

Abstract
Analysis of a nuclear family with three affected offspring identified an autosomal-recessive form of spondyloepimetaphyseal dysplasia characterized by severe short stature and a unique constellation of radiographic findings. Homozygosity for a haplotype that was identical by descent between two of the affected individuals identified a locus for the disease gene within a 17.4 Mb interval on chromosome 15, a region containing 296 genes. These genes were assessed and ranked by cartilage selectivity with whole-genome microarray data, revealing only two genes, encoding aggrecan and chondroitin sulfate proteoglycan 4, that were selectively expressed in cartilage. Sequence analysis of aggrecan complementary DNA from an affected individual revealed homozygosity for a missense mutation (c.6799G → A) that predicts a p.D2267N amino acid substitution in the C-type lectin domain within the G3 domain of aggrecan. The D2267 residue is predicted to coordinate binding of a calcium ion, which influences the conformational binding loops of the C-type lectin domain that mediate interactions with tenascins and other extracellular-matrix proteins. Expression of the normal and mutant G3 domains in mammalian cells showed that the mutation created a functional N-glycosylation site but did not adversely affect protein trafficking and secretion. Surface-plasmon-resonance studies showed that the mutation influenced the binding and kinetics of the interactions between the aggrecan G3 domain and tenascin-C. These findings identify an autosomal-recessive skeletal dysplasia and a significant role for the aggrecan C-type lectin domain in regulating endochondral ossification and, thereby, height.
Main Text
Many of the determinants of normal human growth have been defined by studying short-stature disorders, the skeletal dysplasias.1 Frequently, the disease genes in these disorders have identified molecules essential for normal skeletal development as well as functional domains within the proteins they encode. The functions of the skeletal-dysplasia disease genes are as diverse as the conditions with which they are associated, and the gene products include regulatory molecules, cytoskeletal proteins, centrosomal proteins, and extracellular-matrix structural proteins along with their posttranslational modifiers. The disease genes are often selectively expressed in chondro-osseous tissues, or they can be more widely expressed with a greater effect on cartilage and/or bone resulting from the distinctive biology of skeletal tissues. Together, these genes define the size and shape of the craniofacial, axial, and appendicular skeletal elements, the extent of linear growth, and the long-term stability of cartilage and bone.
Figure 1 shows the pedigree of a family, studied under an institutional review board-approved protocol, with a distinctive skeletal-dysplasia phenotype. The three affected individuals were initially ascertained as young children and shared a consistent set of phenotypic findings that were present at all ages (Table 1). Craniofacial abnormalities included relative macrocephaly, severe midface hypoplasia with almost absent nasal cartilage, relative prognathism, and slightly low-set, posteriorly rotated ears (Figure 1). They had short necks and barrel chests and exhibited a mild lumbar lordosis. Their extremities showed rhizomelia and mesomelia with no bowing of any segment. Hand findings included significant brachydactyly with short, broad thumbs, horizontal nails, and telescoping interphalangeal joints. Their childhoods were only complicated by extreme short stature, without orthopedic or other systemic complications. Radiographs (Figure 2) revealed findings consistent with a previously undescribed form of spondyloepimetaphyseal dysplasia (SEMD). Long bones exhibited generalized irregular epiphyses with widened metaphyses, especially at the knees. Spinal abnormalities included significantly flattened vertebral bodies (platyspondyly) and multiple cervical-vertebral clefts. Hand radiographs showed proportionate brachydactyly with accessory carpal ossification centers.
Figure 1.
