

Abstract
There are many thousands of hereditary diseases in humans, each of which has a specific combination of phenotypic features, but computational analysis of phenotypic data has been hampered by lack of adequate computational data structures. Therefore, we have developed a Human Phenotype Ontology (HPO) with over 8000 terms representing individual phenotypic anomalies and have annotated all clinical entries in Online Mendelian Inheritance in Man with the terms of the HPO. We show that the HPO is able to capture phenotypic similarities between diseases in a useful and highly significant fashion.
Main Text
Analysis of the phenotypic correlates of gene mutations has long been an essential method for discovering biological functions of genes, and more recently, computational analysis of mouse phenotypes related to gene mutations has become possible with tools such as the Mammalian Phenotype Ontology.1,2 Phenotypic analysis has played a central role in the mapping of disease genes and many other fields, and humans are particularly good at recognizing human phenotypic traits and anomalies. However, there are a number of unresolved issues surrounding the computational description and analysis of human phenotypes.
It seems intuitively clear that certain hereditary disorders are phenotypically similar to one another because of shared phenotypic features. For instance, one might say that Marfan syndrome (MIM 154700) and congenital contractural arachnodactyly (MIM 121050) are similar because they share a range of skeletal abnormalities, and in fact the genes mutated in these syndromes, FBN1 and FBN2, belong to the same gene family and share a number of functional similarities.3 The observation that many genetic conditions show overlapping features led to the concept of disease families.4,5 Phenotypic similarities within disease families may be related to dysfunction of a regulatory network, such as a signaling pathway or a biochemical module, as has been demonstrated for Noonan syndrome (MIM 163950) and related disorders.6 Thus, phenotypic analysis is of great importance for our understanding of the physiology and pathophysiology of cellular networks because it can offer clues about groups of genes that together make up pathways or modules, in which dysfunction can lead to similar phenotypic consequences. A number of recent works have suggested the enormous potential of correlating phenotype to features of genetic or cellular networks on a genome-wide scale.7–9
The great majority of human Mendelian syndromes have been described in detail in the Online Mendelian Inheritance in Man (OMIM) database,10 and hierarchical systems based on the clinical descriptions in OMIM have been generated by text mining.11–13 However, computational analysis of the data contained in OMIM has so far been hampered by the lack of a controlled vocabulary including consistent annotations with well-defined relationships to one another. For instance, the descriptions “generalized amyotrophy,” “generalized muscle atrophy,” “muscular atrophy, generalized,” and “muscle atrophy, generalized” are used to describe different diseases in OMIM and might not be easily recognized as synonyms with a purely computational approach. Also, although the clinical-synopsis entries in OMIM are grouped according to organ system, the hierarchical structure does not itself reflect that (for instance) “Hypoplastic philtrum” and “Smooth philtrum” are more closely related to one another than to “Hypoplastic nasal septum” (all three of these descriptions are in the NOSE category of OMIM's clinical synopsis).
An ontology is a data model that represents concepts, attributes, and relationships in the form of a directed acyclic graph. The Gene Ontology (GO), especially, has proven to be extremely useful for the exploratory analysis of microarray and other forms of high-throughput data.14 A number of considerations suggest that an ontological description of human phenotypes has distinct advantages; this prompted us to develop an ontology to describe human phenotypic abnormalities.
The Human Phenotype Ontology (HPO) was constructed with the goal of covering all phenotypic abnormalities that are commonly encountered in human monogenic diseases. To this end, the “omim.txt” file was downloaded from the OMIM database.10 We developed a suite of Java programs and Perl scripts to parse omim.txt in order to extract the textual descriptions of each disease as listed in the Clinical Synopsis section, which is ordered in a hierarchical fashion. For instance, in the description of Marfan syndrome, aortic root dilatation is listed under the category CARDIOVASCULAR, subcategory Vascular. We then generated a list of these features, sorted according to the frequency of occurrence. For instance, aortic root dilatation is listed for a number of diseases other than Marfan syndrome, including Ehlers-Danlos syndrome, type I (MIM 130000). On the other hand, Medial rotation of the medial malleolus is used only once (for Marfan syndrome) in all of omim.txt.
