

Abstract
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous inherited disorder arising from dysmotility of motile cilia and sperm. This is associated with a variety of ultrastructural defects of the cilia and sperm axoneme that affect movement, leading to clinical consequences on respiratory-tract mucociliary clearance and lung function, fertility, and left-right body-axis determination. We performed whole-genome SNP-based linkage analysis in seven consanguineous families with PCD and central-microtubular-pair abnormalities. This identified two loci, in two families with intermittent absence of the central-pair structure (chromosome 6p21.1, Zmax 6.7) and in five families with complete absence of the central pair (chromosome 6q22.1, Zmax 7.0). Mutations were subsequently identified in two positional candidate genes, RSPH9 on chromosome 6p21.1 and RSPH4A on chromosome 6q22.1. Haplotype analysis identified a common ancestral founder effect RSPH4A mutation present in UK-Pakistani pedigrees. Both RSPH9 and RSPH4A encode protein components of the axonemal radial spoke head. In situ hybridization of murine Rsph9 shows gene expression restricted to regions containing motile cilia. Investigation of the effect of knockdown or mutations of RSPH9 orthologs in zebrafish and Chlamydomonas indicate that radial spoke head proteins are important in maintaining normal movement in motile, “9+2”-structure cilia and flagella. This effect is rescued by reintroduction of gene expression for restoration of a normal beat pattern in zebrafish. Disturbance in function of these genes was not associated with defects in left-right axis determination in humans or zebrafish.
Introduction
Primary ciliary dyskinesia (PCD, MIM 242650) refers to a heterogeneous group of genetic ciliopathies characterized by ultrastructural defects in the axonemal structure of “9+2” motile cilia and sperm flagella.1 The incidence is estimated at 1:15,000–30,000,2 with higher incidence in certain consanguineous and isolated populations.3,4 Clinical features reflect the distribution of dysmotile cilia in the body and include neonatal respiratory distress, chronic respiratory infections, sinusitis, and bronchiectasis, due to deficient cilia function in the upper and lower airways. Male and female subfertility occurs as a result of defective sperm flagella and oviduct cilia, respectively. There is occasional hydrocephalus as a result of deficient ependymal cilia.5,6 In most families, there is apparent randomization of left-right axis development, proposed to result from defective function of embryonic nodal cilia.7,8 This manifests in about half of patients as situs inversus or more severe laterality defects, such as cardiovascular abnormalities.
PCD is usually recessively inherited, and five PCD genes have been identified: DNAI1 (MIM 604366), DNAH5 (MIM 603335), DNAH11 (MIM 603339), DNAI2 (MIM 605483)9 and TXNDC3 (MIM 607421) (reviewed by Bush et al.10). These encode axonemal dyneins and are associated with reduction or loss of axonemal outer dynein arms, which are the multisubunit axonemal ATPase complexes that generate the force for cilia motility and govern beat frequency. DNAH5 and DNAI1 are a relatively more common cause of disease, underlying an estimated 28% (DNAH511) and 2%–10% (DNAI112,13) of total cases. The other genes are so far only associated with single or rare PCD cases; therefore, the genetic basis of at least 60% of PCD cases is not yet known. Several additional PCD loci have been mapped, including those on chromosomes 19q13,14 16p12,3 15q13-q15,3 and 15q24-q25,15 but these genes remain to be isolated (reviewed by Geremek and Witt16). The possibility of involvement of PCD genes in sensory- and retinal-cilia functions is suggested by reports of syndromic forms, similar to PCD, that have motile-cilia dysfunction and additional features, including retinitis pigmentosa,17 polycystic kidney disease,18 and mental retardation.19
Patients with ultrastructural abnormalities affecting the central microtubular pair represent a well-recognized subgroup in whom laterality defects have not been observed.20 This is assumed to reflect the 9+0 ultrastructure of nodal cilia, which may be unchanged by mutations affecting a structural element that they do not possess (in most species). Using two distinct family groups affected by PCD associated with central-microtubular-pair defects, we performed SNP-based whole-genome linkage analysis to identify the disease-causing genes.
Subjects and Methods
Families, Clinical Information, and Controls
Informed consent was obtained from patients and family members in accordance with protocols approved by the University College London Hospital NHS Trust ethical committee and collaborating institutions. The diagnosis of PCD was based on the exclusion of cystic fibrosis, immunodeficiency, and tuberculosis and the presentation of classic clinical features. These features included the following: reduced exercise tolerance; chronic wet cough; recurrent respiratory infections; nasal symptoms, including rhinorrhea, rhinitis, nasal blockage, and sinusitis; in addition to glue ear and consequent hearing problems. Bronchiectasis also occurred. Low weight and short stature were noted in the Bedouin and Pakistani families.
The pedigrees are presented in Figure 1A. Family UCL146 from the United Arab Emirates (UAE) was previously reported, showing one patient with a collapsed lower-left pulmonary lobe.20 The family originated from the Bedouin Bani Tameem tribe, and the parents are first cousins. There is an unusual intermittent loss of the central pair in this family, confirmed by longitudinal-section electron microscopy in all three affected children, such that cilia cross-sections show a small proportion with 9+0 structure. Family UCL152 from Israel is also Bedouin, with multiple consanguineous unions within the extended pedigree. Transmission electron microscopy (TEM) performed on one affected member showed a normal axoneme ultrastructure. Family UCL152 was included in the study despite an apparently normal ultrastructure because of respiratory symptoms consistent with a diagnosis of PCD, as described above, and dysmotility of the respiratory cilia, as described below. Furthermore, dysmotile sperm was also reported in two males, and one female required in vitro fertilization treatment as a result of subfertility.
Figure 1.
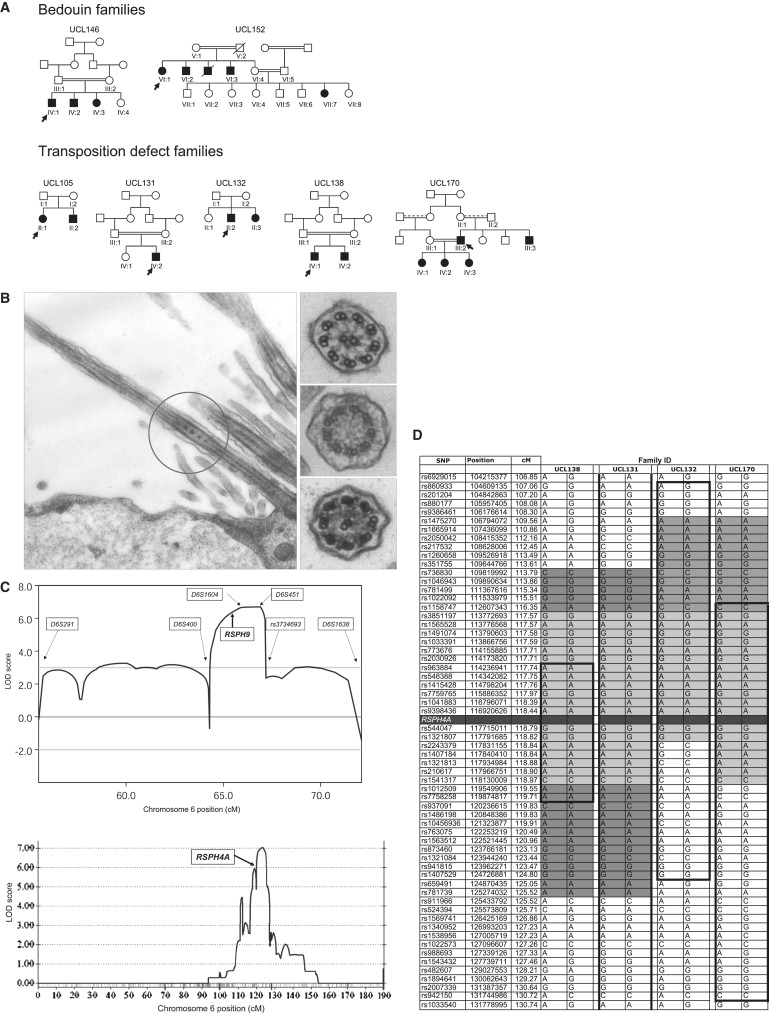
Central-Pair Agenesis in PCD Patients and Linkage Analysis
(A) PCD central-pair-defect pedigrees. Black indicates affected; double line indicates consanguineous union, with a dashed upper line if the exact degree of relatedness is unknown; arrow indicates proband.