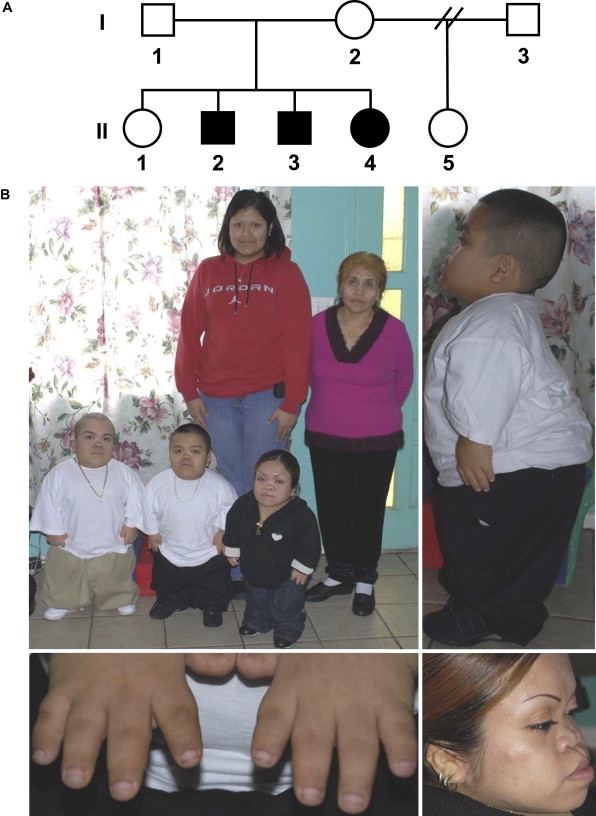
Clinical Phenotype
(A) Pedigree. Filled symbols identify affected individuals.
(B) In the top left image, the back row from left to right shows II-1 (23 years old [yo]) and I-I (58 yo), respectively, and the front row from left to right shows II-2 (19 yo), II-3 (16 yo), and II-4 (26 yo), respectively. Note the relative macrocephaly and lordosis in II-2 in the top right image, the telescoping fingers of II-3 in the lower left image, and the midface hypoplasia, relative prognathism, and low-set ears in II-4 in the lower right image.
Table 1.
Summary of the Clinical Findings among the Three Affected Individuals
| Clinical Findings | II-4 | II-2 | II-3 |
|---|---|---|---|
| Height | 26 in/66 cm | 28 in/68 cm | 27 in/71 cm |
| U/L segment ratios | 1.1 | 1.2 | 1.2 |
| Relative macrocephaly | 56 cm | 58 cm | 58 cm |
| Slightly low-set posteriorly rotated ears | + | + | + |
| Severe midface hypoplasia/absent nasal bridge | + | + | + |
| Relative prognathism | + | + | + |
| Short neck | + | + | + |
| Hoarse voice | − | + | + |
| Bronchospasm | − | + | − |
| Barrel-shaped chest | + | + | + |
| Lumbar lordosis | + | + | + |
| Rhizomelia | + | + | + |
| Mesomelia | + | + | + |
| Brachydactyly (hand length) | 7.5 in/18.5 cm | 8 in/20 cm | 8 in/20 cm |
| Broad thumbs | + | + | + |
| Horizontal nail beds | + | + | + |
| Joint laxity, especially hands | + | + | + |
Figure 2.
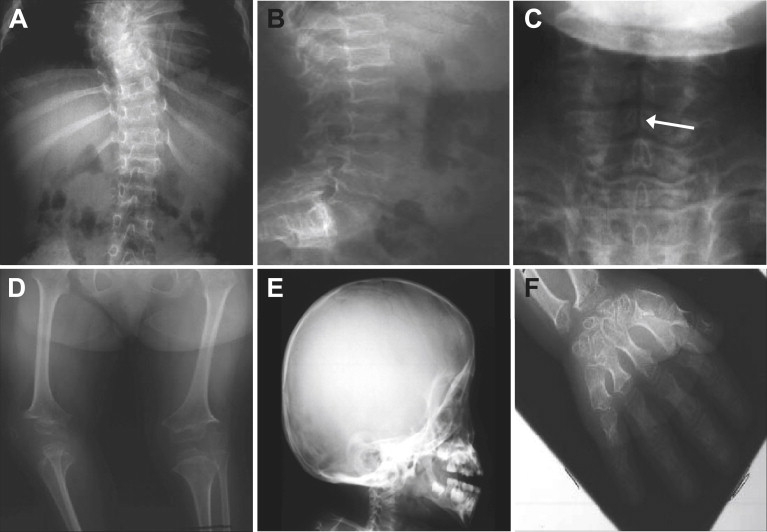
Radiographic Phenotype
(A), (B), and (D)–(F) are from II-4 at 9.5 yo, and (C) is from II-2 at 1 yo. Radiographs show (A) mild scoliosis, (B) platyspondyly, (C) clefts of the cervical-vertebral bodies (indicated by arrow), (D) shortening of the limbs with small irregular epiphyses and widened metaphyses, (E) an enlarged skull with midface hypoplasia, and (F) brachydactyly with extra carpal ossification centers.
