2 consisting of prolonged relaxation time of the posterior periventricular white matter; this pattern was already present when the patients were 5 years old. With age, the posterior limbs of the internal capsules and the corticospinal tracts became involved, and there was thinning of the corpus callosum and the pons with volume depletion of the cerebellum resulting in ventriculomegaly (Figure 3). Notably, the cervical spinal cord was entirely spared.
Figure 2.
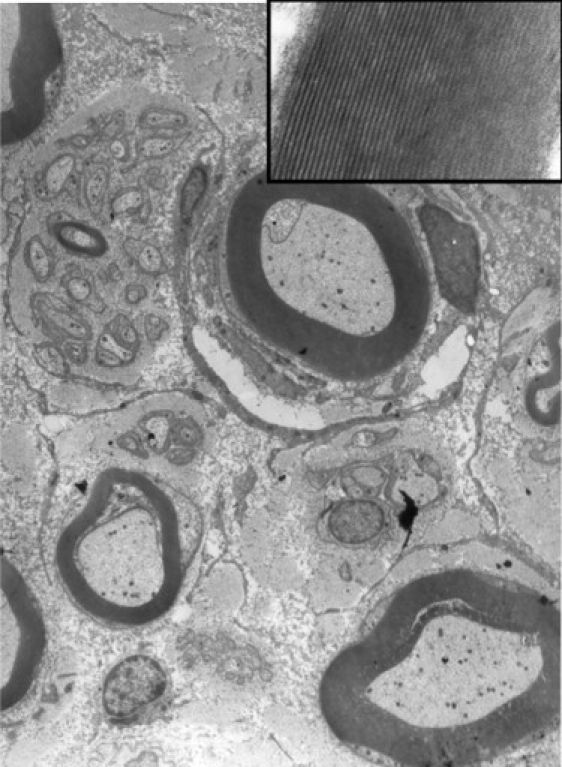
Electron Micrograph of the Sural Nerve Showing Normal Structure and Distribution of Myelinated and Unmyelinated Nerve Fibers
The inset shows details of a myelin sheath to show normal compaction and periodicity of myelin lamellae.
Figure 3.
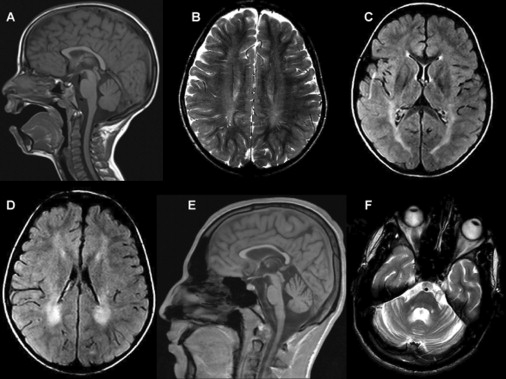
Brain MRI of Patient 2762 at 7 Years of Age and Patient 1211 at 18 Years of Age
(A) Sagittal T1 weighted image.
(B) Axial T2 weighted image.
(C and D) Axial FLAIR images showing prolonged relaxation times in the periventricular white matter, more prominent posteriorly, and to a lesser extent in the posterior limbs of the internal capsules. There is no significant loss of volume. Images in (A)–(D) are of patient 2762 at 7 years of age.
(E and F) Sagittal T1 weighted (E) and axial T2 weighted (F) images showing loss of volume of the corpus callosum and to a lesser extent of the cerebellum and pons. Images are of patient 1211 at 18 years of age.
