

Abstract
Members of the evolutionarily conserved T-box family of transcription factors are important players in developmental processes that include mesoderm formation and patterning and organogenesis both in vertebrates and invertebrates. The importance of T-box genes for human development is illustrated by the association between mutations in several of the 17 human family members and congenital errors of morphogenesis that include cardiac, craniofacial, and limb malformations. We identified two unrelated individuals with a complex cranial, cervical, auricular, and skeletal malformation syndrome with scapular and pelvic hypoplasia (Cousin syndrome) that recapitulates the dysmorphic phenotype seen in the Tbx15-deficient mice, droopy ear. Both affected individuals were homozygous for genomic TBX15 mutations that resulted in truncation of the protein and addition of a stretch of missense amino acids. Although the mutant proteins had an intact T-box and were able to bind to their target DNA sequence in vitro, the missense amino acid sequence directed them to early degradation, and cellular levels were markedly reduced. We conclude that Cousin syndrome is caused by TBX15 insufficiency and is thus the human counterpart of the droopy ear mouse.
Main Text
We studied two unrelated girls of German (patient 1) and Turkish (patient 2) ancestry. The girls shared an identical phenotype consisting of short stature and macrocephaly. Patient 1 had a birth length of 43 cm (−2.4 standard deviation [SD]) and head circumference of 38 cm (+ 0.83 SD) and an adult height of 115 cm (−8.18 SD) and a head circumference of 61.5 cm (+4.12 SD). Patient 2 at her most recent follow-up at 10 years of age had a height of 105 cm (−4.41 SD) and a head circumference of 53 cm (50th percentile). Their dysmorphic features included frontal bossing, narrow palpebral fissures with deep set globes and hypertelorism, strabismus, low-set ears with posterior rotation and dysplasia of the conchae, narrow auditory canals and hypoacusis, a short neck with redundant skinfolds, and a low hairline (Figure 1). The habitus of both girls was characterized by macrocephaly, fixed flexion at the elbow joints, a short neck, and leg shortening caused by bilateral dislocation of the hips and hip flexion. The main radiographic features were hypoplastic scapulae and iliac bones, short femurs, humeroradial synostosis, and moderate brachydactyly. In addition, the skull base was abnormally shaped, resulting in markedly low attachment of the ears and in caudal displacement of the occipital bone (Figure 2). Both girls had normal intelligence and attended normal school (at age 12 years; patient 2) or college (at age 19 years; patient 1). Both parent couples were consanguineous (second and first cousins, respectively) and showed none of the clinical and radiographic signs seen in their daughters. Patient 1 had been given a diagnosis of campomelic dysplasia (MIM 114290) because of scapular and iliac hypoplasia; patient 2 had been diagnosed with Kosenow scapuloiliac dysostosis (MIM 169550). However, their phenotype did not fit either campomelic dysplasia or Kosenow scapuloiliac dysostosis; sequence analysis of the SOX9 gene (MIM 608160) was normal. We found that the dysmorphic pattern in these girls corresponded closely to a condition described by Cousin et al. in 19821 as “Dysplasie pelvi-scapulaire familiale avec anomalies épiphisaires, nanisme, et dysmorphies” (familial pelvis-scapular dysplasia with epiphyseal anomalies, dwarfism, and dysmorphisms; listed in OMIM as Cousin syndrome or pelviscapular dysplasia; MIM 260660). Cousin syndrome has not been reported again since the original description, although some patients reported as having Kosenow syndrome may in fact have had Cousin syndrome.2
Figure 1.
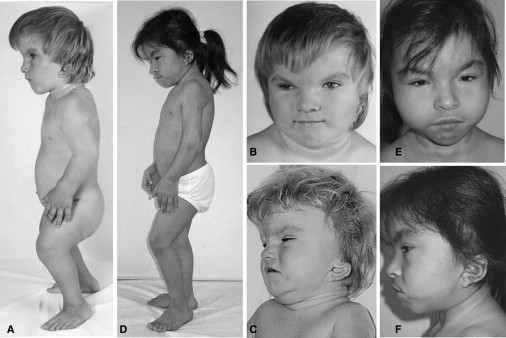
Clinical Features of the Two Girls with Cousin Syndrome and TBX15 Mutations
Patient 1 is age 5 yr in (A) and (B) and age 3 yr in (C); patient 2 is age 10 yr in (D), (E), and (F). The main features are bossing, malar hypoplasia, and a small chin; narrow palpebral fissures; a short neck with redundant skinfolds; low-set, posteriorly rotated, and dysplastic external ears; apparent femoral shortening because of femoral head dislocation; and short stature.