At the time of the most recent examination, their ages ranged from 16–26 years of age. They all exhibited extreme short stature, 26–28 in (66–71 cm) total height, with upper/lower (U/L) segment ratios ranging from 1.1–1.2. Their clinical findings were largely similar to those observed in childhood, except that one of the affected individuals complained of bronchospasm that worsened with respiratory infections, and he had a hoarse voice. The parents both originated in a small village in the state of Zacatecas in Mexico, but consanguinity was denied. The mother and father were 150 cm and 152 cm tall, respectively. A half sister (II-5) was 150 cm tall. The parents and half sibling were proportionate and had neither dysmorphic facial findings nor brachydactyly (normal metacarpophalangeal profiles; data not shown). They denied any complaints of joint pain or limitations and had no difficulty with physical activities. An unaffected full sibling (Figure 1, II-1) was 178 cm tall.
To assess the genetic relationship between the parents, we compared whole-genome single-nucleotide polymorphism (SNP) data. This analysis revealed several 10–20 cM blocks of shared alleles (data not shown), implying an ancestral relationship and supporting the hypothesis that their affected children might be homozygous for the same mutation, identical by descent. We tested this hypothesis by comparing whole-genome SNP genotypes between two of the three affected individuals to look for regions of shared homozygosity. A single interval, located between 72.0 and 89.4 Mb on chromosome 15, met this requirement (Figure 3), suggesting that the phenotype was indeed recessively inherited, that the mutation was within a gene in this interval, and that the affected individuals should be homozygous for the mutation. The unaffected sibling, II-1, was not homozygous for this interval.
Figure 3.
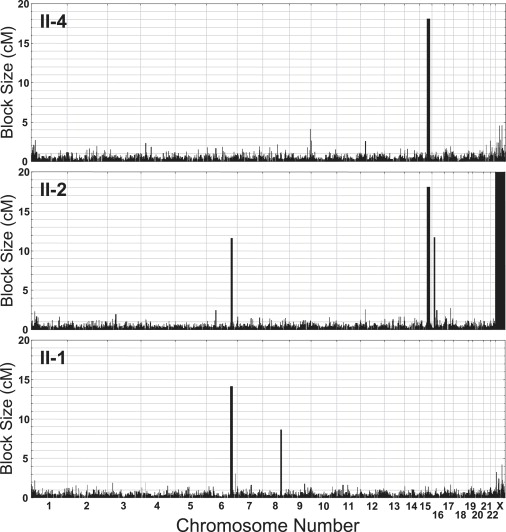
Identity by Descent Mapping of the SEMD-Aggrecan-Type Locus
Homozygosity maps for two of the affected siblings (II-4 and II-2) and one unaffected sibling (II-1) from the pedigree. Each plot shows the size of homozygous blocks for each chromosome across the genome, in centiMorgans, as inferred from SNP genotype data. The large blocks represent recent homozygosity by descent, suggesting ancestrally consanguineous parents. The only block that fits a consanguineous-recessive disease model is the chromosome 15 locus, which has the identical block of homozygosity in the affected siblings that is not shared identically with the unaffected sibling.