(B) Transmission electron micrographs of nasal ciliary epithelium from individuals with a central-pair defect. Left panel, Bedouin UCL146 IV:1 longitudinal section with intermittent central-pair loss, confirming the previous report20 of 9+2 (normal) and 9+0 cross-sections. Right panels, cross-sections from UK-Pakistani UCL170 IV:1, indicative of complete central-pair loss (transposition defect), showing 9+2 (upper panel), 9+0 (middle panel), or 8+1 (lower panel) ultrastructure.
(C) Linkage mapping of families with central-pair defects, to two loci, on chromosome 6p21.1 and 6q22.1. Upper panel, multipoint MERLIN linkage analysis of UCL146 and UCL152 on 6p21.1 across D6S291–D6S1638, with the use of information from Illumina and deCODE scans and in-house microsatellite genotyping. The overlapping shared region of IBD from D6S400 to rs3734693 is reflected by a significant multipoint LOD score > 3, which rises to a peak of 6.7 across D6S1604–D6S451. The location of RSPH9 is shown. Lower panel, multipoint GENEHUNTER linkage analysis of families with transposition defect on 6q22.1 across chromosome 6, with the use of Illumina scan information. The location of RSPH4A is shown, located centromeric to the peak homogeneity LOD score of 7.0, which was generated across markers rs873460–rs941815.
(D) Identical-by-descent homozygosity and founder effect in RSPH4A families. Chromosome 6q22.1 disease-chromosome haplotypes for each of the four UK-Pakistani families are shown, displaying extended homozygosity across the RSPH4A locus. Bold, black lines and boxes indicate the linked region in each pedigree, defined by recombination events or loss of homozygosity. The minimal critical region containing RSPH4A is rs2030926–rs937091 (defined by recombinations in UCL138). Homozygous allele sharing occurs among affected individuals (gray shading), with a putative minimal common ancestral haplotype spanning the RSPH4A gene, across markers rs1158747–rs2243379 (light gray).
Family UCL105 is a nonconsanguineous UK-Northern European family. Families UCL131, UCL132, UCL138, and UCL170 are all of Pakistani origin, now residing in the north of England. The parents in all of these families except family UCL132 are first cousins; any consanguinity in family UCL132 was not known to the family. TEM in affected children from all five families showed a classic ciliary transposition defect indicative of complete central-pair loss, as described in Stannard et al.20
Cilia-motility studies on nasal-biopsy samples collected from affected patients in all seven families showed an abnormal circular movement with a close to normal beat velocity, except for UCL152, which was recorded as having “abnormal motility.” Visualizing a circular pattern is dependent on viewing at the correct orientation, and this can account for some variability in recording.
Control DNA consisted of UK-Northern European samples from panels 1 and 2 of the European Collection of Cell Cultures (ECACC) Human Random Control Collection, anonymized UK-West Midlands Pakistani individuals (UK-Pakistani), Bedouin samples purchased from the National Laboratory for the Genetics of Israeli populations, unrelated UAE males previously reported,21 and additional unrelated members of other pedigrees (13 Pakistani and 26 Arabic), collected for mapping and polymorphism studies.
Linkage Analysis and Autozygosity Mapping
Genome-wide linkage analysis was performed in all families with the 0.64 cM density Illumina Linkage IVb 6008 SNP panel and in family UCL152 with the 8 cM density deCODE 500 microsatellite panel. SNP genotyping was performed at the Turku Centre for Biotechnology, Finland or at deCODE Genetics, Reykjavik, Iceland. Multipoint linkage analysis with the use of MERLIN 1.0.1 (within easyLINKAGE)22,23 or GENEHUNTER 2.1r524 was performed under the assumption of autosomal-recessive inheritance, 0.9 penetrance, 0.007 disease-allele frequency, and 0.00001 phenocopy rate, with allele frequencies set according to Illumina and deCODE map information.
Candidate-Gene Identification and Sequence and Protein Analysis
All known and predicted genes were identified at the RSPH9 locus with the UCSC, NCBI, and Ensembl genome browsers, then prioritized for sequencing with the assumption that PCD genes should be conserved in distantly related ciliated organisms but absent in nonciliated organisms. For identification of this class of gene, a protocol of serial BLAST searches25 was developed to interrogate draft genome sequences of twelve organisms, three nonciliated (A.thaliana, S.pombe, S.cerevisiae) and nine ciliated (C.reinhardtii genome, proteome,26 and deflagellation data set;27 T. brucei proteome;28 T. brucei/L.major combined genomes;29 C. intestinalis genome; T. thermophilia genome; C. elegans proteome (sensory cilia only); and a Foxj1 mouse ciliogenesis microarray of genes downregulated in this cilia-less mouse [S. Brody, Washington University, personal communication]). The process of database formatting, local BLAST searching, and data parsing was automated with a custom perl script (Blast2 matrix; available on request from R.D.E.: r.d.emes@hfac.keele.ac.uk). For this experiment, significant alignment results were determined with the use of an E value of ≤ 10−5 cutoff. The C6orf206 (RSPH9) protein was conserved in eight ciliate databases but not in C.elegans (sensory cilia only) or the three nonciliate databases.
Three prioritized positional candidate genes on chromosome 6p21.1, TBCC (MIM 602971), KNSL8, and C6orf206 (initially annotated as a mitochondrial ribosomal gene and since renamed as RSPH9), in addition to the RSPH4A gene on chromosome 6q22.1, were sequenced with genomic DNA from linked patients and primers designed to span the coding exons and splice sites. Primer sequences for RSPH9 and RSPH4A are shown in Table S1 (available online), and all other primer sequences are available on request. Sequence alignments to analyze conservation of mutated residues were generated using ClustalW and Boxshade. Protein domain analysis was conducted using SMART, PFAM, Genthreader and Scansite prediction tools.
Restriction-digest tests (RSPH9 p.Lys268 del, MboII [shown in Figure S3]; RSPH4A p.Gln154X, BsmAI; p.Pro87Ser, SacII; p.Gln109X, BspCNI) or sequencing (RSPH4A p.Arg490X) confirmed the inheritance pattern of all mutations in all extended kindreds.