Homozygosity mapping with the DNA of three patients from families 1 and 2, individuals 2761, 1211 and 122, on GeneChip Human Mapping 250K Nsp Array of Affymetrix, was performed as previously described.5 All experiments involving DNA of the patients, their relatives, healthy controls, and patients' cells were approved by the Hadassah Ethical Review Committee. Multiple genomic stretches of successive homozygous SNPs, longer than 4 Mb, were present in the DNA of the three patients but there was only one homozygous region of identical haplotype. This region spanned ∼16 Mb and consisted of 1309 consecutive SNPs (59.21–75.17 Mb, rs11076397–rs7500366) on chromosome 16. By using polymorphic microsattelite markers in nonaffected family members, we could narrow it down to ∼13 Mb (62.15–75.17 Mb) (Figure 1A). Within this region there were 186 ORFs that were prioritized according to their tissue expression. The sequence of GABARAPL2, CG1-38, and PLEKHG4 was determined, but no mutation identified. We then sequenced the seven exons of FA2H and their flanking intronic sequences and identified a homozygous intronic mutation, c.786+1G→A. The mutation was present in the seven patients of families 1 and 2 and was not present in a homozygous form in any of the available unaffected relatives, nor was it present in 110 ethnic-matched control individuals. In cDNA produced from fibroblasts and fresh blood of the patients, a single FA2H fragment, 426 bp shorter than the normal fragment because of exon 5 and 6 skipping, was amplified (Figures 4A and 4B).
Figure 4.
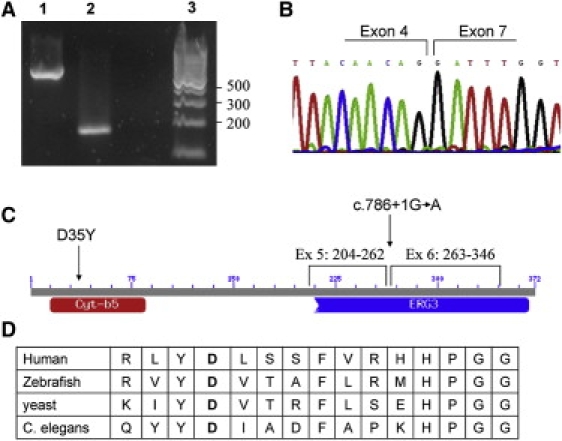
Mutations in the FA2H Gene
(A) A cDNA fragment of the FA2H gene from the patients of family 1 and 2 who are homozygous for the c.786+1G→A mutation. Lane 1 represents the control, lane 2 represents the patient, and lane 3 represents the DNA ladder marker.
(B) Sequence of the short cDNA fragment demonstrating exon 5 and 6 skipping.
(C) Schematic representation of the conserved domains of the FA2H protein. Cyt-b5 stands for cytochrome b5-like heme-binding domain and ERG3 is the sterol desaturase domain. The locations of the residues encoded by exon 5 and 6 and of the two mutations are shown.
(D) Conservation throughout evolution of the aspartic acid at codon 35 (in bold), which is mutated in patients from family 3.
In family 3, the two patients 2327 and 2328 had two identical regions on chromosome 16: 51.20-58.80 Mb and 70.74-77.79 Mb (rs3104798–rs10500429 and rs3852784–rs382888, respectively). Within these regions there were 118 ORFs that were prioritized according to their tissue expression. The sequence of the CAPNS2, PLLP, BOAT, GABARAPL2, and CNTNAP4 genes were determined but no mutation was identified. Sequencing the seven exons of the FA2H gene revealed a homozygous mutation c.103G→T at exon 1 changing a highly conserved aspartic acid at codon 35 to tyrosine (D35Y) (Figure 4D). In this family, the parents were heterozygous for the mutation and a healthy sister, asymptomatic at 6 years of age, had the two normal alleles (Figure 1C).
The human FA2H gene encodes fatty acid 2-hydroxylase, a 372-amino-acid-long, 43 kDa membrane-bound protein. It uses free fatty acids as a substrate and the product, free 2-hydroxy fatty acids, is then incorporated into ceramide, the precursor of GalC. The FA2H protein contains two conserved domains, a cytochrome b5-like heme-binding domain that spans residues 15–85 and accounts for the redox activity of FA2H, including its ability to hydroxylate fatty acids, and a sterol desaturase domain at residues 210–367 (Figure 4C).6,7 We therefore concluded that the c.786+1G→A splice-site mutation with the consequent skipping of exon 5 and 6 (residues 204–346) probably abolishes the catalytic activity; however, a proof of pathogenicity was required for the missense mutation at codon 35. To this end, we transiently transfected COS7 cells with pcDNA3-hFA2H-mut, which contained the D35Y mutation generated by site-directed mutagenesis.8 Western-blot analysis with anti-human FA2H polyclonal antibody6 revealed a normal immune reactive protein. To determine fatty acid 2-hydroxylase activity in vitro, we quantified conversion of [3,3,5,5-D4] tetracosanoic acid to 2-hydroxy [3,3,5,5-D4] tetracosanoic acid by gas chromatography-mass spectrometry as previously described.9 This analysis showed that the activities of cells expressing the mutant FA2H were indistinguishable from the COS7 cells transfected with an empty vector, whereas expression of the wild-type FA2H resulted in over 2-fold increase above the endogenous level, suggesting that the D35Y mutation inactivated FA2H.
