

Abstract
Mutations of the lipid phosphatase FIG4 that regulates PI(3,5)P2 are responsible for the recessive peripheral-nerve disorder CMT4J. We now describe nonsynonymous variants of FIG4 in 2% (9/473) of patients with amyotrophic lateral sclerosis (ALS) and primary lateral sclerosis (PLS). Heterozygosity for a deleterious allele of FIG4 appears to be a risk factor for ALS and PLS, extending the list of known ALS genes and increasing the clinical spectrum of FIG4-related diseases.
Main Text
FIG4, also known as SAC3, is a phosphoinositide 5-phosphatase that regulates the cellular abundance of PI(3,5)P2, a signaling lipid located on the cytosolic surface of membranes of the late endosomal compartment.1 PI(3,5)P2 mediates retrograde trafficking of endosomal vesicles to the trans-Golgi network.2,3 Inactivation of FIG4 in mice results in massive degeneration of neurons in sensory and autonomic ganglia, motor cortex, striatum, and other regions of the central nervous system, with extensive vacuolization of neurons and other cells.4 Sciatic-nerve-conduction velocity is reduced and motor neurons in the ventral spinal cord are vacuolated in homozygous null mice.
Mutations of the human FIG4 gene on chromosome 6q21 are responsible for the recessively inherited disorder CMT4J (MIM 611228), a severe form of Charcot-Marie-Tooth disease (CMT) with early onset and involvement of both sensory and motor neurons.4 CMT4J patients are compound heterozygotes with a loss-of-function allele in combination with the partial loss-of-function mutation I41T. FIG4 mutations were found in four families, and childhood onset occurred in three families. One family exhibited adult onset and primarily motor symptoms that resembled amyotrophic lateral sclerosis (ALS) (MIM 105400), except for reduced nerve-conduction velocity and segmental demyelination indicative of Schwann cell involvement.5 Because mutation of FIG4 affects motor-neuron function in humans and mice, we tested FIG4 as a candidate gene for ALS.
ALS is a severe neurological disorder, with onset between 40 and 80 years of age, characterized by selective neurodegeneration of lower and upper motor neurons in the spinal cord, brainstem, and cortex.6,7 PLS (MIM 611637) is a related motor-specific disorder that primarily affects corticospinal upper motor neurons.6,7 Ninety percent of ALS cases are sporadic (SALS), and 10% have a familial history (FALS), usually with dominant inheritance. ALS is genetically heterogeneous, and known genes explain < 5% of cases.6,7 PLS is usually sporadic, and genetic defects have not been identified. To evaluate the role of FIG4, we screened DNA from 473 patients, including 364 sporadic cases and 109 familial cases.
All patients and controls were of European ancestry. SALS cases included individuals from National Institute of Neurological Disorders and Stroke panels NDPT025 (long-term ALS survivors), NDPT026 (bulbar onset), and NDPT029 (upper-limb onset) (see Web Resources), as well as 92 SALS patients from the Massachusetts General Hospital who had onset at 53 ± 15 years (mean ± SD), a disease duration of 4.8 ± 4 years, and a male to female ratio of 2:1. The site of disease onset was 23% bulbar, 43% upper extremities, 28% lower extremities, and 7% multiple sites, The FALS patients had onset at 55 ± 15 years (median 55 years) and disease duration of 3.4 ± 3.2 years, with a male to female ratio of 1:1.3. The FALS patients were previously tested for mutations in SOD1; their site of disease onset was 27% bulbar, 31% upper extremities, 37% lower extremities, and 5% multiple sites. All procedures were carried out in accordance with the ethical standards of the Institutional Review Boards at the Massachusetts General Hospital and the University of Michigan.
To screen for pathogenic mutations in the coding sequence and splice sites, we amplified the 23 exons of FIG4 from 473 patient genomic-DNA samples. Eleven exons were sequenced directly for all patients (exons 2, 7, 8, 9, 10, 17, 18, 19, 20, 21, and 23). The other 12 exons were first screened by heteroduplex analysis (conformation-sensitive gel electrophoresis),8 and exons with abnormal mobility were sequenced. Small deletions or duplications could have been missed by our screening methods. All variants were confirmed in at least two independent PCR and sequencing reactions. For the detection of possible second-site mutations, all 23 exons were sequenced for the individuals in Table 1.
Table 1.