The HPO was then constructed with OBO-Edit15 in order to define terms and the links between them on the basis of the list of descriptions from omim.txt. For all descriptions that occurred more than once in omim.txt, we created a term in the HPO. One of the main difficulties in using data from OMIM in computational analysis is that OMIM does not use a controlled vocabulary, and it can be difficult to recognize synonyms by computational means. Therefore, we manually curated each term, taking advantage of domain knowledge in human genetics (PNR, DH, SM) in order to merge synonyms into unique HPO terms. For instance, the three OMIM descriptions Carpal bone hypoplasia, Hypoplasia of carpal bones, and Hypoplastic carpal bones were fused into the single term HP:0001498, Carpal bone hypoplasia. We additionally adapted the Smith-Waterman algorithm16 to map additional descriptions that were used only once in omim.txt as synonyms or children of HPO terms. However, each mapping proposed by this algorithm was examined by hand before incorporation into the HPO. Domain knowledge was also used to define more general terms, such as Aplasia/hypoplasia of the outer ear, to group more specific terms, as well as to define links between individual terms.
Each term in the HPO describes a phenotypic abnormality, such as Atrial septum defect. Terms are related to parent terms by “is a” relationships. The structure of the HPO, which allows a term to have multiple parent terms, enables different aspects of phenotypic abnormalities to be explored (see Figure 1 for an example). The HPO itself does not describe individual disease entities but, rather, the phenotypic abnormalities associated with them. The majority of the HPO terms describe organ abnormalities, but separate ontologies are provided for describing the mode of inheritance and the onset and clinical course. We have used the HPO terms to annotate all entries of OMIM with a clinical-synopsis section. Clinical entities are annotated to the most specific terms possible. The true-path rule17 applies to the terms of the HPO. That is, if a disease is annotated to the term Atrial septal defect, then all of the ancestors of this term, such as Abnormality of the cardiac septa, also apply. The structure of the HPO, therefore, allows flexible searches for disease entities according to phenotypic abnormalities, with a broad or narrow focus.
Figure 1.
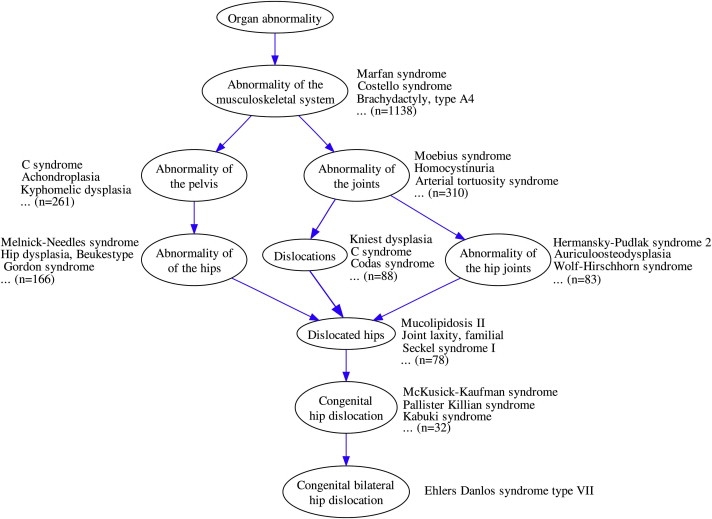
The Human Phenotype Ontology
The HPO term Bilateral congenital hip dislocation and all paths to the root that emanate from this term are shown. Some of the annotated disease entries from OMIM, as well as the total number of annotated diseases, are shown next to the terms. Note that because of the true-path rule, a disease that is directly annotated to a specific term is also implicitly annotated to all ancestors of that term. For instance, Ehlers Danlos syndrome type VII is directly annotated to Bilateral congenital hip dislocation and is thereby implicitly annotated to Abnormality of the hips, Dislocations, and the other terms shown in the figure.
