

Abstract
In patients with autosomal-recessive retinitis pigmentosa (arRP), homozygosity mapping was performed for detection of regions harboring genes that might be causative for RP. In one affected sib pair, a shared homozygous region of 5.0 Mb was identified on chromosome 6, within the RP25 locus. One of the genes residing in this interval was the retina-expressed gene EGFL11. Several genes resembling EGFL11 were predicted just centromeric of EGFL11. Extensive long-range RT-PCR, combined with 5′- and 3′- RACE analysis, resulted in the identification of a 10-kb transcript, starting with the annotated exons of EGFL11 and spanning 44 exons and 2 Mb of genomic DNA. The transcript is predicted to encode a 3165-aa extracellular protein containing 28 EGF-like and five laminin A G-like domains. Interestingly, the second part of the protein was found to be the human ortholog of Drosophila eyes shut (eys), also known as spacemaker, a protein essential for photoreceptor morphology. Mutation analysis in the sib pair homozygous at RP25 revealed a nonsense mutation (p.Tyr3156X) segregating with RP. The same mutation was identified homozygously in three arRP siblings of an unrelated family. A frame-shift mutation (pPro2238ProfsX16) was found in an isolated RP patient. In conclusion, we identified a gene, coined eyes shut homolog (EYS), consisting of EGFL11 and the human ortholog of Drosophila eys, which is mutated in patients with arRP. With a size of 2 Mb, it is one of the largest human genes, and it is by far the largest retinal dystrophy gene. The discovery of EYS might shed light on a critical component of photoreceptor morphogenesis.
Introduction
Retinitis pigmentosa (RP [MIM 268000]) is made up of a clinically and genetically heterogeneous group of diseases characterized by night blindness and constriction of the visual field, leading to severe visual impairment due to progressive degeneration of photoreceptors and, often, blindness. To date, 21 genes have been described as causing autosomal-recessive RP (arRP) and five loci have been identified for which the causative gene is still unidentified (RetNet web resource). Genes that cause arRP encode proteins that exert their function in different pathways within the retina, such as the phototransduction cascade (CNGA1, CNGB1, PDE6A, PDE6B, RGR, RHO, SAG [MIM ∗123825, ∗600724, ∗180071, +180072, ∗600342, +180380, and ∗181031, respectively]) or vitamin A metabolism (ABCA4, LRAT, RLBP1, RPE65 [MIM ∗601691, +604863, ∗180090, and +180069, respectively]). Others encode proteins that have a structural or signaling function (CRB1, RP1, TULP1, USH2A [MIM +604210, ∗603937, ∗602280, and +608400, respectively]), play a role in transcriptional regulation (NR2E3, NRL [MIM ∗604485 and +162080, respectively]), or play a role in phagocytosis of the RPE (MERTK [MIM +604705]), or their exact role still awaits discovery (CERKL, PRCD, PROM1 [MIM ∗608381, ∗610598, and ∗604365, respectively]).1 USH2A is the most frequently mutated gene, causing ∼8% of arRP, whereas most other genes account for only 1% of arRP cases.1 Altogether, these 21 known genes are estimated to account for 30% of arRP cases,2 indicating that more genes await discovery. Mutations at the RP25 locus [MIM %602772] might also represent a significant cause of arRP, given that 10%–25% of Spanish arRP families were previously shown to map to this locus.3,4 Previous studies have excluded mutations in 60 genes at the RP25 locus.5–15 Recently, the RP25 locus was significantly reduced by linkage studies in additional Spanish families and the identification of a 100–200 kb deletion in one of the linked families, but the causative gene has not yet been identified.3,8
Homozygosity mapping has proven to be an effective approach in the search for genes16–18 and in the discovery of mutations in known arRP genes.19 The purpose of this study was to identify retinal dystrophy genes, utilizing homozygosity mapping with SNP microarray technology. Genome-wide homozygosity mapping in a large series of outbred arRP patients revealed a region on chromosome 6q12-q11.1 that was homozygous in two affected siblings and was fully situated within the previously defined RP25 locus.4 We characterized an exceptionally large gene variant in this region, and we found it to be specifically expressed in the retina. Sequence analysis revealed a homozygous nonsense mutation in these siblings, segregating with RP in the family. Subsequently, the same mutation was detected in an unrelated family with arRP, whereas another mutation was identified in an isolated RP patient.