Figure 2.
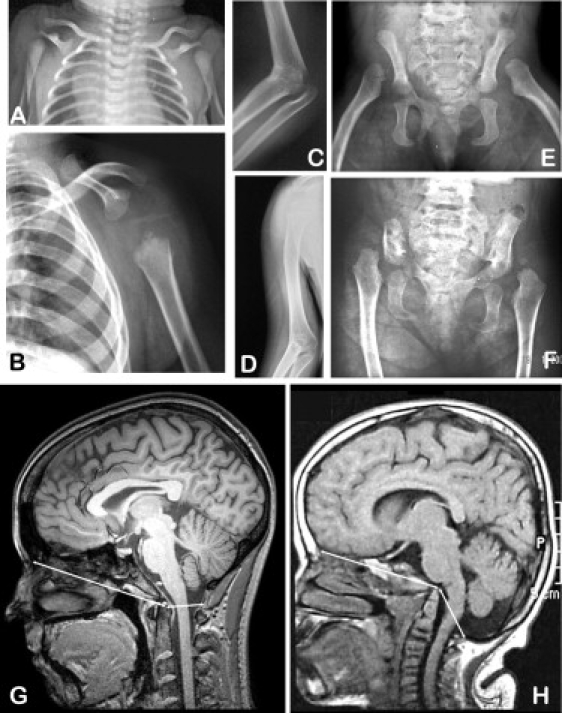
Radiographic Findings in Cousin Syndrome
(A, C, and E) Patient 1. (B, D and F) Patient 2. Skeletal features seen in the two patients include aplasia of the blade of the scapula, humeroradial synostosis, marked hypoplasia of the iliac bones, and dislocation of the femoral heads. (G) A sagittal section of a cranial MRI of a normal woman aged 19 years. (H) A corresponding section from patient 2 at age 12 years. From the tip of the odontoid process in the center of each panel, the arrows extend anteriorly to the frontal bone and posteriorly to the posterior margin of the foramen magnum. Caudal displacement of the occipital bone is evident in the patient; note also the redundant skin fold over the posterior aspect of the neck. The cranial and skeletal features are remarkably similar to those seen in Tbx15-ablated mice.5
Guided by the findings of scapular hypoplasia and abnormal cranio-facio-cervical morphology, we identified the Tbx15-deficient mouse phenotype as a possible homolog to Cousin syndrome. Murine Tbx15 deficiency occurs as the result of a deletion involving several exons of Tbx15 in the spontaneous mouse mutant, droopy ear (de)3, and has been reproduced by targeted disruption of the gene.4,5 First described in 1959, droopy ear exhibits a complex craniofacial malformation including small, widely spaced eyes with short palpebral fissures, a broad nasal area, a shortened skull held in an elevated position, misshapen and rotated external ears, and an abnormal coat-color patterning.3,6 The skeletal phenotype of Tbx15-inactivated mice includes small overall size, hypoplastic scapulae, moderate shortening of several long bones, and a dysmorphogenesis of cranial bones and cervical vertebrae, including vertical displacement of the supraoccipital bone, a small basiooccipital bone, a small foramen magnum, and changes in the shape of the squamosum and of the first and second vertebrae.4,5 These changes were reminiscent of those observed in the two patients studied.