We will now show that an ontological similarity measure defines a useful and highly significant phenotypic-similarity metric among hereditary diseases listed in OMIM. In the HPO, as in GO, terms that are closer to the root of the ontology represent more general concepts than do terms that are farther away from the root. (For instance, in Figure 1, the term Abnormality of the joints is more general than the term Congenital hip dislocation.) The information content of each node in the HPO can be estimated through its frequency among annotations of the entire OMIM corpus. In our implementation, the information content of a term is based on the frequency of annotations to that term among the 4779 diseases in the OMIM database that were annotated to terms of the HPO. Intuitively, if two diseases are both annotated to a term with high information content, such as Calcific stippling, then the degree of similarity calculated on the basis of this should be higher than that calculated when the two diseases are both annotated to a more general term, such as Abnormality of the musculoskeletal system, which has a lower information content.
For each term t of the HPO, the information content is quantified as the negative logarithm of its probability: −logp(t), in which the probability of a term is taken to be its frequency among annotations to all 4779 annotated diseases. If a disease is annotated to any term t in the HPO, it must also be annotated to all the ancestors of t. Therefore, the higher in the ontology a term is located, the lesser its information content. Resnik18 introduced a similarity measure for two terms in an ontology that is based on their shared information content, which is given by the information content in the set of their common-ancestor nodes. In the case of HPO, a term might have multiple parent terms, so that a pair of terms might have more than one path of common ancestors. Denoting the set of all common-ancestor terms of terms t1 and t2 as A(t1, t2), we define the similarity between two terms, t1 and t2, as
| (1) |
which defines the probability of the minimum common ancestor of t1 and t2. Individual diseases are usually annotated to multiple phenotypic features. In order to calculate the similarity between two diseases, d1 and d2, we adapt a method previously developed for estimating protein similarity with GO,19 whereby each feature of d1 is matched with the most similar feature of d2 and the average is taken over all such pairs of features:
| (2) |
Because Equation (2) is not symmetric with respect to d1 and d2, the final similarity metric is defined as the mean of Equation (2) taken in both orientations:
| (3) |
This metric can now be used to define the similarity between two diseases, each of which is annotated to multiple terms of the HPO.
We used this measure to define similarity between the diseases listed in the OMIM database. We analyzed 727 diseases belonging to one of 21 physiological-disorder classes.7 Phenotypic relationships between these diseases are shown by the linking of all pairs of diseases exceeding a threshold similarity score (Figure 2A). Although generated independently of the disorder classes, the resulting phenotypic network clearly displays clusters corresponding to many of the 21 classes. It is also apparent that some of the clusters show interconnections between one another, which were not visible in the disease map based only on shared disease genes.7 For instance, hematological disorders are strongly connected to immunological disorders, bone disorders are strongly connected to skeletal disorders, and neurological, muscular, and psychiatric disorders are multiply linked to one another. Also, diseases that cluster together with diseases from a different physiological class share important phenotypic similarities with that class. For instance, all four diseases that are from the metabolic class and are located in the muscle cluster (Enolase-beta deficiency, MCardle disease, dimethylglycine dehydrogenase deficiency, and elevated serum creatine phosphokinase) show important muscular symptoms (Figure 2A). Analysis of randomized networks showed that the observed correlation between network connections and disease class is highly significant (Figure 2B). Thus, this phenotypic network, as defined by the HPO, is made up of dense clusters of shared phenotypic features that show characteristic patterns of interconnections between selected areas of the phenotypic continuum.
Figure 2.