The interval on chromosome 15 contained 193 annotated and 103 unannotated genes. To prioritize the genes for mutation analysis, we used whole-genome microarray data to determine the cartilage selectivity of gene expression2 under the hypothesis that the cartilage-specific nature of the phenotype would be consistent with a mutation in a gene selectively expressed in the target tissue. There were two highly cartilage-selective genes in the interval (Figure 4), chondroitin sulfate proteoglycan 4 (CSPG4 [MIM 601172]) and aggrecan (ACAN [MIM 155760]). These were the only genes in the interval that were highly expressed in cartilage (at least 5-fold higher in cartilage than in the noncartilage tissues) and that were not expressed in the noncartilage tissues (defined by an analog of coefficient of variation of below 1 in the noncartilage tissues).
Figure 4.
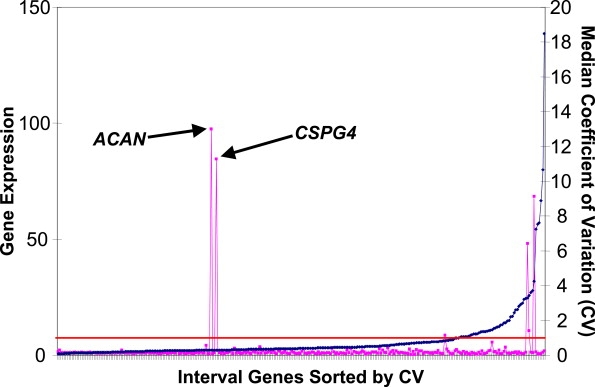
Prioritization of Genes within the Chromosome 15 Region with Respect to their Cartilage-Selective Gene Expression
For each gene in the interval, cartilage gene expression was defined by the average expression in cartilage relative to the average expression in a panel of noncartilage tissues, indicated by pink squares with the scale on the left. The median derived coefficient of variation (CV; shown in blue), an indirect measure of expression in noncartilage tissues, was plotted for each gene with the scale indicated on the right. The genes in the interval were sorted by lowest CV (left) to highest CV (right). The threshold CV of 1 is marked by a horizontal red line.
Aggrecan is a member of the lectican family of proteoglycans and is among the most abundant proteoglycans in the cartilage extracellular matrix. Mutations in the ACAN gene have been identified in skeletal-dysplasia phenotypes in humans3 and other species,4–6 so the coding region of ACAN was tested for mutations (for primer sequences, please see Table S1, available online). Sequence analysis of complementary DNA (cDNA) from II-2, derived from RNA isolated from cultured skin fibroblasts, revealed homozygosity for a point mutation (NM_013227.2: c.6799G → A) that predicts a p.D2267N substitution in the C-type lectin domain (CLD) within the G3 globular domain of the protein (Figure 5). The sequence change was confirmed by sequence analysis of amplified genomic DNA, and the other two affected individuals were also homozygous for this change. The parents and half sibling (II-5) were heterozygous for the sequence change, whereas II-1 did not inherit the putative mutant allele. The sequence change was not found among 240 ethnically matched control chromosomes, indicating that it is not a common polymorphism in the population and consistent with the D2267N change causing the disorder.
Figure 5.