In Situ Hybridization
The Rsp9 probe corresponded to the area from nt 317 in exon 2 to nt 837 in exon 5, amplified from mouse Riken cDNA clone 1700027N10 (RZPD German Resource Centre) with primers 5′-TGGTGAGTGGCCGTTTCAT-3′ and 5′-CCATGTTCTTCTCTCCTGTGC-3′, and the product was cloned into pCR4 by TOPO-TA cloning (Invitrogen). The mouse Dnah5 probe in pBluescript SKII+30 was kindly provided by H. Omran, University Hospital Freiburg. Sense and antisense digoxygenin-labeled riboprobes were prepared with the use of a digoxigenin RNA-labeling kit (Roche) and purified on Chroma Spin-100 columns (BD Biosciences), prior to in situ hybridization on whole-mount embryos or 7 μm paraffin-wax sections. Randomly bred CD1 mice served as a source of mouse embryos and fetuses. Litters were generated by timed matings, and the day that a copulation plug was found was designated embryonic day 0.5 (E0.5). Embryos were collected at E7.5, E18.5, and E19.5. Whole-mount in situ hybridization was performed as described previously.31 In situ hybridization on sections was performed as described previously32 and photographed with an MZFLIII, DC500 imaging system (Leica). Sense-strand controls yielded no specific hybridization signal.
Zebrafish Morpholino Injections
Morpholinos (MOs) were designed against the splice-donor sites of zebrafish rsph9 exon 2 (MOex2: 5′-GGTGTAAGGCTTTTACCGTGACCTC-3′) and exon 3 (MOex3: 5′-GCTGTAAGTATACCTCCAAAGCTTC-3′) (GeneTools). One- to two-cell-stage embryos were injected with 6.25 ng of MO in Danieau buffer (5 mM HEPES pH7.6, 58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mMCa(NO3)2). Green fluorescent protein (GFP) mRNA was coinjected for determining correct MO distribution. Siblings from the same pool as the MO-injected embryos served as negative controls. Effective doses were determined for each MO with the use of a concentration in which MO-injected embryos developed normally. Higher doses causing detectable detrimental effects on development were not pursued further.
Primers 5′-AGCGAATAGATCGAGATGGAC-3′ and 5′-TGGAGATTGTGTCGCTGAAG-3′ were used for confirming missplicing, by RT-PCR, of RNA isolated with TRIzol (Invitrogen) from ten control or MO-injected embryos, at 24 and 72 hr postfertilization (hpf). PCR products were gel purified (Invitrogen) and sequenced to reveal missplicing events.
For motility studies, 7–9 control and MO-injected embryos at 72 hpf, confirmed as alive by observation of heartbeat beforehand, were analyzed individually under glass coverslips. Olfactory-pit cilia were viewed under a water-immersion objective lens (×50) on an inverted Nikon Diaphot microscope in a humidified (80%) and temperature-controlled (28°C) chamber. High-speed (500 frames per second [fps]) video sequences of the zebrafish olfactory pits were captured (Trouble-shooter 500, Lake Image systems, UK). The stored sequences were then replayed in slow motion (Midas 2.0 player, Xcitex) and the cilia beat frequency (CBF) was calculated as previously described,33 with the use of the following formula: fps (500) / number of frames elapsed for five beat cycles × beat cycles counted (5) = CBF (Hz). The immotility index (II) and dysmotility index (DI) were determined by a count of the number of static (II) or dysmotile (DI; abnormal movement + static) cilia as a percentage of the total number present in the video sequences. The mean ± SEM was calculated for 7–9 embryos, and data were tested by one-way ANOVA, with individual data sets compared by an unpaired Student's t test with Bonferroni correction for repeated-measures.
Zebrafish Phenotype Rescue
Full-length mouse Rsph9 mRNA was subcloned into pßUT3 vector, and mRNA was transcribed with the mMessage machine kit (Ambion) and T3 polymerase, then titrated for determination of the maximum effective injection dose. One-cell-stage embryos were coinjected with 150 pg mRNA and 6.25 ng of MO via methods described above, and olfactory-cilia movement was assessed at 72 hpf as described above. The controls were WT and MO-treated embryos.
Chlamydomonas Strains and Maintenance
WT (CC-1732 and cw15), pf17 parental (CC-1035), and progeny (CC-1332, CC-1143, CC-2645, and CC-262) strains were obtained from the Chlamydomonas Genetics Center (see Web Resources). Cells were grown and expanded via standard protocols in Tris-acetate phosphate (TAP) medium,34 under light (∼45 μE/m2/s), at 25°C.
Identification of Chlamydomonas pf17-Strain Mutation
The precise genetic lesion in Chlamydomonas mutant strain pf17 had not previously been identified. The five coding exons, intron-exon boundaries, and 5′ and 3′ UTRs of RSP9 were sequenced in genomic DNA isolated, via a method adapted from Goldschmidt-Clermont et al.,35 from WT Chlamydomonas and mutant pf17. This revealed a single-bp deletion in exon 2 (c.131delG) present in pf17 but not in the WT, predicting a premature stop codon after Ser45 in the 269 residue protein. This change abolishes a BspEI restriction site, and we confirmed that the mutation causes the paralyzed flagella phenotype and had not arisen since original isolation of pf17, by BspEI restriction digest of a 1.1 kb exon 2 PCR product, amplified with primers 5′-CGCAGCTCACTTATCTCTTCCT-3′ and 5′-AGCACACGCCTCATCCAATAG-3′, from genomic DNA isolated from the WT, the original pf17 mutant strain CC-1035, and its four pf17 progeny strains: CC-1332, CC-1143, CC-2645 and CC-262. The c.131delG mutation identified in mutant pf17 was present in the original pf17 and all progeny strains, but not in the WT control (data not shown).
Chlamydomonas RSP9 Vectors
Transformation vectors for creating the pf17-T and pf17-Tmut strains were made as follows: The WT genomic RSP9 gene was amplified from genomic DNA with pFusion Taq (Invitrogen) and PCR primers P1 (5′-AGATTCCACACCTCACGGATAC-3′) and P2 (5′-ACCAGTCAAACTTTCGAACCAG-3′) to include the 5′ and 3′ UTR and approximately 400 bp of the upstream sequence incorporating the tub-box sequence motif known to enhance transcription after deflagellation,36 then cloned into pBluescript SKII−. A mutated RSP9 was created through introduction of a 3 bp deletion into this WT DNA construct, mimicking the human c.801_803delGAA; p.Lys268del mutation. The resulting mutation in Chlamydomonas RSP9, a deletion of the homologous residue, is c.780-783delCGC; p.Arg261del (Figure S2A). This was done with the use of a primer with the relevant 3 bp missing and a BmgBI restriction site at the 5′ end (P3: 5′-CGAGCTGACGTGGGGCAGCCTGTACGTGGGCGACGGCCTGAACAACGACC-3′) and the use of a reverse primer (P4: 5′-GTATGTTGTGTGGAATTGTGAGCGG-3′) complementary to pBluescript, downstream of a unique NotI site. The pBluescript WT RSP9 construct was amplified with P3-P4, and the product was subcloned into pCR4 by TOPO-TA cloning (Invitrogen) and then excised by BmgBI and NotI double digestion. pBluescript WT RSP9 was separately BmgBI and NotI double digested for elimination of the WT RSP9 insert, then these DNAs were ligated, creating a pBluescript plasmid containing mutated RSP9.