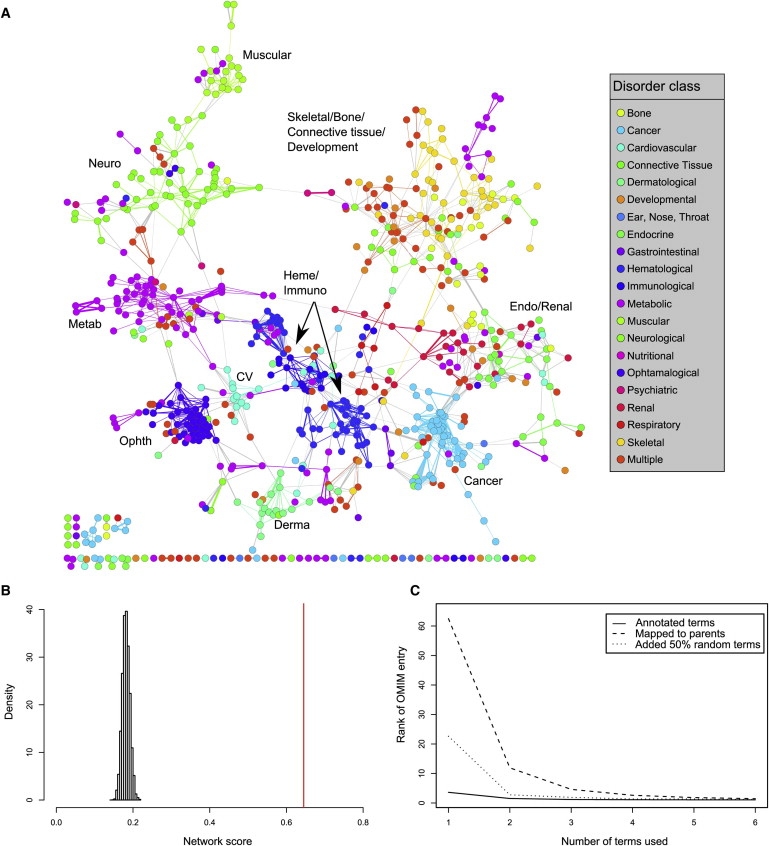
Applications of the HPO
(A) Visualization of the human phenome. Each of 727 diseases listed in OMIM for which a disorder class was defined is shown as a node in the graph and is colored according to membership in a set of 21 predefined disorder classes, defined on the basis of the physiological system.7 The organic layout algorithm of Cytoscape27 was used for showing the clustered structure of the phenotypic network. Connections between nodes are shown starting from a similarity score of 4.5, whereby the thickness of the connection reflects the degree of phenotypic similarity. Abbreviations are as follows: CV, cardiovascular; derma, dermatological; endo, endocrinological; heme, hematological; immuno, immunological; metab, metabolic; neuro, neurological; ophth, ophthalmological.
(B) Analysis of randomized phenotypic networks. In order to estimate the probability that this result could be due to chance, we created 10,000 random networks in which edges were randomly rewired 2000 times.28 The mean network score of the random nets was 0.182 ± 0.0098. Thus, the actual score of 0.645 was 47.2 standard deviations above the mean random score.
(C) Searching the HPO. All 2116 diseases listed in OMIM with at least six HPO annotations were identified and included in the analysis. For each disease, between 1 and 6 of the most specific terms to which the disease is annotated in the HPO were used for the search (“Annotated terms”). The set of clinical features determined in this way was then used for querying the entire set of OMIM diseases for the best match. The average rank of the original disease among the diseases identified by the search for different proportions of removed terms is shown. In separate experiments, each of these terms was mapped to a parent term or 50% unrelated (“noise”) terms were added (rounded up for odd numbers of terms; i.e., one noise term was added to searches with one term, two noise terms to searches with three terms, and three noise terms to searches with five terms).
Next, we explored the utility of using the HPO in a clinical setting. Combinations of features are often used in medical genetics in the search for a clinical diagnosis. This can be a challenging undertaking, given the large number of hereditary disorders and the range of partially overlapping clinical features associated with them. Clinicians may be able to describe clinical features in varying levels of detail. Also, an individual patient with a hereditary disease might not show all of the features that are potentially associated with a disorder, or he or she may have additional features unrelated to the disorder. Optimally, diagnostic algorithms will allow searches at varying levels of detail, weigh specific features more highly than general features, and not be overly sensitive to the fact that individual features might not be present in an individual patient. An ontological approach, therefore, appears to be particularly appropriate in this setting. To simulate this kind of search process, we used the 2116 diseases in OMIM annotated to at least six HPO terms to simulate a clinical-search process by selecting only 1–6 of the terms associated with a given disease and then searching in the original database for similar diseases. Optimally, the search algorithm will assign the original disease the highest rank or at least place it among the first few diseases. As can be seen in Figure 2C, there was excellent performance, even when only two search terms were used, with relatively small reductions in accuracy when 50% of random noise terms were added to the search terms or when each of the terms was mapped to a (less specific) parent term. This suggests that the HPO is able to capture phenotypic similarity at various levels of granularity and that the calculation of similarity is not overly sensitive to noise, completeness, or specificity of the set of phenotypic terms used for the search.