Peripheral blood DNA was obtained with appropriate fully informed consent from the two individuals with Cousin syndrome and from their parents. Human studies were approved by the ethical review boards of the two hospitals (Freiburg and Ankara). The TBX15 genomic sequences were determined with PCR amplifications of individual exons by standard methods; primer sequences were designed with the TBX15 genomic sequences available at Ensembl (see Web Resources; accession number ENSG00000092607). We studied the consequences of the mutatons by inserting the mutations into TBX15 expression vectors. TBX15 was amplified from normal human fibroblast cDNA, oligonucleotides were based on transcript ENST00000207157 (TBX15S, 496 amino acids), and transcript ENST00000369429 (TBX15L, 602 amino acids). PCR products were inserted into pCRII-TOPO (Invitrogen), the disease-associated mutations TBX15-1042 delA, TBX15-1044 delA, and the deletion mutant TBX15-ΔC (amino acids 1–344, stop codon at base pair 1045) were introduced by site-directed mutagenesis. Wild-type and mutated ORFs were fully sequence verified and subcloned into pBABE-puro,7 pBABE-EGFP,8 and pCDNA3 (Invitrogen). N-terminal tags for the FLAG (pBABE), EGFP (pBABE), and the Myc epitope (pCDNA3, pSP64) were fused in frame by PCR or subcloning. For in vitro expression, TBX15 cDNAs were subcloned in the vector pSP64 (Promega) that was modified to contain a 5′ β-globin leader and a 3′ β-globin trailer, suitable for the TNT®SP6 High-Yield Protein Expression System (Promega). We performed the electrophoretic mobility shift assay (EMSA) as described9 to test the DNA binding ability of mutant TBX15 protein. Oligonucleotides used to generate binding sites were BS.dirF 5′-GATCCGGAGGTGTGAAGGTGTGAAAGGA-3′ and BS.dirR 5′-GATCTCCTTTCACACCTTCACACCTCCG-3′. Cell culture, gene transfer, and expression analysis were performed as described.8,9 High-titer retroviral gene transfer via BING packaging cells10 was used for transduction of human chondrosarcoma (HCS, ATCC) and human fibrosarcoma cells (HT1080, ATCC). Viral supernatants were titered prior to infection to give an MOI (multiplicity of infection) of 10 for each construct. Transduced cell populations were analyzed either 48 hr after infection or after selection with 5 μg/ml puromycin for 72 hr. Whole-cell lysates and subcellular protein fractions were obtained 48 hr after transfection by lysis in 1× SDS sample buffer or by hypotonic lysis,8 followed by sonification and immunoblot analysis. Patients' fibroblasts derived from skin biopsies were expanded to passage five; RIPA buffer was used for extracting proteins for immunoblot analysis. Antibodies included anti-FLAG M5 (Sigma), anti-GFP (Santa Cruz), anti-β-actin (Sigma), and a polyclonal serum generated by immunization of rabbits with a GST fusion protein containing the amino acids 1–300 of TBX15. For direct fluorescent detection of EGFP fusion proteins, cells were cultured on chamber slides, fixed in 4% paraformaldehyde for 10 min at room temperature, washed thrice with PBS, and counterstained with DAPI antifade (Q Biogene). Images were captured with a Leica (DMRXA) microscope with a cooled CCD (Sensys Photometrics). For proteasome inhibition, MG-132 (Sigma) was added to the culture medium at 25 μM prior to the collection of cell lysates.
To test the possible homology between Cousin syndrome and the mouse Tbx15 deficiency, we obtained DNA from the two individuals with Cousin syndrome and their parents after having obtained informed consent and then searched directly for TBX15 genomic sequence variations (primer information and PCR conditions are available upon request). We identified two different single-nucleotide deletions that segregated in each family and were homozygous in the affected girls, heterozygous in their parents, and absent in a panel of over 216 control chromosomes. Both deletions occurred at codon 344; in family 1, deletion of adenosine 1042 (c.1042 delA at the first position within codon 344) led to a frame shift with 78 missense amino acids followed by a stop codon; in family 2, the deletion occurred at the third position of the same codon (c.1044 delA) an led to 77 missense amino acids and the stop codon (Figure 3A); both missense stretches include five cysteine residues. Both mutations predicted the abolition of the C-terminal 152 amino acids of the principal short and long isoforms of the mature TBX15 protein (Figure 3C; for the reference sequence of TBX15 isoforms, see Web Resources). Semiquantitative PCR and sequencing of cDNA isolated from patients' and controls' fibroblasts showed that the mutant mRNA was expressed at a level that was the same as or higher than that of wild-type mRNA (Figure 3B).
Figure 3.