Chlamydomonas Transformation
Immotile pf17 (CC-1035) mutant Chlamydomonas were transformed with the WT or mutated RSP9 vectors, with the use of a modified protocol from Kindle.37 pf17 was first backcrossed with cell-wall-less mutant cw15, for ease of transformation. Backcrossed pf17 cultures (as well as cell-wall-less cw15 control cultures) were then used. Cells were resuspended to 2 × 108 cells/ml, and 300 μl was transferred to 5 ml tubes containing ∼0.3 g of 0.4 mm diameter washed glass beads (BDH). 1 μg plasmid DNA and 4 μg plasmid pSI103, which confers paramomycin antibiotic resistance for selection of transformant colonies, were added together, and the mixture was vortexed for 15 s and grown overnight with slow shaking to allow for recovery and gene expression. Cells were plated on TAP 2% agar supplemented with 20 μg/ml paramomycin and inverted in the light at 22°C. Transformed cell-wall-less pf17 colonies were visible after 7–10 days and were then selected by viewing 100 colonies of each and monitoring them for rescued motility. The pf17 strain transformed with WT RSP9 was called pf17-T, and pf17 transformed with mutant RSP9 was called pf17-Tmut.
The correct incorporation of WT and mutant RSP9 constructs into the genetic material of these transformed strains was confirmed in genomic DNA by restriction digest, making use of the BspEI site destroyed by the original pf17 RSP9 c.131delG mutation. In addition, the c.780_783delCGC mutation mimicking that of human RSPH9 patients destroys an FspI site. BspEI and FspI digests showed that the control cw15 strain carried WT RSP9, the pf17-T strain carried both WT and c.131delG RSP9, and the pf17-Tmut strain carried no WT RSP9 but did carry c.131delG and c.780_783delCGC mutated RSP9. Thus, the WT and mutant RSP9 constructs had incorporated correctly into the genetic material of pf17-T and pf17-Tmut.
Chlamydomonas-Motility Analysis
Cell-wall-less cw15, pf17, pf17-T, and pf17-Tmut were picked into TAP medium, grown overnight, and analyzed under a water-immersion objective lens (×50) on an inverted Nikon Diaphot microscope in a humidified (80%) and temperature-controlled (30°C) chamber. High-speed (500 fps) video sequences were captured (Trouble-shooter 500, Lake Image systems, UK), and the stored sequences were replayed in slow motion (Midas 2.0 player, Xcitex) for measuring flagella beat frequency (FBF) using the formula published by Chilvers et al.44: fps (500) / number of frames elapsed for five beat cycles × beat cycles counted (5) = FBF (Hz). Chlamydomonas flagella do not beat with a planar motion as cilia do but, rather, can change the beat direction to facilitate directional movement, and motility was too variable to allow for calculation of a meaningful DI as was performed for zebrafish; therefore, only an II was calculated, when the beat pattern was symmetrical. This was determined by a count of the number of static flagella as a percentage of the total number present in the video sequences. The means ± SEM were calculated for 21 (cw15), 26 (pf17), 20 (pf17-T), and 21 (pf17-Tmut) embryos for FBF and for 10 (cw15), 5 (pf17), 7 (pf17-T), and 10 (pf17-Tmut) embryos for II. Data were tested by one-way ANOVA, and individual data sets were compared with the use of an unpaired Student's t test with Bonferroni correction for repeated measures.
Results
We undertook genome-wide linkage analysis and subsequent positional-candidate-gene analysis in seven PCD families with central-microtubular-pair defects: two Bedouin, four UK-Pakistani and one UK-Northern European (Figure 1A). Five of the families were consanguineous. No patients displayed laterality defects. For those in whom ciliary movement was studied, the ciliary beat frequency was within the normal range. However, the beat pattern was recorded as being circular rather than having the normal forward and backward planar motion,33 for all seven families except one Bedouin family, in which the beat pattern was recorded as “abnormal motility” (detailed in Subjects and Methods). One of the two Bedouin families, UCL146, had an unusual intermittent absence of the central pair, resulting in cilia cross-sections with both a 9+2 and a 9+0 ultrastructure20 (Figure 1B), and the second family, UCL152, had an apparently normal ultrastructure. The other five families, UCL105, UCL131, UCL132, UCL138, and UCL170, had a classic “transposition” defect, consisting of complete absence of the central pair and ciliary transposition. As previously described,20 in this defect, a proportion of cilia cross-sections have an absent central pair (9+0) and a proportion have an 8+1 arrangement, in which the central pair is absent and one peripheral microtubule doublet with attached dynein arms is transposed to the center (Figure 1B).
Identical-by-descent (IBD) regions of homozygosity shared among the affected individuals were identified in the two family groups. The full results of the whole-genome linkage scan are shown in Figure S1. For the two Bedouin families, a 4.8 Mb region in UCL146, between rs1738240 and rs945131, overlapped with a 10.8 Mb region in UCL152, between D6S291 and D6S452 (not shown). Higher-resolution genotyping using known or in-house-designed microsatellites defined a common 1.9 Mb critical region of IBD between markers D6S400 and rs3734693 on chromosome 6p21.1, with a peak multipoint LOD score of 6.7 across D6S1604–D6S451 (Figure 1C). Across this 1.9 Mb IBD region, the two families shared alleles at only two microsatellite markers, located either side of RSPH9, such that any linkage disequilibrium between the families is small (0.6 Mb maximum) (not shown).
All five transposition families were linked, with the use of only Illumina data, without additional genotyping, to a single 6.7 Mb region from rs2030926 to rs937091 on chromosome 6q22.1, defined by recombination events in family UCL138 (Figures 1C and 1D). There was a peak multipoint LOD score of 7.0 across rs873460–rs941815. No other genomic region was significant for linkage (Figure S1). The four UK-Pakistani families shared an IBD region and a common 5.2 Mb haplotype across markers rs1158747–rs2243379, suggesting a founder effect. This was shared among all ten affected individuals, including those of UCL132, a family that appears to have previously unknown consanguinity and ancestral sharing with the other three UK-Pakistani families (Figure 1D). The UK-Northern European family (UCL105) was consistent for linkage across the 57 Mb (rs1014976–rs1385732) spanning this region but had no significant marker homozygosity or allele sharing with the Pakistani families (not shown).
The linked region in the two Bedouin families harbored the positional candidate gene RSPH9. A homozygous 3 bp deletion (c.801_803delGAA) was identified in all seven affected Bedouin individuals, predicting in-frame loss of the C-terminal Lys268 (p.Lys268 del) (Figures 2A and 2B). This residue is largely conserved across distant phyla (Figure S2A). Screening of population-matched controls revealed that this change was not carried on 126 Bedouin and Arabic control chromosomes. However, screening of chromosomes from a collection of 160 unrelated control UAE males21 showed that three individuals were heterozygous for this amino acid deletion, a result that was not regarded as surprising given the high incidence of consanguineous unions in the culturally isolated UAE population.38 The overall frequency of this change in all of the control chromosomes screened was 0.7%. These findings therefore suggest that RSPH9 mutations probably cause PCD.
Figure 2.
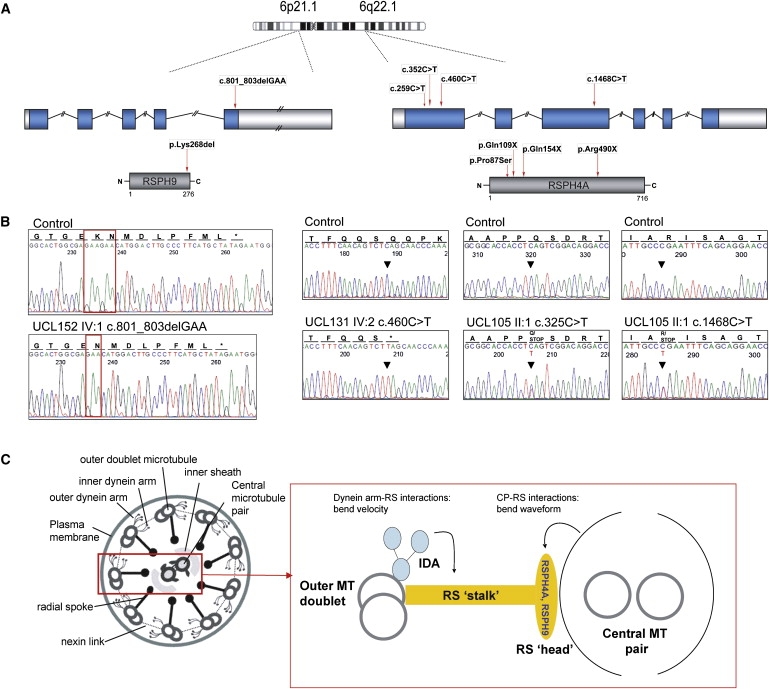
RSPH9 and RSPH4A Mutations
(A) Chromosome 6 location of RSPH9 (left) and RSPH4A (right), their genomic structure showing the 5′ and 3′ UTRs (white), introns and exons (blue), their derived proteins (grey), and mutations.