Previous efforts at computational analysis of phenotypic data in human hereditary disease have involved various strategies for automated text mining of OMIM. A number of works have used MeSH terms or Unified Medical Language System (UMLS) concepts to map phenotypic concepts from medline or OMIM.13,20–22 Most of these works used the text-mined concepts to create feature vectors in order to describe diseases. That is, a feature vector for a given disease contains one entry for each concept, set to 1 if the concept is found in the disease and to 0 otherwise. Similarity is often measured by some variation of the normalized cosine angle between normalized feature vector pairs.22 Although these works were successful, we contend that a manually curated ontological approach to computational phenotype analysis offers a number of advantages. One difficulty with text-mining approaches is that the MeSH and UMLS indexing terms are not specifically designed for the needs of describing human hereditary diseases and their phenotypes. For instance, only 34% of 1700 diseases with a specific phenotype associated with a specific gene in OMIM could be specifically mapped to concepts in the UMLS.23 One advantage of the HPO is that the terms and structure of the ontology are based on medical knowledge rather than on text-mining systems. The HPO will be refined and extended in the future. An ontology as a data structure has several distinct advantages over other kinds of data structure that have been used for phenotypic analysis. One of the main reasons for the success of GO is the greater flexibility and descriptive power of ontologies compared to hierarchical systems and feature vectors,14 including the ability to relate concepts (terms) to multiple parents and to allow descriptions and queries at different levels of granularity and completeness. Additionally, a number of computational algorithms have been developed for ontological analysis after the success of GO (e.g., 19,24,25) and can now be applied to human phenotype data.
The value of data is greatly increased as sources of data are enabled to be integrated with one another. We have designed the HPO using the widely used OBO format.26 This file, together with a flat file with annotations of 4779 diseases listed in OMIM to the terms of the HPO, is freely available for download from the HPO web site, which also describes the background and goals of the project. The HPO is participating in the OBO Foundry project,26 and the files are available for download there as well. We anticipate that the HPO will continue to evolve over many years and are currently recruiting collaborators to refine specific areas of the HPO, to improve annotations to disorders listed in OMIM, and to extend the annotations to other hereditary disorders, such as microdeletion syndromes and chromosomal aberrations. We plan to include additional information, including frequency and severity of features, in the annotation files. It is hoped that the HPO will provide a basis for computational biomedical research involving human phenotype analysis, allowing the human phenome to be related to the molecular pathophysiology of the cell by directly linking the human phenome to sources of data such as protein-protein interactions, metabolic and signal-transduction pathways, and gene coexpression. Additionally, we anticipate that the HPO will provide a unified basis for clinical research in medical genetics by providing a standardized vocabulary for the description of phenotypes.
Acknowledgments
This work was supported by the Berlin-Brandenburg Center for Regenerative Therapies (BCRT) (Bundesministerium für Bildung und Forschung, project number 0313911) and the Deutsche Forschungsgemeinschaft (SFB 760).