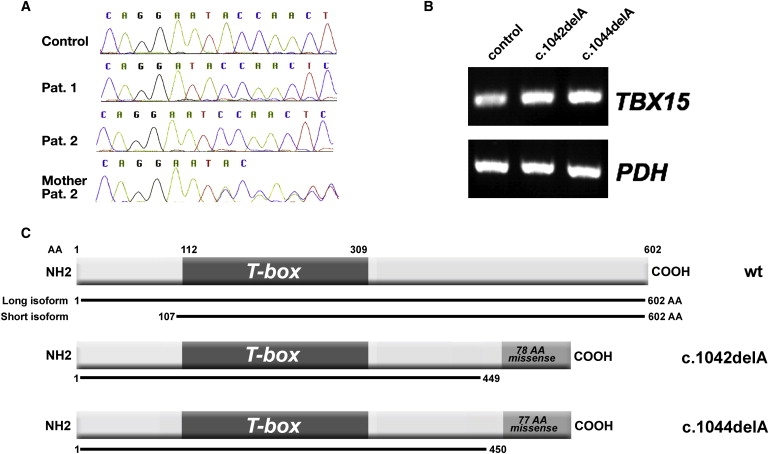
Genomic TBX15 Mutations
(A) The genomic TBX15 sequences in patient 1, patient 2, the mother of patient 2, and a control. Patient 1 is homozygous for c.1042 delA, and patient 2 is homozygous for c.1044 delA in the same codon. (B) The TBX15 mRNA levels in an age-matched control and patients' fibroblasts of the same passage by semiquantitative RT-PCR; expression of the mutant alleles was verified by cDNA sequencing. (C) A schematic representation of the two physiological transcripts of the TBX15 protein (differing in the start codon) as well as of the two predicted mutant ORFs; in the mutant proteins, the normal amino acid sequence is interrupted after amino acid 449 or 450 and is followed by a stretch of 78 or 77 missense amino acids. The DNA-binding T-box is preserved in both mutant variants.
We then investigated whether the mutations affected the ability of mutant TBX15 protein to bind their target DNA sequences in an EMSA after incubation of in vitro-synthesized wild-type and mutant TBX15 proteins (Figure 4B) with oligonucleotides corresponding to the mouse Tbx15 target DNA sequence that was identified previously9 (Figure 4A). Target DNA binding of the mutant proteins was similar to that of wild-type protein (Figure 4C), in keeping with the notion that the DNA-binding region of the protein, the T-box, was unaffected by the mutations.
Figure 4.
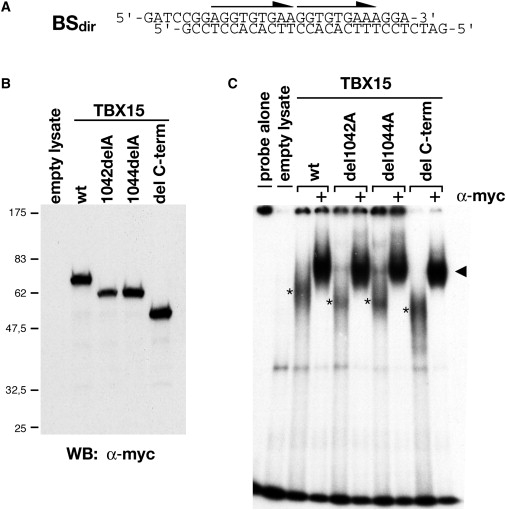
In Vitro DNA Binding Analysis of Mutant TBX15 Proteins
(A) The previously identified7 binding site of mouse Tbx15 (BSdir) that was tested in EMSAs. Arrows indicate the orientation of T-half sites. (B) Assays were performed with equal amounts of in vitro-synthesized Myc-tagged TBX15 proteins, as determined by anti-Myc immunoblot. All proteins were expressed with the expected molecular weights. (C) No differences in DNA binding to the site BSdir are observed between the C-terminally truncated form TBX15-del C-term protein and the wild-type form of TBX15 or the c.1042 delA and c.1044 delA mutants in a gel-shift assay. Equally strong complexes were detected for all proteins (asterisks). Specificity of binding was confirmed by addition of anti-Myc antibody, which resulted in the formation of a prominent supershifted complex (black arrowhead).