Heretofore, FA2H was considered to be the only enzyme active toward straight-chain fatty acids in human. There are three types of fatty acid 2-hydroxylases known to date: (1) the di-iron-containing monooxygenases, such as FA2H, which has no known homologs in human and mouse,10 (2) cytochrome P450 that catalyzes the H2O2-dependent 2-hydroxylation of fatty acids in bacteria but is absent in eukaryotes,11 and (3) the 2-oxoglutarate-dependent oxygenase, phytanoyl-CoA 2-hydroxylase, which is localized to the peroxisome and does not use straight-chain fatty acids.12 Consistent with the lack of known FA2H homologs in human, FA2H is highly expressed where 2-hydroxy fatty acids are found: in the brain (primarily in oligodendrocytes), in peripheral myelin, and in skin keratinocytes.10,13 Furthermore, FA2H was shown to be the major fatty acid 2-hydroxylase in mouse brain given that anti-FA2H antibodies inhibited most of the fatty acid 2-hydroxylase activity.10 The fact that peripheral myelin was spared and that only the brain was affected in our patients was therefore unexpected and suggested a second fatty acid 2-hydroxylating activity in human. To clarify this issue, we measured the fatty acid 2-hydroxylation in cultured skin fibroblasts of the patients and controls by using deuterated tetracosanoic acid as a substrate and discovered that the amount of labeled 2-hydroxy-tetracosanoic acid was slightly higher in the patients than in the controls (0.178 pmol/min/mg protein in a family 1 patient, 0.127 pmol/min/mg protein in a family 3 patient, and a mean value of 0.113 pmol/min/mg protein in two separate normal fibroblast cell lines). Because family 1's patients are homozygous for a deleterious mutation at the FA2H gene, these results are supportive of an additional fatty-acid-hydroxylation activity that is probably expressed in human fibroblasts but not in human brain and may explain peripheral myelin sparing.
Myelinogenesis in human brain starts prenatally at ∼20 weeks of gestation and continues at a significant rate till 5–6 years of age.14 The galactolipids are major constituents of the lipid fraction of myelin and half of their fatty acids are hydroxylated at their C2 position.15,16 The proportion of 2-hydroxy to nonhydroxy galactolipids in rat and mouse brain increases 6- to 8-fold with age and parallels the expression of myelin protein genes.10,15 These data suggest a role for 2-hydroxylation in myelin maturation; studies in semi-synthetic 2-hydroxyl galactolipids revealed that this modification contributes to the hydrogen-bonding network formed between neighboring lipid molecules.17 That deleterious mutations in the FA2H gene manifested clinically only when patients were 4–6 years of age is consistent with this timeline of myelin formation and with the need for 2-hydroxy galactolipids at a later stage of this process.