(B) Electropherograms indicate the normal sequence (top traces) and mutations (bottom traces).
(C) “9+2” cilia axoneme model (cross-section) with putative RSPH9 and RSPH4A location, based on Chlamydomonas homology. Abbreviations are as follows: CP, central microtubular pair; RS, radial spoke; IDA, inner dynein arm; MT, microtubule.
The linked region in the five families with a transposition defect harbored the positional candidate gene RSPH4A, also located within the region spanned by the ancestral UK-Pakistani haplotype (Figure 1D). Four C-to-T transition-sequence variants were identified (Figures 2A and 2B). All ten affected UK-Pakistani individuals were homozygous for a nonsense mutation, p.Gln154X (c.460C→T), accompanied by a second upstream missense variant, p.Pro87Ser (c.259C→T). Patients in the Northern European family UCL105 were compound heterozygous for nonsense mutations pGln109X (c.325C→T) and p.Arg490X (c.1468C→T). Cosegregation of all mutations with the disease status in all extended kindreds was confirmed and was found to be in accordance with haplotypes (Figure S3, Subjects and Methods). Screening of population-matched controls showed that they did not carry any of the identified RSPH4A mutations. These controls comprised 154 UK-Pakistani chromosomes screened for the p.Pro87Ser and p.Gln154X mutations; 170 and 354 UK-Northern European control chromosomes were also screened for the p.Pro87Ser and p.Gln154X mutations, respectively. In addition, 348 and 368 UK-Northern European chromosomes were screened for the p.Gln109X and p.Arg490X mutations, respectively.
Both RSPH9 and RSPH4A are predicted to encode radial spoke head proteins (Figure 2C), on the basis of homology with proteins of known function in the biflagellate alga Chlamydomonas reinhardtii and other ciliates.39 The RSPH9 protein was identified as 28% identical to biflagellate alga Chlamydomonas reinhardtii radial spoke head 9 (RSP9) protein.39 The RSPH4A protein was identified as 31% and 30% identical, respectively, to two similar Chlamydomonas proteins, RSP4 and RSP6 (Figure S2B).
Radial spokes are regularly spaced along cilia, sperm, and flagella axonemes and have a multisubunit “stalk” and “head” that form a signal-transduction scaffold between the central pair and the dynein arms (Figure 2C). Available evidence suggests that they regulate dynein-induced movement and govern cilia and flagella waveform. Central pair–radial spoke interactions determine bend direction and shape (waveform), whereas radial spoke–inner dynein arm interactions influence velocity.39–41
Further investigation of orthologs of RSPH9 were undertaken in three model organisms: mouse, zebrafish (Danio rerio), and Chlamydomonas, for determination of the pathogenic potential of the in-frame Lys268del change. We determined the tissue distribution of Rsph9 gene expression in mouse. In situ hybridization showed specific expression in nasal, lung, trachea, and brain ventricle epithelium at E18.5–19.5 and at the embryonic node at E7.5. This was a similar pattern to that of Dnah5,6,30 which is restricted to regions where motile cilia are located (Figure 3).
Figure 3.
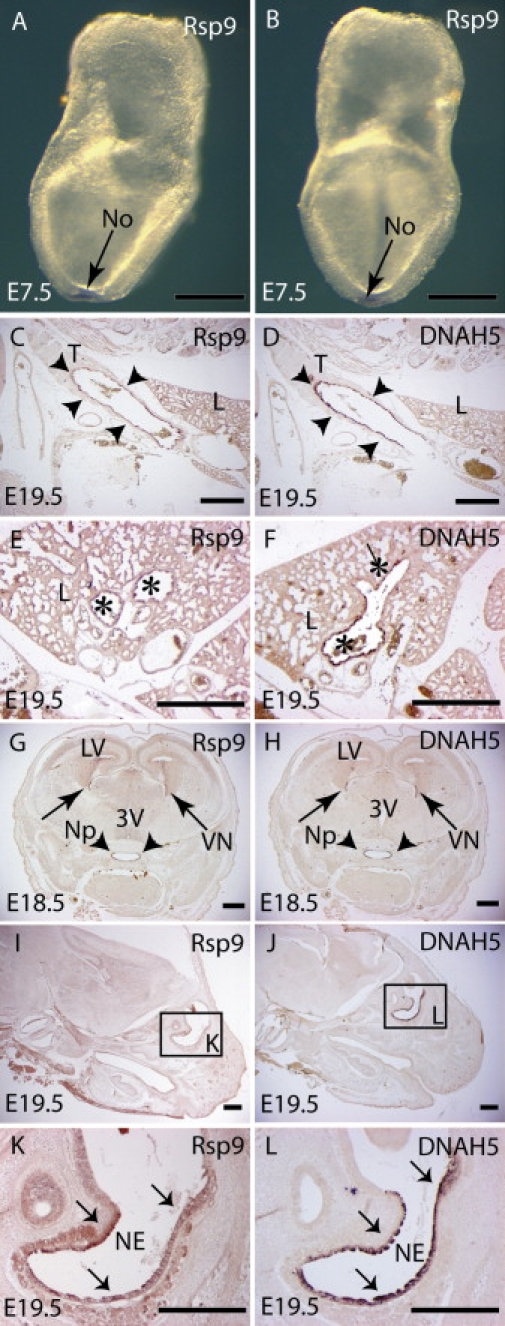
Expression of Rsph9 in the Node and Ciliated Epithelia
Whole-mount in situ hybridization of Rsph9 reveals specific expression in the node (No) of E7.5 mouse embryos (arrow); side (A) and anterior (B) views. Expression of Rsph9 (C, E, G, I, K) and Dnah5 (D, F, H, J, L) detected by in situ hybridization on sagittal (C–F and I–L) and coronal (G and H) sections at E18.5 (G and H) and E19.5 (C–F and I–L). Similar expression of both genes was detected in the epithelia lining the trachea (T) (arrowheads in C and D), the bronchi (asterisks in E and F), and the nasopharynx (Np) and neuroepithelium of the lateral ventricles (VN) (G and H). Expression was also prominent in the olfactory epithelium (I and J, magnified in K and L). Abbreviations are as follows: L, lung; LV, lateral ventricle; NE, nasal epithelium; 3V, third ventricle. Scale bar represents 500 μm.