Given that the DNA-binding ability of the mutant proteins was conserved, we then focused on the localization and stability of mutant TBX15 proteins. Immunoblot analysis of cytoplasmic and nuclear extracts of patients' fibroblasts with antiserum raised against the amino terminal part of the long isoform of TBX15 (TBX15L) showed an almost complete loss of the TBX15L signal in the patients, whereas bands of the expected size were detected in the nuclear fraction of both age-matched controls (Figure 5A). For further studies, the wild-type and mutant cDNA sequences were cloned into expression vectors. Figure 5B shows expression of FLAG-tagged TBX15 proteins in human chondrosarcoma (HCS) cells. The wild-type protein was readily detectable and showed the expected predominantly nuclear localization; no TBX15 protein was detected after transduction with either of the two mutants, in spite of adequate mRNA levels (data not shown). When EGFP-tagged TBX15 protein isoforms were analyzed by direct fluorescence microscopy, the wild-type protein gave a strong signal in both the cytoplasm and the nucleus (short isoform, Figure 6B) or exclusively in the nucleus (long isoform, Figure 6A); the mutant constructs gave a barely detectable signal, indicating that protein stability was compromised. Similar results were obtained when Myc-tagged TBX15 proteins and anti-Myc immunofluorescence detection were used in HeLa and HEK293 cells (data not shown).
Figure 5.
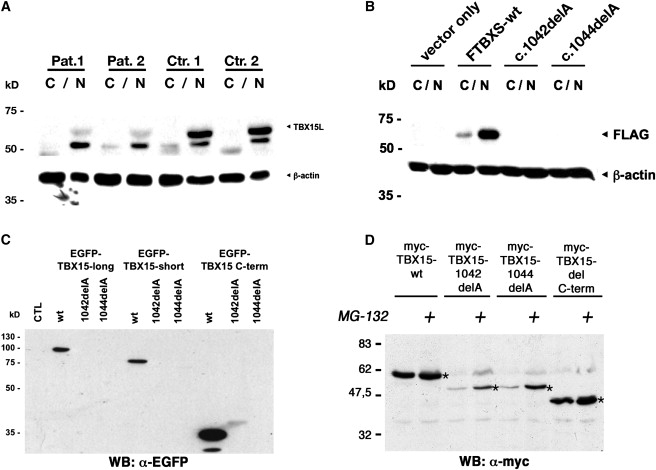
Immunoblot Analysis of TBX15 Protein in Patients' Fibroblasts and in Cell Transfection Experiments
(A) Analysis of TBX15 proteins in subcellular fractions (C = cytoplasmic extract, N = nuclear extract) of patients' and control fibroblasts. A polyclonal antiserum directed against the amino-terminus of the long isoform of TBX15 (TBX15L) was used for the immunoblots, which show a strong reduction of the signal for the long isoform of TBX15 in both patients. The nature of the band that has an apparent molecular weight of approximately 50 kDa and migrates below TBX15L is unclear but may be due to cross-reactivity of the antiserum with the related protein TBX18. Membranes were reprobed with an antibody against β-actin so that equal loading could be ensured.
(B) Disease-associated mutations confer instability to transgenic TBX15. Analysis of TBX15 proteins in subcellular fractions (C = cytoplasmic extract, N = nuclear extract) of human chondrosarcoma (HCS) cells overexpressing either FLAG-tagged wild-type short variant TBX15 or analogous constructs carrying the disease-associated mutations. An anti-FLAG immunoblot shows the presence of the wild-type recombinant protein only.
(C) Disease-associated mutations confer instability to all isoforms of TBX15 and other proteins. Analysis of TBX15 proteins in human chondrosarcoma (HCS) cells overexpressing either EGFP-tagged wild-type TBX15 or constructs carrying the disease-associated mutations is shown. In addition, EGFP was fused directly to either the wild-type C-terminal portion of TBX15 (152 amino acids) or to the two stretches of 78 or 77 missense amino acids predicted by the human mutations. Whole-cell lysates were analyzed by anti-GFP immunoblot. Only lanes loaded with the wild-type fusion constructs gave a signal of the expected size.
(D) The C-terminal sequences resulting from disease-associated TBX15 mutations confer instability by directing proteins to proteosomal degradation. Protein analysis of Myc-tagged wild-type TBX15, c.1042 delA, c.1044 delA, and ΔC proteins in HeLa cells probed in an anti-Myc immunoblot. Expression of TBX15 mutants c.1042 delA and c.1044 delA is strongly reduced in comparison to that of the TBX15-wt and Tbx15ΔC proteins (asterisks), indicating that the missense amino acids, and not the truncation, affect protein stability. Incubation with the proteasome inhibitor MG-132 (25 μM) leads to a marked stabilization of TBX15-c.1042 delA, and c.1044 delA proteins. No more slowly migrating bands that could represent poly-ubiquitinylated proteins were observed.
Figure 6.