Many forms of genetic leukodystrophy, such as adrenoleukodystrophy (MIM 300100), Krabbe disease (MIM 245200), and metachromatic leukodystrophy (MIM 250100), are not associated with defective myelin composition but are rather caused by a toxic effect of the accumulating material on the myelin-forming cells.18,19 Mutations in the PLP1 gene that encodes the major structural protein of central myelin, protein lipid protein 1, are the only known deficiency of a myelin structural component in human. The clinical phenotypes range from the severe connatal leukodystrophy, Pelizaeus-Merzbacher disease (PMD [MIM 312080]), to a milder x-linked spastic paraparesis (SPG2 [MIM 312920]).20 To our knowledge, FA2H deficiency is the first identified deficiency in the lipid compartment of myelin in human. We speculate that the relatively milder phenotype of complete FA2H deficiency, compared to the severe phenotype of complete PLP1 deficiency (PMD), might be partly attributed to the timeline of myelin assembly; the bulk of myelin lipids are added at a late stage of myelin assembly and their entry is directed by the already present proteins.21 We propose that the severe FA2H mutation associated with leukodystrophy and its allelic, milder mutation, associated with spastic paraparesis are analogous to PMD and SPG2 as well as to MITCHAP60 disease (MIM 612233) and SPG13 (MIM 605280).22 In these three allelic couples, the severe mutations cause leukodystrophy, whereas the milder mutations cause spastic paraplegia.
We conclude that mutations in the FA2H gene are associated with a progressive neurologic disorder manifested by spasticity, dystonia, and white matter degeneration. This is the seventeenth locus associated with an autosomal-recessive complex spastic paraparesis,23 and SPG35 may even be an allelic form.24 Nonetheless, because of the prominent additional symptoms and the role of FA2H in myelinogenesis, FA2H deficiency should be regarded as a new form of leukodystrophy.
Given the long presymptomatic period in our patients and the full penetrance of their disease, venues of therapy should be sought. Administration of 2-OH fatty acids could be considered with caution because of the potential toxicity of 2-OH fatty acids outside of the ER, the site of GalC synthesis.25 Notably, other myelin sphingolipids, sphingomyelin, and complex glycolipids, which are synthesized in the Golgi apparatus, do not contain 2-OH fatty acids. The observation of minor expression of FA2H in primary microglia10 raises the possibility of hematopoeitic stem cell transplantation. The safety and efficacy of these options should be clarified in an animal model.
Acknowledgments
We gratefully acknowledge the collaboration of the patients' families. This work was supported by the Joint Research Fund of the Hebrew University and Hadassah Medical Organization and by the National Institute of Health grant NS060807.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Morell P., Quarles R.H. Myelin Formation, Structure and Biochemistry. In: Siegel G.J., Agranoff B.W., Fisher S.K., Albers R.W., Uhler M.D., editors. Basic Neurochemistry. Molecular, Cellular and Medical Aspects. Sixth Edition. Lippincott-Raven; New York: 1999. pp. 69–94. [Google Scholar]
- 2.Campagnoni A.T. Molecular biology of myelination. In: Kettenmann H., Ransom B.R., editors. Neuroglia. Oxford University Press; New York: 2004. pp. 253–263. [Google Scholar]
- 3.Norton W.T., Cammer W. Isolation and characterization of myelin. In: Morell P., editor. Myelin. Plenum Press; New York: 1984. pp. 147–195. [Google Scholar]
- 4.Webster H.D.F., Sternberger N.H. Morphological features of myelin formation. In: Bauman N., editor. Neurological mutations affecting myelination. INSERM Symposium. Vol. 14. Elsevier; 1980. pp. 73–86. [Google Scholar]
- 5.Edvardson S., Shaag S., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-tRNA synthetase gene is associated with ponto-cerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alderson N.L., Rembiesa B.M., Walla M.D., Bielawska A., Bielawski J., Hama H. The human FA2H gene encodes a fatty acid 2-hydroxylase. J. Biol. Chem. 2004;279:48562–48568. doi: 10.1074/jbc.M406649200. [DOI] [PubMed] [Google Scholar]