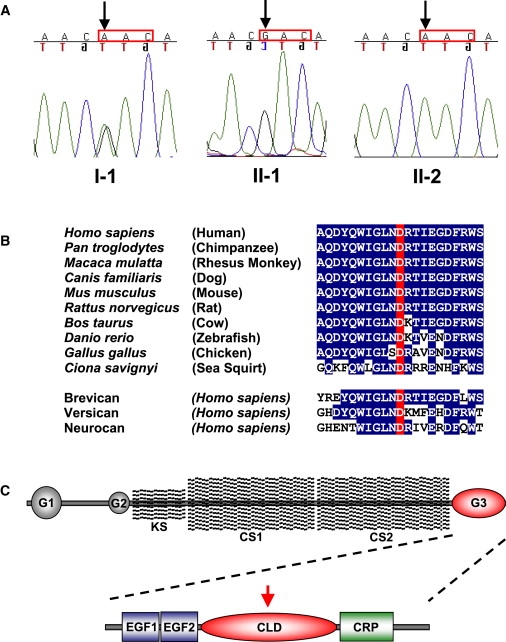
ACAN Mutation Analysis and Evolutionary Conservation of Aspartic-Acid Residue 2267
(A) Sequence traces derived from three family members, indicating the single-nucleotide change with the respective codon highlighted (red box).
(B) Partial protein sequence of aggrecan showing the region containing Asp2267 among aggrecan orthologs as well as paralogous members of the lectican family of proteins.
(C) Diagram of the aggrecan molecule showing N-terminal G1 and G2 globular domains, a highly repetitive central region modified with keratan sulfate (KS) and chondroitin sulfate (CS) side chains, and the C-terminal G3 globular domain where the mutation is located. In the expanded diagram of the G3 domain shown below, the red arrow indicates the location of the mutation in the C-type lectin (CLD) motif. Alternatively spliced epidermal growth factor-like (EGF) and complement regulatory protein-like (CRP) motifs are also shown.
The D2267 aspartic-acid residue exhibits a very high degree of conservation throughout evolution. It has been identified in all known aggrecan orthologs, including those in mouse, chick, zebrafish, and sea squirt (Figure 5). Thus, this residue has been completely conserved since at least 550 million years ago, a time when sea squirts and vertebrates shared a common ancestor,7 suggesting functional significance. Brevican, versican, and neurocan, three other members of the lectican family of proteoglycans, also contain this conserved aspartic-acid residue in their G3 globular domains, supporting a functionally important role in paralogous proteins. The evolutionary conservation also suggests that the sequence change causes the disorder.
The crystal structure of the CLD of rat aggrecan complexed with the fibronectin type III repeats of tenascin-R8 (Research Collaboratory for Structural Bioinformatics Protein Data Bank structure 1TDQ) also suggests a functional role for D2267. The structure shows that the aggrecan-tenascin interaction is highly calcium-ion dependent, requiring coordination of three calcium ions in the CLD. Coordination of calcium-1 requires five amino acids including D2267, the residue found to be mutated in this family (known as Asp64 in Lundell et al.8). According to the structure, the calcium ions function as an integral part of the CLD coordination loops, ordering these regions such that they mediate specific interactions with fibronectin type III repeats 4 and 5 of tenascin-R. These findings suggest that the mutation might disrupt interactions between aggrecan and tenascin or, perhaps, other matrix molecules.
To determine the effect of the D2267N mutation on the secretion and interactions of the CLD of aggrecan, we expressed a cDNA fragment containing the entire CLD in mammalian cells and purified the recombinant protein. In brief, we performed PCR-based site-directed mutagenesis on a wild-type human aggrecan G3 domain cDNA clone to introduce the D2267N mutation. We subcloned the resulting PCR products into a pCEP4 vector modified to contain a signal sequence and a carboxyl-terminal His tag with a proximal enterokinase cleavage site.9 The purified DNA was then transfected into 293-EBNA cells (human embryonic kidney cells; Invitrogen) with LipofectAMINE 2000 reagent (Invitrogen). Transfected cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, and, after 24 hr in culture, the transfected cells were selected by the addition of Hygromycin B (50 μg/ml). After cell confluency, the culture medium was replaced with serum-free medium, and the cells were incubated in this medium for a further 48 hr before the conditioned medium was collected. SDS-PAGE and western blotting were used for analyzing the cell-culture media and cell lysates of the wild-type and mutant cell lines (Figure 6A). Two recombinant forms of both WT and mutant (glycosylated) aggrecan G3 domain were used in these studies, one containing the epidermal growth factor (EGF) repeats (long form) that have been shown to enhance tenascin-C binding,10 and one without the EGF modules (short form). Analysis of the media and cell-lysate samples confirmed that the WT proteins were efficiently secreted, as has previously been shown for recombinant ACAN protein fragments containing folded globular motifs.10 The mutated proteins were also efficiently secreted into the culture media, but they migrated more slowly on SDS-PAGE, indicating a greater mass than the equivalent WT proteins. The diffuse migration of the mutated proteins suggested that they might be glycosylated, a hypothesis supported by the observation that the mutation introduced a potential N-glycosylation site (Asn2267-Arg-Thr) into the protein sequence.