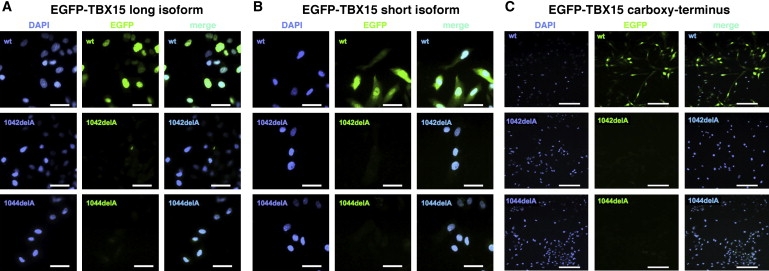
Fluorescence Studies of EGFP-Tagged TBX15 Constructs
Intracellular localization of EGFP-tagged TBX15 proteins in HCS cells as detected by direct EGFP fluorescence microscopy. The TBX15 protein localizes exclusively (long variant [A]) or predominantly to the nucleus (short variant [B]; compare nuclear staining with DAPI), whereas TBX15-c.1042 delA and c.1044 delA are barely detectable, both in the context of the long and of the short variant. A similar loss of fluorescent signal was observed when the C terminus of the disease-associated protein variants was fused to EGFP (C). HCS cells were transduced with titered recombinant virus at the same MOI for all constructs. Scale bars represent 15 (A and B) or 25 (C) μm.
To elucidate the mechanisms leading to instability of the mutant proteins, we transfected HeLa cells with wild-type human TBX15, the two mutants identified in the Cousin syndrome patients, or a synthetic mutant harboring the truncation of the protein after amino acid residue 344. Cellular levels of the C-truncated protein were similar to that of the wild-type, whereas levels of the two human mutants were significantly reduced; these could be partially restored by addition of the proteasome inhibitor, MG-132 (Figure 5D). Thus, simple truncation of 152 amino acids at the C terminus did not affect stability, but the presence of the 78 (versus 77) missense amino acids in the mutants directed the TBX15 protein to proteasomal degradation. To confirm this interpretation, we generated constructs in which EGFP was fused directly onto either the wild-type carboxy-terminal portion of TBX15 or the two stretches of missense amino acids predicted by the human mutations. Immunoblot analysis showed a strong signal for the wild-type C terminus, but the two mutant C termini were not detectable (Figure 5C). Upon direct fluorescence microscopy, a signal was detected with the constructs containing the wild-type but not the mutant isoforms of TBX15 (Figures 6A and 6B). When the constructs consisting of EGFP and the C-terminal portions only were analyzed, fluorescence was detected with the constructs containing the wild-type C terminus but not with the constructs containing the mutant C termini, confirming that the stretches of missense amino acids produced by the frameshift mutation conferred instability to the EGFP protein (Figure 6C).
In summary, genomic sequencing indicated that two similar mutations segregated in the two families; these mutations predicted the synthesis of TBX15 protein that had an intact T-box but lacked a significant portion of the C-terminal region and had instead a stretch of 78 (or 77) missense amino acids that included five cysteine residues. The subsequent set of experiments indicated that although TBX15 protein synthesized from the two mutant alleles retained the ability to bind their DNA targets in vitro, cellular levels of TBX15 were drastically reduced or abolished because of proteasome-associated degradation directed by the C-terminal missense stretch. A similar mutation in the TNSALP gene (MIM 171760), causing the addition of a stretch of 80 missense amino acids including four cysteine residues, has been shown to cause proteasomal degradation of the mutant alkaline phosphatase protein and to result in severe hypophosphatasia (MIM 241500)11. Thus, the two affected subjects were homozygous for mutations that resulted in functional TBX15 deficiency, confirming the initial hypothesis of a shared molecular basis for the changes seen in the craniocervical bones, the scapula, and the pelvis in the subjects with Cousin syndrome and in Tbx15-deficient mice.
T-box genes encode a family of transcription factors that regulate a variety of developmental processes in metazoa ranging from hydra to humans. Mammalian genomes are known to harbor 17 family members involved in patterning and differentiation processes during gastrulation and organogenesis. Notably, mutations in human TBX genes, TBX1, TBX3, TBX4, TBX5, TBX19(TPIT), TBX20, and TBX22, are known to cause congenital disorders with craniofacial, endocrine, limb, and cardiac malformation.12–19 The identification of Cousin syndrome as a TBX15-related disorder in humans further underscores the importance of the T-box gene family for human development and disease and adds an important piece to the mosaic of our understanding of human genetic anomalies of the skeleton.20 It also allows for molecular diagnosis and genetic counseling; in fact, prior to our studies, the two families reported here had been given other diagnoses and had been counseled for dominant inheritance.