- 7.Eckhardt M., Yaghootfam A., Fewou S.N., Zöller I., Gieselmann V.A. Mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem. J. 2005;388:245–254. doi: 10.1042/BJ20041451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 9.Alderson N.L., Walla M.D., Hama H. A novel method for the measurement of in vitro fatty acid 2-hydroxylase activity by gas chromatography-mass spectrometry. J. Lipid Res. 2005;46:1569–1575. doi: 10.1194/jlr.D500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Alderson N.L., Maldonado E.N., Kern M.J., Bhat N.R., Hama H. FA2H-dependent fatty acid 2-hydroxylation in postnatal mouse brain. J. Lipid Res. 2006;47:2772–2780. doi: 10.1194/jlr.M600362-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Matsunaga I., Sumimoto T., Ueda A., Kusunose E., Ichihara K. Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis: Substrate structure required for alpha-hydroxylation. Lipids. 2000;35:365–371. doi: 10.1007/s11745-000-533-y. [DOI] [PubMed] [Google Scholar]
- 12.Croes K., Foulon V., Casteels M., Van Veldhoven P.P., Mannaerts G.P. Phytanoyl-CoA hydroxylase: Recognition of 3-methyl-branched acyl-CoAs and requirement for GTP or ATP and Mg2+ in addition to its known hydroxylation cofactors. J. Lipid Res. 2000;41:629–636. [PubMed] [Google Scholar]
- 13.Uchida Y., Hama H., Alderson N.L., Douangpanya S., Wang Y., Crumrine D.A., Elias P.M., Holleran W.M. Fatty acid 2-hydroxylase, encoded by FA2H, accounts for differentiation-associated increase in 2-OH ceramides during keratinocyte differentiation. J. Biol. Chem. 2007;282:13211–13219. doi: 10.1074/jbc.M611562200. [DOI] [PubMed] [Google Scholar]
- 14.Kinney H.C., Karthigasan J., Borenshteyn N.I., Flax J.D., Kirschner D.A. Myelination in the developing human brain: biochemical correlates. Neurochem. Res. 1994;19:983–996. doi: 10.1007/BF00968708. [DOI] [PubMed] [Google Scholar]
- 15.Hoshi M., Williams M., Kishimoto Y. Characterization of brain cerebrosides at early stages of development in the rat. J. Neurochem. 1973;21:709–712. doi: 10.1111/j.1471-4159.1973.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto Y., Radin N.S. Isolation and determination methods for brain cerebrosides, hydroxy fatty acids, and unsaturated and saturated fatty acids. J. Lipid Res. 1959;1:72–78. [Google Scholar]
- 17.Boggs J.M., Koshy K.M., Rangaraj G. Influence of structural modifications on the phase behavior of semi-synthetic cerebroside sulfate. Biochim. Biophys. Acta. 1988;938:361–372. doi: 10.1016/0005-2736(88)90134-4. [DOI] [PubMed] [Google Scholar]
- 18.Hein S., Schönfeld P., Kahlert S., Reiser G. Toxic effects of X- linked adrenoleukodystrophy (X-ALD)-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum. Mol. Genet. 2008;17:1750–1761. doi: 10.1093/hmg/ddn066. [DOI] [PubMed] [Google Scholar]
- 19.Igisu H., Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- 20.Saugier-Veber P., Munnich A., Bonneau D., Rozet J.M., Le Merrer M., Gil R., Boespflug-Tanguy O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet. 1994;6:257–262. doi: 10.1038/ng0394-257. [DOI] [PubMed] [Google Scholar]
- 21.Simons M., Trotter J. Wrapping it up: The cell biology of myelination. Curr. Opin. Neurobiol. 2007;17:533–540. doi: 10.1016/j.conb.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Magen D., Georgopoulos C., Bross P., Ang D., Segev Y., Goldsher D., Nemirovski A., Shahar E., Ravid S., Luder A. Mitochondrial Hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008;83:30–42. doi: 10.1016/j.ajhg.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: an update. Curr. Opin. Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 24.Dick K.J., Al-Mjeni R., Baskir W., Koul R., Simpson M.A., Patton M.A., Raeburn S., Crosby A.H. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology. 2008;71:248–252. doi: 10.1212/01.wnl.0000319610.29522.8a. [DOI] [PubMed] [Google Scholar]
- 25.Lippel K., Mead J.F. Alpha-oxidation of 2-hydroxy tetracosanoate in the rat. Lipids. 1969;4:129–134. doi: 10.1007/BF02531931. [DOI] [PubMed] [Google Scholar]