We investigated rsph9 knockdown in zebrafish. Two different rsph9 splice-site MOs directed against the exon 2 (MOex2) and exon 3 (MOex3) splice-donor sites were used for disruption of gene expression (Figure S4). Both gave a dose-dependent phenotype of dysmotile olfactory-pit cilia with a normal beat frequency but an ineffective circular beat pattern (66% cilia dysmotile) (Figure 4A and Movies S1–S3). Statistical comparison between data sets indicated that the II and DI for both sets of MO-injected embryos were significantly higher than that observed for controls (p < 0.001). Morphants had a more static fluid flow than did WT, and this allowed debris to accumulate in the pits (Figure 4B and Movies S1–S3). Their beat pattern resembled that of cilia in RSPH patients, confirming that ablating gene expression causes similar cilia-dysmotility defects. This effect was rescued by coinjection of mouse Rsph9 mRNA, which restored the normal beat pattern (Figure 4A).
Figure 4.
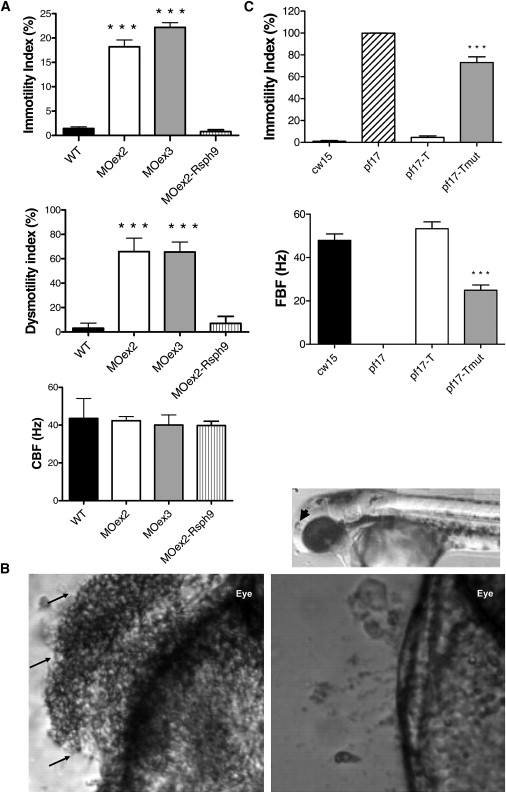
Loss of Zebrafish and Chlamydomonas RSPH9 Gene Function, Causing Dysmotility of Cilia and Flagella
(A) Olfactory-pit cilia movement in WT zebrafish and rsph9 morphants. Top panel: 18.2% (Moex2), 22.2% (MOex3), and 0.8% (MOex2 + rsph9 mRNA) of cilia in morphants were immotile (1.4% in WT). Middle panel: 65.9% (MOex2), 65.6% (MOex3), and 7% (MOex2 + rsph9 mRNA) of cilia were dysmotile (3.1% in WT), this number including that which was immotile in addition to dysmotile; i.e., displaying an ineffective circular beat pattern. Bottom panel: cilia beat frequency was unaffected at 42.3 Hz (MOex2), 40.0 Hz (MOex3), and 39.8 Hz (MOex2 + rsph9 mRNA) (WT 43.6 Hz). Means ± SEM from seven WT, eight MOex2, nine MOex3 and five MOex2 plus coinjected rsph9 mRNA embryos are shown. Triple asterisk indicates p < 0.001.
(B) Accumulation of debris in rsph9 zebrafish morphant olfactory pit. Top panel, 72 hpf zebrafish, indicating nasal pit in relation to the eye (arrowhead). Bottom left panel: representative example of 72 hpf rsph9 MOex3 morphant zebrafish with debris accumulation evident in the nasal pit (arrows). Bottom right panel: WT zebrafish showing normal debris clearance due to fluid vortex created by cilia beating. MOex2 showed the same defect (not shown).
(C) Chlamydomonas flagella movement in WT (cw15) and in pf17, pf17-T and pf17-Tmut strains. Top: flagella motility was within the normal range at 4.6% in pf17-T, but in pf17-Tmut, 73.0% of flagella were immotile, compared to 1.0% in WT and 99.8% in pf17. Bottom: flagella beat frequency was within the normal range at 53.5 Hz in pf17-T, but reduced to 24.9 Hz in pf17-Tmut (WT 47.9 Hz, pf17 0.0 Hz). Shown are means ± SEM from 5–10 cells (immotility index) or 20–26 cells (flagellar beat frequency [FBF]). Triple asterisk indicates p < 0.001.
Comparison at 24 and 48 hpf between morphant zebrafish (n = 163 exon 2, 163 exon 3) and WT zebrafish (n = 270) showed that laterality was unaffected. Three of the exon 3 morphants, none of the exon 2 morphants, and one WT fish displayed situs inversus, which was a nonsignificant difference. Situs inversus presents occasionally as a well-recognized “background” zebrafish phenotype (L.R and S.W.W., unpublished data).
The Chlamydomonas mutant strain pf17 has a mutation in RSP9, the ortholog of human RSPH9, resulting in immotile flagella. The entire radial spoke head complex is absent, and there is central-pair displacement, rather than loss.42 We first determined that pf17 carries a single-bp deletion, c.131delG, in RSP9, predicting an early premature stop codon (p.Ser45AlafsX3). We then used the presumed Rsp9 null background of the pf17 strain to investigate the effects of the human RSPH9 p.Lys268 del mutation. We stably transformed pf17 with the WT RSP9 gene to create the strain pf17-T. pf17-T regained a normal beat velocity and pattern, indicating complete phenotype rescue (Figure 4C and Movies S4–S7). We then stably transformed pf17 with a mutated version of RSP9 carrying the equivalent of the 3 bp p.Lys268del deletion to create the strain pf17-Tmut. Statistical comparison between data sets indicated that the FBF for pf17-Tmut was significantly reduced and the II significantly increased, compared to controls (p < 0.001). Therefore, pf17-Tmut showed only partial rescue, with a beat velocity at half the WT level and flagella either immotile (73% immotile) or displaying a slowed, disorganized beat ineffective for normal movement (Figure 4C and Movies S4–S7). Differences in flagella and cilia movement preclude rigorous comparisons of beat pattern. Thus, recreation of the human RSPH9 mutation in Chlamydomonas provides direct evidence of its pathogenic effect on motility of cilia and flagella.
Discussion
We have identified mutations in two genes encoding radial spoke head proteins, RSPH9 and RSPH4A, in PCD families that have defects of the central microtubular pair. This is the first report of PCD genes that cause disease associated with cilia-axoneme defects other than a loss or reduction of the outer dynein arms. Using model organisms, we have also shown that Rsph9 is expressed in ciliated epithelia and that gene knockdown and mimicking of the human RSPH9 p.Lys268 del mutation recapitulates its detrimental effect on cilia motility.
Analysis of Chlamydomonas RSP9 mutant strains indicates that the human RSPH9 p.Lys268 del mutation is likely to be hypomorphic, given that some flagella function is retained in these mutants, in contrast to those with a null allele. RSPH9 residue Lys268 is conserved in all mammals, but not in some of the nonvertebrates and ciliates tested. This could reflect a functional distinction among the cilia of different species, or it could be that the loss of this amino acid, rather than its specific chemical properties, is what confers the disease-causing effect. Although the p.Lys268 del mutation was detected on 0.7% of control chromosomes, it was never present in a homozygous state in the control individuals, whereas sequencing of the entire open reading frame in affected patients showed that they were all homozygous for this single mutation. The population frequency of the mutant allele is not significantly different from what is expected of a pathogenic recessive mutation. Additionally, the majority of control chromosomes were sampled from the highly consanguineous UAE population, from which one family carrying the mutation (UCL146) originates. These observations support the idea that the p.Lys268 del mutation is disease causing.