Tbx15 is known to be required for the condensation of mesenchymal precursor cells in early development of the skeleton.4,5,21 In the mouse embryo, Tbx15 is expressed first in mesenchyme of the limb buds and subsequently in the developing zeugopodal elements. In the cranium, expression begins in the mesenchyme near the surface ectoderm and then can be detected in various craniofacial elements that include the mandibular process of the first mandibular arch and the dorsal edge of the hyomandibular cleft, which will eventually become the external auditory meatus; the surface of the second branchial arch as well as the maxillary and the mandibular portions of the first branchial arch surrounding the developing mouth; and the area that is dorsal to each optic prominence and that later extends to surround most of the eye.4,5,21 No expression is seen in internal organs between embryonic days 8.5 and 12.5, whereas data from EST libraries suggest an expression in adult tissues of bone, brain, intestine, liver, muscle, testis, and thymus. Although no expression data in the human are available, the mouse expression pattern fits reasonably well with the phenotype observed in Cousin syndrome.
The target genes of TBX15 and of its closely related family member TBX18 (MIM 604613) have remained elusive so far. Scapular and pelvic hypoplasia are features of campomelic dysplasia (MIM 114290) caused by SOX9 (MIM 608160) mutations; scapular hypoplasia with humeroradial synostosis is a feature of Antley-Bixler syndrome (MIM 207410, 201750) associated with mutations in either FGFR2 or POR; the phenotypic relationship suggests that TBX15 may act within this contextual frame and/or share target genes with SOX9, FGFR2 (MIM 176943), and POR (MIM 124015). Kosenow syndrome (scapuloiliac dysostosis, MIM 169550) is a dominantly inherited condition with hypoplasia of the scapula and pelvis but no craniocervical malformation; its molecular basis is unknown. If Cousin syndrome is caused by homozygosity for recessive TBX15 null mutations, there may be a phenotype associated with dominant mutations within the T-box region, and Kosenow syndrome would be a possible candidate.
Acknowledgments
We thank the families for their gracious participation in this study. We are particularly grateful to subject 1, a language student whose strive for independence and ability to comment on her condition with philosophy and humor make her a leading figure in the German Association for Little People and their Families (BKMF, www.bkmf.de). Work in the Freiburg Skeletal Dysplasia Centre and laboratory (www.skeldys.org) was supported by the SKELNET project of the German Bundesministerium für Bildung und Forschung (BMBF), by the European Community EuroGrow consortium, and by individual grants of the Deutsche Forschungsgemeinschaft (DFG). Work in the laboratory of A.K. in Hannover was supported by individual grants of the DFG and the DFG-funded Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy).
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Ensembl Human Gene View, www.ensembl.org; for human TBX15, see
www.ensembl.org/Homo_sapiens/geneview?gene=ENSG00000092607; the short isoform (TBX15S; 496 amino acids) corresponds to transcript ENST00000207157, and the long isoform (TBX15L; 602 amino acids) corresponds to transcript ENST00000369429.