Rare Variants of FIG4 in Patients with ALS and PLS
| Sample | Amino Acid | Exon | Nucleotide | Controls (Frequency) | Polyphen Score | Sift Score | Comments | Predicted Deleterious | El Escorial Diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| SALS E12 | p.R183X | exon 6 | c.547C→T | 0/558 | n.a. | n.a | Truncation before active site | Yes | ALS definite |
| SALS 8533 | p.Q403X | exon 11 | c.1207C→T | 0/558 | n.a. | n.a. | Truncation before active site | Yes | ALS possible |
| FALS G07 | p.R23fsX30 or p.del(23–55) | exon 2 splice site | c.67−1G→T | 0/558 | n.a | n.a. | Truncation or deletion of 33 aa | Yes | ALS suspected |
| ND 09489 | p.S424_K462 del insR | exon 12 splice site | c.1386+5G→T | 0/395 | n.a. | n.a | Deletion in active site domain or truncation | Yes | ALS definite |
| FALS G03 | p.D53Y | exon 2 | c.157G→T | 0/558 | 3 | 2 | Impaired in yeast | Yes | ALS definite |
| SALS B12 | p.D48G | exon 2 | c.143A→G | 0/558 | 2 | 1 | Functional in yeast | No | PLS possible |
| SALS H11 | p.R388G | exon 11 | c.1162A→G | 0/558 | 1 | 1 | Impaired in yeast | No | PLS |
| SALS E12 | p.I411V | exon 11 | c.1231A→G | 0/558 | 1 | 1 | Functional in yeast | No | ALS definite |
| ND 11318 | p.Y647C | exon 17 | c.1940T→G | 0/395 | 2 | 1 | Not conserved in yeast | unclear | ALS definite |
| FALS A04 | p.I902T | exon 23 | c.2705T→C | 0/558 | 2 | 2 | Not conserved in yeast | unclear | ALS definite |
PolyPhen scores are as follows: 1, benign; 2, possibly damaging; 3, probably damaging. SIFT scores are as follows: 1, tolerated; 2, affects protein structure. The first five of the ten variants and R388G are highly likely to be pathogenic. “n.a.” indicates not applicable (not missense). For clinical descriptions of these patients, see Table S1 SALS, plate 1p1. FALS, plate 1p2.
Variants detected in the patients were tested in 395–558 ethnically matched controls. Control samples from the Coriell Institute include 192 samples from the neurological normal control panels NDPT006 and NDPT009 and 163 neurologically normal individual samples that did not overlap with the panels. A set of 111 controls older than 60 years of age without personal or family history of neurological disease was previously described.9 Spouses of ALS patients provided 92 controls. SNPs detected in patients and controls are presented in Table S2, available online.
We identified ten unique nonsynonymous variants of FIG4 in nine patients, including six with SALS and three with FALS (Table 1). Seven patients carried a diagnosis of definite or probable ALS, and two patients carried a diagnosis of PLS, with average age of onset of 56 ± 14 years (mean ± SD) and average duration of 9 ± 11 years. Clinical findings for these patients are presented in Table S1. There was a striking prominence of corticospinal findings. Subtle changes in personality were mentioned in two cases.
Each mutation was found in a single patient and was not present in controls (Table 1) or in the dbSNP, indicating that they are not common polymorphisms. The variants include two protein-truncation mutations, two mutations in consensus splice sites, and six missense mutations (Table 1). Sequence chromatograms and evolutionary conservation are presented in Figure S1.
The two protein-truncation mutations, R183X and Q403X, are located upstream of the SAC phosphatase active site10,11 and result in loss of FIG4 phosphatase activity.
The exon 2 splice acceptor mutation alters the invariant −1G nucleotide, which prevents correct splicing. This mutation created a novel out-of-frame consensus acceptor site 2 bp downstream of the original site (Figure S1). Splicing to the new site is strongly predicted from analysis of human mutations at the −1 position.12 This outcome would result in the protein truncation R23fsX30. Skipping of exon 2 would result in an in-frame deletion of 33 evolutionarily conserved amino acids, likely to interfere with protein function.
The splice-site mutation in the donor site of exon 12 changes the important +5G residue that is the site of many human mutations.13 The predicted outcome is skipping of exon 12, resulting in an in-frame deletion of 39 amino acid residues from the SAC phosphatase domain.13 Alternatively, readthrough into intron 12 would generate the in-frame stop codon K463fsX474. Cell lines from these patients were not available for analysis of splice products.
The six missense mutations were analyzed with the protein-prediction programs PolyPhen and SIFT (see Web Resources). D53Y was most strongly predicted to be deleterious (Table 1). Four of the missense mutations change amino acid residues that are conserved in yeast; we tested their ability to rescue the enlarged vacuole in a Fig4Δ null yeast strain. To be functional in the yeast assay, the variant protein must bind the other proteins in the PI(3,5)P2-regulatory complex, become localized to the vacuolar membrane, and retain phosphatase activity.14 Consistent with the predictions, D53Y is a deleterious allele, with less activity than wild-type FIG4 (p < 0.001) and less activity than the CMT4J mutant allele I41T, which was included for comparison (p < 0.02) (Figure 1A). R388G also has significantly less activity than the wild-type allele (p < 0.02). The variants D48G and I411V were close to the wild-type in function, and their pathogenicity remains uncertain (Figure 1A). Mutations D48G and D53Y are located at two ends of a predicted β-sheet domain, consistent with an effect on protein interaction.