We expect that the three premature nonsense mutations that we have identified in RSPH4A are more likely to be null alleles, because they are predicted to result in premature protein truncation, but this requires further functional work for confirmation. These RSPH4A nonsense mutations predict a disruption of the “radial spoke domain” (Figure S2B). No other functional domains could be identified by computer modeling in either of the two radial spoke head proteins, although RSPH4A orthologs are noted as proline rich,43 thereby preventing significant genotype-phenotype predictions.
Determination of ultrastructural changes in the axoneme is limited by methodological constraints. In particular, observations reflect a sampling of total tissue such that local changes in ciliary structure might be missed. The observed structural changes appear to vary according to species, organelle type, protein involved, and specific mutation. Truncation mutations in RSPH4A in human motile cilia are associated with a loss of the central pair, yet for an RSP9 truncation mutation in Chlamydomonas pf17, the central pair is retained but displaced. The different contribution of RSPH4A and RSPH9 proteins to the spoke-head structure is not clear. Furthermore, it is not yet known whether differences in central-pair structural constraints and waveform between cilia and flagella could explain the disparity or whether the human patients might have more intact and functionally preserved radial spoke heads than the Chlamydomonas mutants. In humans with a single-aa deletion in RSPH9, the structural consequences are distinct, with only a localized loss of the central-pair microtubules, perhaps reflecting the predicted milder mutation. Of the two families with the RSPH9 p.Lys268 del mutation, a normal central-pair ultrastructure was in fact recorded in family UCL152, but in view of the shared mutation in common with UCL146, it seems likely that the minor central-pair defect observed in UCL146 could have missed detection in UCL152 without the more detailed sampling that was undertaken in UCL146, which included generation of longitudinal sections.20
The functional consequences that we have observed arising from RSPH9 and RSPH4A defects in patients and model organisms support a more significant role for radial spoke heads and central-pair microtubules in determining cilia beat waveform rather than velocity, although velocity may also be affected. This is consistent with the observed retention of the force-generating dynein arms that govern velocity in the cilia axonemes of central-pair-defect patients. The natural movement of 9+2 motile cilia and 9+0 nodal cilia differs: the 9+0 cilium has a circular motion, rather than the planar “whiplash” movement of 9+2 cilia, with effective and recovery strokes.7,44 In zebrafish and RSPH9 and RSPH4A patients, altered function of the radial spoke heads caused 9+2 cilia motility to resemble this simpler rotary 9+0 cilia movement. This is consistent with previous evidence that a disconnection of radial spoke head central-pair interactions in 9+2 cilia would lead to a change from the normal planar motion to abnormal movement.39,41 For example, an antibody to sea urchin RSPH4A was previously shown to affect sperm-flagella beat pattern but not velocity, changing the movement from planar to circular.45
The normal laterality observed in RSPH9 and RSPH4A patients and rsph9 zebrafish morphants is consistent with the notion that radial spoke proteins are not essential for nodal ciliary function. Their role in waveform appears more important in central-pair-containing 9+2 cilia. Our observation from in situ hybridization, that Rsph9 is expressed in mouse nodal cilia (which lack a central pair), suggests either redundancy or an alternative, perhaps structural, role. The precise function of the radial spoke head proteins at the embryonic node is likely to be relevant to the molecular basis of the difference in 9+0 node cilia and 9+2 motile cilia waveforms and remains of considerable interest. However, many unresolved questions remain concerning the correlation of structure and function in different cilia types.
In summary, our observations provide new insights into the role of radial spoke head proteins in the structure and function of cilia and flagella and in the molecular genetic basis of primary ciliary dyskinesia. RSPH9 and RSPH4A represent good candidate disease-causing genes for cases of PCD with central-pair defects and also for cases in which the axonemal dynein arms are retained and the patients do not display laterality defects. Characterization of RSPH9 and RSPH4A and continuing elucidation of the molecular basis of PCD provide new opportunities for noninvasive diagnosis and the possibility of new therapies in what we have shown is, at least in some model organisms, a reversible (rescuable) molecular defect.
Supplemental Data
Supplemental Data include four figures, one table, and seven movies and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Chlamydomonas Center, http://www.chlamy.org/
Ensembl, http://www.ensembl.org/
European Collection of Cell Cultures, http://www.ecacc.org.uk/
National Laboratory for the Genetics of Israeli Populations, http://nlgip.tau.ac.il/
Online Mendelian Inheritance in Man (OMIM), http://ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu
Acknowledgments
We are grateful to the families for their involvement in this study. We thank the physicians involved in patient recruitment and sampling, in particular Robert Mueller, Maggie Meeks, Astrid Weber, and Yannick Crow. We thank Sandra Strautnieks for providing Arabic control samples. We are grateful to Peter Scambler for critical reading of the manuscript. We thank Lucille Fressynet, Chloe Cheung, Chloe McCann, Kate Everett, and Barry Choiza for technical assistance. We thank Elspeth Bruford at the HUGO Gene Nomenclature Committee, European Molecular Biology Laboratory-European Bioinformatics Institute, for much help with radial-spoke nomenclature and renaming. We are grateful to A. Rodaway, King's College London, for the pßUT3 rescue vector, to H. Omran for the Dnah5 in situ probe, and to the following for release of prepublication proteomic and genomic data sets, in addition to bioinformatics help: G. Pazour, K. Gull, P. McKean, and S. Brody. This work was supported by the Medical Research Council (UK), the Wellcome Trust (UK), the Milena Carvajal-Prokartagener Foundation (Switzerland), the PCD Foundation (USA), and the PCD Family Support Group (UK).