References
- 1.Cousin J., Walbaum R., Cegarra P., Huguet J., Louis J., Pauli A., Fournier A., Fontaine G. Dysplasie pelvis-scapulaire familiale avec anomalies epiphysaires, nanisme et dysmorphies: Un nouveau syndrome? Arch. Fr. Pediatr. 1982;39:173–175. [PubMed] [Google Scholar]
- 2.Elliott A.M., Roeder E.R., Witt D.R., Rimoin D.L., Lachman R.S. Scapuloiliac dysostosis (Kosenow syndrome, pelvis-shoulder dysplasia) spectrum: Three additional cases. Am. J. Med. Genet. 2000;95:496–506. doi: 10.1002/1096-8628(20001218)95:5<496::aid-ajmg16>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 3.Candille S.I., Van Raamsdonk C.D., Chen C., Kuijper S., Chen-Tsai Y., Russ A., Meijlink F., Barsh G.S. Dorsoventral patterning of the mouse coat by Tbx15. PLoS Biol. 2004;2:e3. doi: 10.1371/journal.pbio.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuijper S., Beverdam A., Kroon C., Brouwer A., Candille S., Barsh G., Meijlink F. Genetics of shoulder girdle formation: Roles of Tbx15 and aristaless-like genes. Development. 2005;132:1601–1610. doi: 10.1242/dev.01735. [DOI] [PubMed] [Google Scholar]
- 5.Singh M.K., Petry M., Haenig B., Lescher B., Leitges M., Kispert A. The T-box transcription factor Tbx15 is required for skeletal development. Mech. Dev. 2005;122:131–144. doi: 10.1016/j.mod.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Curry G.A. Genetical and development studies on droopy-eared mice. J. Embryol. Exp. Morphol. 1959;7:39–65. [PubMed] [Google Scholar]
- 7.Morgenstern J.P., Land H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trost T.M., Lausch E.U., Fees S.A., Schmitt S., Enklaar T., Reutzel D., Brixel L.R., Schmidtke P., Maringer M., Schiffer I.B. Premature senescence is a primary fail-safe mechanism of ERB2-driven tumorigenesis in breast carcinoma cells. Cancer Res. 2005;65:840–849. [PubMed] [Google Scholar]
- 9.Farin H.F., Bussen M., Schmidt M.K., Singh M.K., Schuster-Gossler K., Kispert A. Transcriptional repression by the T-box proteins Tbx18 and Tbx15 depends on Groucho corepressors. J. Biol. Chem. 2007;282:25748–25759. doi: 10.1074/jbc.M703724200. [DOI] [PubMed] [Google Scholar]
- 10.Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komaru K., Ishida Y., Amaya Y., Goseki-Sone M., Orimo H., Oda K. Novel aggregate formation of a frame-shift mutant protein of tissue-nonspecific alkaline phosphatase is ascribed to three cysteine residues in the C-terminal extension. Retarded secretion and proteasomal degradation. FEBS J. 2005;272:1704–1717. doi: 10.1111/j.1742-4658.2005.04597.x. [DOI] [PubMed] [Google Scholar]
- 12.Bamshad M., Lin R.C., Law D.J., Watkins W.C., Krakowiak P.A., Moore M.E., Franceschini P., Lala R., Holmes L.B., Gebuhr T.C. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat. Genet. 1997;16:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 13.Basson C.T., Bachinsky D.R., Lin R.C., Levi T., Elkins J.A., Soults J., Grayzel D., Kroumpouzou E., Traill T.A., Leblanc-Straceski J. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 14.Merscher S., Funke B., Epstein J.A., Heyer J., Puech A., Lu M.M., Xavier R.J., Demay M.B., Russell R.G., Factor S. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 15.Yagi H., Furutani Y., Hamada H., Sasaki T., Asakawa S., Minoshima S., Ichida F., Joo K., Kimura M., Imamura S. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1342–1343. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 16.Wilson V., Conlon F.L. The T-box family. Genome Biol. 2002;3:3008.1–3008.7. doi: 10.1186/gb-2002-3-6-reviews3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naiche L.A., Harrelson Z., Kelly R.G., Papaioannou V.E. T-box genes in vertebrate development. Annu. Rev. Genet. 2005;39:219–239. doi: 10.1146/annurev.genet.39.073003.105925. [DOI] [PubMed] [Google Scholar]
- 18.Vallette-Kasic S., Brue T., Pulichino A.M., Gueydan M., Barlier A., David M., Nicolino M., Malpuech G., Déchelotte P., Deal C. Congenital isolated adrenocorticotropin deficiency: An underestimated cause of neonatal death, explained by TPIT gene mutations. J. Clin. Endocrinol. Metab. 2005;90:1323–1331. doi: 10.1210/jc.2004-1300. [DOI] [PubMed] [Google Scholar]
- 19.Kirk E.P., Sunde M., Costa M.W., Rankin S.A., Wolstein O., Castro M.L., Butler T.L., Hyun C., Guo G., Otway R. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007;81:280–291. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 21.Agulnik S.I., Papaioannou V.E., Silver L.M. Cloning, mapping, and expression analysis of TBX15, a new member of the T-Box gene family. Genomics. 1998;51:68–75. doi: 10.1006/geno.1998.5278. [DOI] [PubMed] [Google Scholar]