Figure 1.
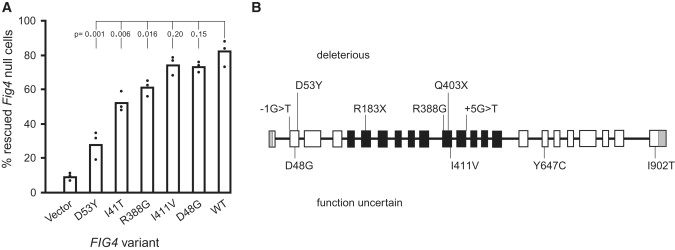
Rescue of Vacuole Formation in Null Fig4Δ Yeast
(A) Patient missense mutations were introduced into yeast Fig4p and tested for their ability to correct the enlarged vacuole in a Fig4Δ null strain of yeast as previously described.4,8 The pathogenic CMT4J variant I41T was included for comparison. The low level of PI(3,5)P2 in Fig4Δ cells results in enlarged vacuoles, and introduction of wild-type Fig4p restores both PI(3,5)P2 levels and vacuole morphology.4,8 Bars represent the mean from three independent experiments, containing 100–300 cells per experiment. p values for each mutant compared with the wild-type were calculated with the unpaired t test.
(B) Locations of patient mutations.
Overall, six of the ten variants are clearly deleterious: the two stop codons, the two consensus splice-site variants, and the missense mutations D53Y and R388G (Figure 1B). The SALS patient E12 carried two variants, R183X and I411V. Because I411V retained full function in the yeast assay, it is unlikely to contribute to disease. The other three missense variants in Table 1 could be pathogenic as well. DNA was not available from family members for analysis of cosegregation of the FALS variants. The missense mutations were identified in single patients, and detection in additional patients would support their pathogenicity.
In our previous work on CMTJ4 families, we observed two parents and one sibling who were heterozygous carriers of null alleles of FIG4 but did not exhibit clinical symptoms.4 These individuals were younger than our patients with late-onset ALS and could be presymptomatic or reflect incomplete penetrance. Heterozygous missense mutations of FIG4 could exert their effects either through partial loss of function or by a dominant-negative mechanism via competition with the wild-type protein for incorporation into the multimeric PI(3,5)P2-regulatory complex.14 The variable age at onset in CMT4J families, from early childhood to adulthood, suggests that genetic background and/or environmental exposures modify the clinical course. These factors are also thought to influence manifestation of ALS caused by mutations in other genes. The known role of FIG4 in motor-neuron survival and the impaired function of the patient-specific variants described here support the view that these mutations contribute to the development of ALS.
CMT4J patients derive all of their FIG4 activity from one copy of the defective allele I41T; as a result, they have less FIG4 activity than the ALS patients, who have one wild-type allele in addition to their defective allele. Most CMT4J patients have onset in early childhood and a severe course. The adult-onset CMT4J patients differ from ALS patients by their Schwann cell involvement, as indicated by reduced nerve-conduction velocity and sural-nerve demyelination.5 The ALS patients and adult-onset CMT4J patients both exhibit asymmetric progression, absence of sensory symptoms, and the absence of dementia. The ALS and PLS cases have normal conduction velocities and striking corticospinal-tract signs. In the SALS cases, the corticospinal signs were the most salient findings (Table S1). Similarly, in FIG4 null mice, neurodegeneration is much earlier and more extensive in the motor cortex than in spinal motor neurons.4
Phosphoinositides serve as molecular tags for intracellular vesicles and mediate vesicle trafficking. Other genes affecting phosphoinositide signaling are responsible for Charcot-Marie-Tooth type 4B1, 4B2, and 4H, and SPG15, which targets corticospinal motor neurons.15–19 Phosphoinositide metabolism has not been previously implicated in ALS. Human motor neurons could be particularly susceptible to mutations that affect membrane trafficking because of their need to turnover membrane components from long axonal processes during many decades of life.1
Each of the previously identified ALS genes account for only a few percent of cases.6,20 The identification of FIG4 mutations in 1%–2% of ALS patients suggests that FIG4 is another contributor to this genetically heterogenous disease. More extensive screening will be needed for a precise definition of the contribution of FIG4 to ALS and other motor-neuron disorders.