References
- 1.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia, current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rott H.D. Kartagener's syndrome and the syndrome of immotile cilia. Hum. Genet. 1979;46:249–261. doi: 10.1007/BF00273308. [DOI] [PubMed] [Google Scholar]
- 3.Jeganathan D., Chodhari R., Meeks M., Faeroe O., Smyth D., Nielsen K., Amirav I., Luder A.S., Bisgaard H., Gardiner R.M. Loci for primary ciliary dyskinesia map to chromosome 16p12.1–12.2 and 15q13.1–15.1 in Faroe Islands and Israeli Druze genetic isolates. J. Med. Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Callaghan C. Innate pulmonary immunity, cilia. Pediatr. Pulmonol. 2004;26(Suppl.):72–73. doi: 10.1002/ppul.70057. [DOI] [PubMed] [Google Scholar]
- 5.Kosaki K., Ikeda K., Miyakoshi K., Ueno M., Kosaki R., Takahashi D., Tanaka M., Torikata C., Yoshimura Y., Takahashi T. Absent inner dynein arms in a fetus with familial hydrocephalus-situs abnormality. Am. J. Med. Genet. A. 2004;129A:308–311. doi: 10.1002/ajmg.a.30177. [DOI] [PubMed] [Google Scholar]
- 6.Ibanez-Tallon I., Pagenstecher A., Fliegauf M., Olbrich H., Kispert A., Ketelsen U.P., North A., Heintz N., Omran H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- 7.Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 9.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with outer dynein arm defects. Am. J. Hum. Genet., in press. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush A., Hogg C., Mitchison H.M., Nisbet M., Wilson R. Update in primary ciliary dyskinesia. Clin. Pulm. Med. 2008 in press. [Google Scholar]
- 11.Hornef N., Olbrich H., Horvath J., Zariwala M.A., Fliegauf M., Loges N.T., Wildhaber J., Noone P.G., Kennedy M., Antonarakis S.E. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zariwala M.A., Leigh M.W., Ceppa F., Kennedy M.P., Noone P.G., Carson J.L., Hazucha M.J., Lori A., Horvath J., Olbrich H. Mutations of DNAI1 in primary ciliary dyskinesia, evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med. 2006;174:858–866. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Failly M., Saitta A., Munoz A., Falconnet E., Rossier C., Santamaria F., de Santi M.M., Lazor R., Delozier-Blanchet C.D., Bartoloni L., Blouin J.L. DNAI1 Mutations Explain Only 2% of Primary Ciliary Dykinesia. Respiration. 2008;76:198–204. doi: 10.1159/000128567. [DOI] [PubMed] [Google Scholar]
- 14.Meeks M., Walne A., Spiden S., Simpson H., Mussaffi-Georgy H., Hamam H.D., Fehaid E.L., Cheehab M., Al-Dabbagh M., Polak-Charcon S. A locus for primary ciliary dyskinesia maps to chromosome 19q. J. Med. Genet. 2000;37:241–244. doi: 10.1136/jmg.37.4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geremek M., Schoenmaker F., Zietkiewicz E., Pogorzelski A., Diehl S., Wijmenga C., Witt M. Sequence analysis of 21 genes located in the Kartagener syndrome linkage region on chromosome 15q. Eur. J. Hum. Genet. 2008;16:688–695. doi: 10.1038/ejhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 16.Geremek M., Witt M. Primary ciliary dyskinesia, genes., candidate genes and chromosomal regions. J. Appl. Genet. 2004;45:347–361. [PubMed] [Google Scholar]
- 17.Moore A., Escudier E., Roger G., Tamalet A., Pelosse B., Marlin S., Clement A., Geremek M., Delaisi B., Bridoux A.M. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2006;43:326–333. doi: 10.1136/jmg.2005.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeki H., Kondo S., Morita T., Sasagawa I., Ishizuka G., Koizumi Y. Immotile cilia syndrome associated with polycystic kidney. J. Urol. 1984;132:1165–1166. doi: 10.1016/s0022-5347(17)50080-4. [DOI] [PubMed] [Google Scholar]
- 19.Budny B., Chen W., Omran H., Fliegauf M., Tzschach A., Wisniewska M., Jensen L.R., Raynaud M., Shoichet S.A., Badura M. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum. Genet. 2006;120:171–178. doi: 10.1007/s00439-006-0210-5. [DOI] [PubMed] [Google Scholar]
- 20.Stannard W., Rutman A., Wallis C., O'Callaghan C. Central microtubular agenesis causing primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2004;169:634–637. doi: 10.1164/rccm.200306-782OC. [DOI] [PubMed] [Google Scholar]
- 21.Cadenas A.M., Zhivotovsky L.A., Cavalli-Sforza L.L., Underhill P.A., Herrera R.J. Y-chromosome diversity characterizes the Gulf of Oman. Eur. J. Hum. Genet. 2008;16:374–386. doi: 10.1038/sj.ejhg.5201934. [DOI] [PubMed] [Google Scholar]
- 22.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann K., Lindner T.H. easyLINKAGE-Plus–automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 24.Kruglyak L., Lander E.S. Faster multipoint linkage analysis using Fourier transforms. J. Comput. Biol. 1998;5:1–7. doi: 10.1089/cmb.1998.5.1. [DOI] [PubMed] [Google Scholar]
- 25.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST, a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pazour G.J., Agrin N., Leszyk J., Witman G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stolc V., Samanta M.P., Tongprasit W., Marshall W.F. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc. Natl. Acad. Sci. USA. 2005;102:3703–3707. doi: 10.1073/pnas.0408358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broadhead R., Dawe H.R., Farr H., Griffiths S., Hart S.R., Portman N., Shaw M.K., Ginger M.L., Gaskell S.J., McKean P.G. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 29.El-Sayed N.M., Myler P.J., Blandin G., Berriman M., Crabtree J., Aggarwal G., Caler E., Renauld H., Worthey E.A., Hertz-Fowler C. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 30.Olbrich H., Haffner K., Kispert A., Volkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 31.Ybot-Gonzalez P., Copp A.J., Greene N.D. Expression pattern of glypican-4 suggests multiple roles during mouse development. Dev. Dyn. 2005;233:1013–1017. doi: 10.1002/dvdy.20383. [DOI] [PubMed] [Google Scholar]
- 32.Lai C.S., Gerrelli D., Monaco A.P., Fisher S.E., Copp A.J. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–2462. doi: 10.1093/brain/awg247. [DOI] [PubMed] [Google Scholar]
- 33.Chilvers M.A., O'Callaghan C. Analysis of ciliary beat pattern and beat frequency using digital high speed imaging, comparison with the photomultiplier and photodiode methods. Thorax. 2000;55:314–317. doi: 10.1136/thorax.55.4.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris E. Academic Press; San Diego: 1989. The Chlamydomonas Sourcebook. A Comprehensive Guide to Biology and Laboratory Use. [DOI] [PubMed] [Google Scholar]
- 35.Goldschmidt-Clermont M., Girard-Bascou J., Choquet Y., Rochaix J.D. Trans-splicing mutants of Chlamydomonas reinhardtii. Mol. Gen. Genet. 1990;223:417–425. doi: 10.1007/BF00264448. [DOI] [PubMed] [Google Scholar]
- 36.Davies J.P., Grossman A.R. Sequences controlling transcription of the Chlamydomonas reinhardtii beta 2-tubulin gene after deflagellation and during the cell cycle. Mol. Cell. Biol. 1994;14:5165–5174. doi: 10.1128/mcb.14.8.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kindle K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 1990;87:1228–1232. doi: 10.1073/pnas.87.3.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdulrazzaq Y.M., Bener A., al-Gazali L.I., al-Khayat A.I., Micallef R., Gaber T. A study of possible deleterious effects of consanguinity. Clin. Genet. 1997;51:167–173. doi: 10.1111/j.1399-0004.1997.tb02447.x. [DOI] [PubMed] [Google Scholar]
- 39.Yang P., Diener D.R., Yang C., Kohno T., Pazour G.J., Dienes J.M., Agrin N.S., King S.M., Sale W.S., Kamiya R. Radial spoke proteins of Chlamydomonas flagella. J. Cell Sci. 2006;119:1165–1174. doi: 10.1242/jcs.02811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porter M.E., Sale W.S. The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J. Cell Biol. 2000;151:F37–F42. doi: 10.1083/jcb.151.5.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith E.F., Yang P. The radial spokes and central apparatus, mechano-chemical transducers that regulate flagellar motility. Cell Motil. Cytoskeleton. 2004;57:8–17. doi: 10.1002/cm.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang B., Piperno G., Ramanis Z., Luck D.J. Radial spokes of Chlamydomonas flagella, genetic analysis of assembly and function. J. Cell Biol. 1981;88:80–88. doi: 10.1083/jcb.88.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curry A.M., Williams B.D., Rosenbaum J.L. Sequence analysis reveals homology between two proteins of the flagellar radial spoke. Mol. Cell. Biol. 1992;12:3967–3977. doi: 10.1128/mcb.12.9.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chilvers M.A., Rutman A., O'Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gingras D., White D., Garin J., Cosson J., Huitorel P., Zingg H., Cibert C., Gagnon C. Molecular cloning and characterization of a radial spoke head protein of sea urchin sperm axonemes, involvement of the protein in the regulation of sperm motility. Mol. Biol. Cell. 1998;9:513–522. doi: 10.1091/mbc.9.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.