Subjects and Methods
Subjects and Clinical Evaluation
Five patients from three families (II-1 and II-3 from family A, II-3 and II-6 from family B, and II-1 from family C) received the RP diagnosis several years ago through ophthalmologic examination. The examination included evaluation of best-corrected visual acuity and slit-lamp biomicroscopy, followed by indirect ophthalmoscopy and fundus photography after pupillary dilatation. The size and the extent of the visual-field defects were assessed with Goldmann kinetic perimetry (targets V-4e, II-4e, and I-4e to I-1e; for all patients) and Humphrey static perimetry (30-2; only for patient II-3 in family A). Finally, an electroretinogram (ERG) was recorded in all five patients, in accordance with the protocol of the International Society for Clinical Electrophysiology of Vision (ISCEV)20. After the nature of this phenotype-genotype study was explained, an informed consent adhering to the tenets of the Declaration of Helsinki was obtained from all patients and from the unaffected siblings of family A and B. Blood samples from these individuals were then collected for future molecular-genetics testing. The initial results of the molecular-genetics analysis warranted further ophthalmologic investigation in the supposedly unaffected individual II-4 from family B. This investigation included all of the elements of the earlier ophthalmologic examination of the affected individuals, with the exception of the visual-field assessment.
Furthermore, 143 probands with RP and indications of a recessive mode of inheritance participated in this study. Control DNA samples from 276 unrelated Dutch individuals were used.
Homozygosity Mapping and Mutation Analysis
Genomic DNA was isolated from lymphocytes by standard salting-out procedures.21 DNA samples of 145 RP patients, mainly of Dutch origin, were genotyped on either the GeneChip Mapping 250K NspI array, containing 262,000 SNPs, or the GeneChip Genome-Wide Human SNP Array 5.0, which contains 500,568 polymorphic SNPs in addition to 420,000 nonpolymorphic probes for the detection of germline copy-number variations (Affymetrix). Array experiments were performed according to protocols provided by the manufacturer. The 250K SNP genotypes were analyzed with the software package CNAG.22 Data from the 5.0 array were genotyped with Genotype Console software (Affymetrix), whereas regions of homozygosity were calculated with Partek Genomics Solution (Partek).
All 41 coding exons and three noncoding exons of EYS were PCR amplified and analyzed in sense and antisense directions with a dye-termination chemistry (BigDye Terminator, version 3 on a 3730 or 2100 DNA analyzer; Applied Biosystems). Primers for PCR and sequencing of the 44 exons are given in Table S1, available online; PCR conditions are available upon request.
A subset of 131 RP patients, mainly from The Netherlands, and a control panel of 276 ethnically matched unrelated and unaffected individuals were screened for the p.Tyr3156X mutation with the amplification-refractory mutation system (ARMS; primers listed in Table S2).
Characterization of the Genetic Composition of EYS
For characterization of the expression of predicted genes encoding EGF-like and/or laminin A G-like domains (NT_007299.33, NT_007299.34, NT_007299.35, NT_007299.37 and ENST00000237253) in retina, several primers were designed, corresponding to the exons of these gene predictions. Long-range PCRs were performed on human retina Marathon-Ready cDNA (Clontech) with the Advantage cDNA PCR Kit (Clontech), in accordance with the manufacturer's protocol. Nested PCR reactions with the use of a number of these primer combinations resulted in the amplification of PCR products representing parts of a transcript expressed in retina. PCR products were purified on Nucleospin Plasmid Quick Pure columns (Macherey Nagel) and either directly sequenced or cloned in the pCR4-TOPO vector with the use of the TOPO TA Cloning Kit (Invitrogen) for sequencing with T7 and T3 sequencing primers as described above. Primer sequences are listed in Table S3.
To characterize the 5′- and 3′- untranslated regions (UTR) of the detected transcripts, rapid amplification of cDNA ends (5′- and 3′- RACE, Clontech) was performed, in accordance with the manufacturer's protocol, using the Advantage cDNA PCR kit and human retina Marathon-Ready cDNA (Clontech) as a template.
RT-PCR Analysis for Determining Tissue Distribution in EYS
Total RNA from human placenta, adult brain, testis, kidney, and retina and from fetal heart, skeletal muscle, liver, and lung was obtained from Clontech. For cDNA synthesis, 2 μg of total RNA was incubated with 5 ng/μl of random hexamers (pd(N)6, Pharmacia) and 0.3 mM dNTPs (Invitrogen Life Sciences). Subsequently, cDNA was synthesized with the M-MLV Reverse Transcriptase kit (Invitrogen Life Sciences), with a final concentration of 10 mM DTT, 11 U Reverse Transcriptase, and 0.33 U RNAguard (American Biosciences) per reaction. For detecting the distribution patterns of human EYS in various human tissues, RT-PCR was carried out with Advantage Polymerase (Clontech), with the use of various primer pairs equally distributed along the transcript. As a control, β-actin (ACTB) was amplified. To verify that the amplified products indeed corresponded to the EYS transcript, PCR products were purified and sequenced as described above. Primer sequences are listed in Table S4.