Acknowledgments
This study was supported by National Institutes of Health grants GM24872 (M.H.M.), NS050557 (R.H.B.), NS050641 (R.H.B.), T32 GM007544 (C.Y.C.), and NS064015 (L.S.W.) and by the ALS Association (M.H.M. and R.H.B.), the ALS Therapy Alliance, Project ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, and the Al-Athel ALS Foundation. We thank Emily Kauffman for assistance in development of the yeast-vacuole assay. P.S. is supported through the auspices of H. Robert Horvitz, an Investigator of the Howard Hughes Medical Institute. DNA panels from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds), as well as clinical data, were used in this study. The submitters that contributed samples are acknowledged in detailed descriptions of each panel: NDPT025, NDPT026, NDPT029, NDPT006, and NDPT009.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Coriell Institute, http://ccr.coriell.org/Sections/Collections/NINDS/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Single Neucleotide Polymorphism Database (dbSNP), http://www.ncbi.nlm.nih.gov/projects/SNP/
References
- 1.Volpicelli-Daley L., De Camilli P. Phosphoinositides' link to neurodegeneration. Nat. Med. 2007;13:784–786. doi: 10.1038/nm0707-784. [DOI] [PubMed] [Google Scholar]
- 2.Rutherford A.C., Traer C., Wassmer T., Pattni K., Bujny M.V., Carlton J.G., Stenmark H., Cullen P.J. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J. Cell Sci. 2006;119:3944–3957. doi: 10.1242/jcs.03153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y., Zolov S.N., Chow C.Y., Slutsky S.G., Richardson S.C., Piper R.C., Yang B., Nau J.J., Westrick R.J., Morrison S.J. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc. Natl. Acad. Sci. USA. 2007;104:17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow C.Y., Zhang Y., Dowling J.J., Jin N., Adamska M., Shiga K., Szigeti K., Shy M.E., Li J., Zhang X. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X., Chow C.Y., Sahenk Z., Shy M.E., Meisler M.H., Li J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain. 2008;131:1990–2001. doi: 10.1093/brain/awn114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 7.Schymick J.C., Talbot K., Traynor B.J. Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007;16:R233–R242. doi: 10.1093/hmg/ddm215. [DOI] [PubMed] [Google Scholar]
- 8.Escayg A., MacDonald B.T., Meisler M.H., Baulac S., Huberfeld G., An-Gourfinkel I., Brice A., LeGuern E., Moulard B., Chaigne D. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 9.Rainier S., Sher C., Reish O., Thomas D., Fink J.K. De novo occurrence of novel SPG3A/atlastin mutation presenting as cerebral palsy. Arch. Neurol. 2006;63:445–447. doi: 10.1001/archneur.63.3.445. [DOI] [PubMed] [Google Scholar]
- 10.Duex J.E., Tang F., Weisman L.S. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J. Cell Biol. 2006;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes W.E., Cooke F.T., Parker P.J. Sac phosphatase domain proteins. Biochem. J. 2000;350:337–352. [PMC free article] [PubMed] [Google Scholar]
- 12.Vorechovsky I. Aberrant 3′ splices sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2006;34:4630–4641. doi: 10.1093/nar/gkl535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buratti E., Chivers M., Královicová J., Romano M., Baralle M., Krainer A.R., Vorechovsky I. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35:4250–4263. doi: 10.1093/nar/gkm402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin N., Chow C.Y., Liu L., Zolov S.N., Bronson R., Davisson M., Petersen J.L., Zhang Y., Park S., Duex J.E. VAC14 nucleates a protein complex essential for the regulation of PI(3,5)P2 levels in yeast and mouse. EMBO J. 2008 doi: 10.1038/emboj.2008.248. Published online November 27, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolino A., Muglia M., Conforti F.L., LeGuern E., Salih M.A., Georgiou D.M., Christodoulou K., Hausmanowa-Petrusewicz I., Mandich P., Schenone A. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 2000;25:17–19. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- 16.Senderek J., Bergmann C., Weber S., Ketelsen U.P., Schorle H., Rudnik-Schöneborn S., Büttner R., Buchheim E., Zerres K. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum. Mol. Genet. 2003;12:349–356. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- 17.Stendel C., Roos A., Deconinck T., Pereira J., Castagner F., Niemann A., Kirschner J., Korinthenberg R., Ketelsen U.P., Battaloglu E. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am. J. Hum. Genet. 2007;81:158–164. doi: 10.1086/518770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delague V., Jacquier A., Hamadouche T., Poitelon Y., Baudot C., Boccaccio I., Chouery E., Chaouch M., Kassouri N., Jabbour R. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am. J. Hum. Genet. 2007;81:1–16. doi: 10.1086/518428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanein S., Martin E., Boukhris A., Byrne P., Goizet C., Hamri A., Benomar A., Lossos A., Denora P., Fernandez J. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am. J. Hum. Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valdmanis P.N., Rouleau G.A. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008;70:144–152. doi: 10.1212/01.wnl.0000296811.19811.db. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.