Web Resources
The URLs for data presented herein are as follows:
The Gene Ontology (GO), http://www.geneontology.org/
The Human Phenotype Ontology, http://www.human-phenotype-ontology.org
The Obo Foundry, http://obofoundry.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Smith C.L., Goldsmith C.-A.W., Eppig J.T. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6:R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lussier Y.A., Liu Y. Computational approaches to phenotyping: high-throughput phenomics. Proc. Am. Thorac. Soc. 2007;4:18–25. doi: 10.1513/pats.200607-142JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson P.N., Arteaga-Solis E., Baldock C., Collod-Broud G., Booms P., Paepe A.D., Dietz H.C., Guo G., Handford P.A., Judge D.P. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006;43:769–787. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunner H.G., van Driel M.A. From syndrome families to functional genomics. Nat. Rev. Genet. 2004;5:545–551. doi: 10.1038/nrg1383. [DOI] [PubMed] [Google Scholar]
- 5.Oti M., Brunner H. The modular nature of genetic diseases. Clin. Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 6.Gelb B.D., Tartaglia M. Noonan syndrome and related disorders: Dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15 Spec No 2:R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- 7.Goh K.-I., Cusick M.E., Valle D., Childs B., Vidal M., Barabsi A.-L. The human disease network. Proc. Natl. Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman I., Rzhetsky A., Vitkup D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Köhler S., Bauer S., Horn D., Robinson P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamosh A., Scott A.F., Amberger J.S., Bocchini C.A., McKusick V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masseroli M., Galati O., Manzotti M., Gibert K., Pinciroli F. Inherited disorder phenotypes: controlled annotation and statistical analysis for knowledge mining from gene lists. BMC Bioinformatics. 2005;6(Suppl 4):S18. doi: 10.1186/1471-2105-6-S4-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bajdik C.D., Kuo B., Rusaw S., Jones S., Brooks-Wilson A. CGMIM: automated text-mining of Online Mendelian Inheritance in Man (OMIM) to identify genetically-associated cancers and candidate genes. BMC Bioinformatics. 2005;6:78. doi: 10.1186/1471-2105-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Driel M.A., Bruggeman J., Vriend G., Brunner H.G., Leunissen J.A.M. A text-mining analysis of the human phenome. Eur. J. Hum. Genet. 2006;14:535–542. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- 14.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene Ontology: tool for the unification of biology. the Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day-Richter J., Harris M.A., Haendel M., Gene Ontology OBO-Edit Working Group. Lewis S. OBO-Edit–an ontology editor for biologists. Bioinformatics. 2007;23:2198–2200. doi: 10.1093/bioinformatics/btm112. [DOI] [PubMed] [Google Scholar]
- 16.Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 17.Gene Ontology Consortium Creating the Gene Ontology resource: design and implementation. Genome Res. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Resnik P. Using information content to evaluate semantic similarity in a taxonomy. Proceedings of the 14th IJCAI. 1995;1:448–453. [Google Scholar]
- 19.Pesquita C., Faria D., Bastos H., Ferreira A.E.N., Falco A.O., Couto F.M. Metrics for GO based protein semantic similarity: a systematic evaluation. BMC Bioinformatics. 2008;9(Suppl 5):S4. doi: 10.1186/1471-2105-9-S5-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Iratxeta C., Bork P., Andrade M.A. Association of genes to genetically inherited diseases using data mining. Nat. Genet. 2002;31:316–319. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- 21.Hristovski D., Peterlin B., Mitchell J.A., Humphrey S.M. Using literature-based discovery to identify disease candidate genes. Int. J. Med. Inform. 2005;74:289–298. doi: 10.1016/j.ijmedinf.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 22.Lage K., Karlberg E.O., Størling Z.M., Olason P.I., Pedersen A.G., Rigina O., Hinsby A.M., Tümer Z., Pociot F., Tommerup N. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 23.Bodenreider O., Mitchell J.A., McCray A.T. Evaluation of the UMLS as a terminology and knowledge resource for biomedical informatics. Proc AMIA Symp. 2002:61–65. [PMC free article] [PubMed] [Google Scholar]
- 24.Grossmann S., Bauer S., Robinson P.N., Vingron M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y., Rosenfeld R., Simon I., Nau G.J., Bar-Joseph Z. A probabilistic generative model for GO enrichment analysis. Nucleic Acids Res. 2008;36:e109. doi: 10.1093/nar/gkn434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith B., Ashburner M., Rosse C., Bard J., Bug W., Ceusters W., Goldberg L.J., Eilbeck K., Ireland A., Mungall C.J. The OBO foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007;25:1251–1255. doi: 10.1038/nbt1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maslov S., Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296:910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]