Figure 6.
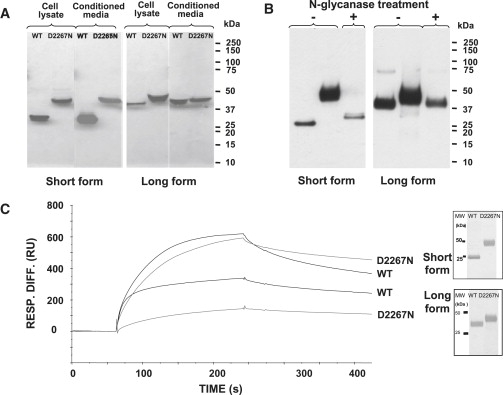
Biochemical Analyses of the Wild-Type and Mutant Aggrecan G3 Proteins
(A) The effect of the D2267N mutation on the secretion of the aggrecan G3 domain. SDS-PAGE and western-blot analysis of conditioned media and cell lysates from 293-EBNA cells transiently transfected with the aggrecan G3 domain constructs (WT and mutant). Recombinant proteins were resolved on 4%–12% Bis-Tris gel under reducing conditions, transferred onto nitrocellulose membrane, and detected by western blotting with an antibody against an in-frame His tag. Recombinant WT G3 domains migrated at approximately 26 kDa for the short form and 40 kDa for the long form, whereas the mutant proteins migrated more slowly, with a mass of ∼45 kDa for both forms. Protein samples from cell lysates are shown in the left panel and from the culture media in the right panel for each form; the size of molecular weight (MW) markers is shown on the right.
(B) N-glycanase digest of the mutated aggrecan G3 proteins. Western-blot analysis is of purified D2267N G3 protein with (digested) or without (undigested) N-glycanase treatment. The undigested D2267N protein appears as a diffuse ∼45 kDa band corresponding to the glycosylated form, which is in contrast to the digested form that becomes a sharp ∼26 kDa band for the short form and ∼40 kDa for the long form (similar to WT protein), indicating that the secreted mutated proteins possess N-glycan chains.
(C) Binding analysis of recombinant aggrecan G3 domains to tenascin-C. The purity of the recombinant proteins was verified by silver staining of SDS-PAGE gel under reducing conditions (inset). The purified proteins were then tested for their ability to bind tenascin-C. Shown are representative sensograms (surface plasmon resonance, SPR) for WT G3 or D2267N G3 proteins at a concentration of 10 μg/ml. The proteins were injected at a flow rate of 30 μl/min for 3 min.
To determine whether the mutated proteins were glycosylated, we first purified the secreted WT and mutant proteins from the conditioned medium by affinity chromatography with the His tag. In brief, the medium was incubated with the TALON affinity column (BD Biosciences) for 1 hr at 4°C. Bound proteins were eluted from the beads with 150 mM imidazole in 20 mM Tris (pH 7.4), 300 mM NaCl. Fractions (1 ml) were collected and analyzed by SDS-PAGE with silver staining (Figure 6C, inset). The fractions containing the aggrecan G3 domain were desalted with PD-10 columns (GE Healthcare) and further purified by anion exchange chromatography on a MiniQ column with the Ettan purifier HPLC system (GE Healthcare). We treated 0.5 μg of recombinant protein with 0.01 U PGNase (QA Bio) for 48 hr at 37°C, and we analyzed the treated protein product by SDS-PAGE and western blotting. After the digestion, the diffuse D2267N proteins appeared sharper and migrated at a similar size to that of the WT proteins (Figure 6B). This indicated that the secreted mutated proteins contained N-glycan chains and suggested that aberrant glycosylation could affect the function of the aggrecan G3 domain.
To determine the effect of the mutation on aggrecan interactions, we used surface plasmon resonance (BIAcore 3000) to study the binding of the aggrecan G3 domain with its known binding partner tenascin-C10 (Figure 6C). In brief, 10 μg/ml of recombinant tenascin-C (Chemicon) was immobilized onto a CM5 sensor chip. G3 aggrecan scans were performed in HEPES buffer containing 2 mM Ca2+. All of the proteins bound to tenascin-C in the presence of 2 mM Ca2+. However, the mutant long form bound at a significantly lower level than its WT counterpart (p < 0.001, n = 3). Tightly bound proteins were then dissociated by the injection of 10 mM EDTA, confirming that the binding was cation dependent. For both the long form (p < 0.05, n = 3) and the short form (p < 0.001, n = 3), the mutant aggrecan dissociated at a slower rate.
Overall, these data indicate that the D2267N mutation does not affect secretion of the G3 domain of aggrecan in vitro. Furthermore, the introduction of an asparagine at residue 2267 created a potential N-glycosylation recognition motif that was glycosylated when the G3 domain was expressed in mammalian cells. However, the pathological relevance of this posttranslation modification in vivo remains undetermined.
The data presented identify the molecular basis of a recessive form of SEMD, here termed SEMD aggrecan type, characterized by extreme short stature, which results from homozygosity for a missense mutation affecting the G3 domain of aggrecan. The D2267N substitution affected a highly conserved residue that contributes to the structure of the C-type lectin domain within the G3 domain of the protein, in part by coordinating binding of one of three calcium ions important for its structure. The substitution also created a potential N-glycosylation site that is used in vitro. These changes appear to influence the binding and dissociation kinetics of interactions between the G3 domain and tenascin-C, at least in vitro. Thus, a model for the mechanism of disease includes the effect of the mutation on the structure of the protein as well as the effect of introducing a novel glycosylation site, both of which could alter the interactions between aggrecan and its binding partners in the cartilage extracellular matrix. The relative contributions of these mechanisms cannot be distinguished by the data presented herein.
In addition to the effect on stature, the radiographs show clefts of the cervical-vertebral bodies and extra ossification centers in the carpal bones of the hands, features also observed in diastrophic dysplasia11 (DTD [MIM 222600]). DTD is a recessive disorder that results from reduced activity of the universal sulfate transporter, encoded by the SCL26A2 (MIM 606718) gene.12 The finding that a mutation in the aggrecan gene produces similar radiographic features suggests that aggrecan is an important target in the developing skeleton affected by the SLC26A2 mutations that produce DTD.
The carrier parents and carrier half sibling had adult heights of approximately 150 cm, whereas a WT sibling of the proband was 178 cm tall. This observation raises the possibility that there is a carrier phenotype of mild, proportionate short stature. This inference is supported by the finding that haploinsufficiency for aggrecan can lead to the mild spondyloepiphyseal dysplasia (SED) Kimberley phenotype,3 as well as similar phenotypes in other species.4–6 However, because homozygosity for null mutations in other species produces lethal phenotypes, and because neither the SEMD-aggrecan-type cases nor carriers manifest an early-onset osteoarthritis phenotype as observed in the SED Kimberly type, the mechanisms of disease in the two disorders are likely to be distinct. Recently, whole-genome association studies of normal variation in human height have implicated the ACAN region on chromosome 15 as a regulator of height.13 Together with the data presented here, these findings suggest that variation in the quantity and/or structure of the aggrecan protein can produce a wide range of human height variation.
Acknowledgments
This work was supported in part by grant HD22657 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health. D.K. is supported by a grant from the Joseph Drown Foundation and a Winnick Family Clinical Scholar award from Cedars-Sinai Medical Center. We thank Arleen Hernandez for assistance with coordinating the clinical studies and Marc Firestein for assistance with the metacarpophalangeal profiles. M.D.B. is the recipient of a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science (Grant 071161/Z/03/Z). BIAcore anaylsis was undertaken in the Biomolecular Analysis Core Facility in the Faculty of Life Sciences at the University of Manchester. The aggrecan cDNA constructs were a kind gift from Tim Hardingham.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Entrez Nucleotide, http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Research Collaboratory for Structural Bioinformatics Protein Data Bank, http://www.rcsb.org
References
- 1.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 2.Funari V.A., Day A., Krakow D., Cohn Z.A., Chen Z., Nelson S.F., Cohn D.H. Cartilage-selective genes identified in genome-scale analysis of non-cartilage and cartilage gene expression. BMC Genomics. 2007;8:165. doi: 10.1186/1471-2164-8-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gleghorn L., Ramesar R., Beighton P., Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 2005;77:484–490. doi: 10.1086/444401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe H., Kimata K., Line S., Strong D., Gao L.Y., Kozak C.A., Yamada Y. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat. Genet. 1994;7:154–157. doi: 10.1038/ng0694-154. [DOI] [PubMed] [Google Scholar]
- 5.Landauer W. Nanomelia, a lethal nutation of the fowl. J. Hered. 1965;56:131–138. doi: 10.1093/oxfordjournals.jhered.a107392. [DOI] [PubMed] [Google Scholar]
- 6.Cavanagh J.A., Tammen I., Windsor P.A., Bateman J.F., Savarirayan R., Nicholas F.W., Raadsma H.W. Bulldog dwarfism in Dexter cattle is caused by mutations in ACAN. Mamm. Genome. 2007;18:808–814. doi: 10.1007/s00335-007-9066-9. [DOI] [PubMed] [Google Scholar]
- 7.Dehal P., Satou Y., Campbell R.K., Chapman J., Degnan B., De Tomaso A., Davidson B., Di Gregorio A., Gelpke M., Goodstein D.M. The draft genome of Ciona intestinalis: Insights into chordate and vertebrate origins. Science. 2002;298:2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- 8.Lundell A., Olin A.I., Mörgelin M., al-Karadaghi S., Aspberg A., Logan D.T. Structural basis for interactions between tenascins and lectican C-type lectin domains: Evidence for a crosslinking role for tenascins. Structure. 2004;12:1495–1506. doi: 10.1016/j.str.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 9.Bengtsson E., Aspberg A., Heinegard D., Sommarin Y., Spillmann D. The amino-terminal part of PRELP binds to heparin and heparan sulfate. J. Biol. Chem. 2000;275:40695–40702. doi: 10.1074/jbc.M007917200. [DOI] [PubMed] [Google Scholar]
- 10.Day J.M., Olin A.I., Murdoch A.D., Canfield A., Sasaki T., Timpl R., Hardingham T.E., Aspberg A. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J. Biol. Chem. 2004;279:12511–12518. doi: 10.1074/jbc.M400242200. [DOI] [PubMed] [Google Scholar]
- 11.De la Chapelle A., Maroteaux P., Havu N., Granroth G. Arch. Fr. Pediatr. 1972;29:759–770. [PubMed] [Google Scholar]
- 12.Hästbacka J., de la Chapelle A., Mahtani M.M., Clines G., Reeve-Daly M.P., Daly M., Hamilton B.A., Kusumi K., Trivedi B., Weaver A. The diastrophic dysplasia gene encodes a novel sulfate transporter: Positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78:1073–1087. doi: 10.1016/0092-8674(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 13.Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.