Bioinformatic Analysis
Genes and gene-prediction tracks were derived from the UCSC Genome Working Draft, March 2006 assembly (hg18). For identification of homologous proteins of human and Drosophila eys, protein blast and tblastn were run under default settings (BLAST web resource). Conserved functional domains within proteins were searched with either the web-based tool SMART23,24 or Pfam.25 Prediction of amino acid residues that might be subject to O-linked glycosylation was carried out with the NetOGlyc 3.1 Server.26
Results
Homozygosity Mapping
In our search for retinal dystrophy genes, homozygosity mapping was conducted in a large series of patients with RP. Genome-wide SNP genotyping revealed two shared homozygous regions ≥ 5 Mb in two affected siblings diagnosed with RP. The largest homozygous region (19.3 Mb) was located on chromosome 2 between SNP_A-1816491 and SNP_A-2053763, whereas the second region, of 5.0 Mb, was located at chromosome 6q12-q11.1 between SNP_A-2144407 and SNP_A-1833968. The region on chromosome 6 overlapped with a well-known and published locus for arRP, namely RP25,4 for which the causative gene has not yet been identified (Figure 1A). Therefore, a search for candidate genes residing within this homozygous interval was conducted. Within the homozygous region shared with the RP25 locus, five genes are known to reside (Figure 1A), of which EGFL11 was found to be expressed in the eye, according to the Unigene database. RT-PCR analysis confirmed abundant expression of this gene in human retina (data not shown).
Figure 1.
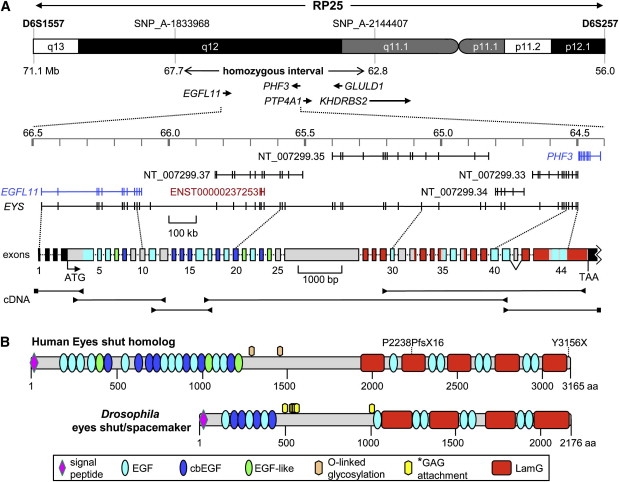
Genomic Structure, cDNA Fragments, and Protein Domains of EYS
(A) Upper panel: the RP25 chromosomal region at 6p12.1-q133, the 5.0 Mb homozygous region identified in family A, and the five known genes within the homozygous region. Exons 1 and 2 of KHDRBS2 reside in the critical region. In the middle, the exon predictions are depicted on the basis of RefSeq (in blue), Genescan (in black), and Ensembl (in red), with the use of the March 2006 UCSC genome build (hg18). Below the genomic-exon annotation is the exon structure of human EYS (exons drawn to scale; intron sizes can be found in the top panel). The complete nucleotide sequence of human EYS cDNA is presented in Figure S2. For details of the exon-intron structure, see Table S5. The 5′- and 3′- UTRs are indicated in black boxes; the colors of the protein-coding exons correspond with those of the protein domains in (B). Lower panel: reverse-transcription PCR fragments of human EYS with retina RNA and EYS-specific primers (arrowheads) or 5′- and 3′- RACE adaptor primers (squares). The 5′- UTR, the open reading frame, and the 3′-UTR altogether measure 10,475 nts (see Table S5). Exon 42 (63 bp) is alternatively spliced in retina RNA (see Figure 2). For details of RT-PCR studies, see Figure S1.
(B) Protein-domain structure of EYS and its Drosophila ortholog (GenBank ID ABH07112.1). Note the conspicuous conservation of the order of EGF-like and laminin A G-like domains between human and Drosophila. The p.Pro2238ProfsX16 frame-shift mutation truncates several EGF-like and laminin A G-like domains, whereas the carboxy-terminal p.Tyr3156X mutation truncates the last ten amino acids of human EYS. Abbreviations are as follows: EGF, epidermal growth factor domain; cbEGF, calcium-binding EGF-like domain; EGF-like, EGF-like domain; LamG, laminin A G-like domain. The asterisk denotes glycosaminoglycan (GAG) attachment sites predicted by Husain and coworkers.27 Two putative O-glycosylation sites are predicted in the human protein (Thr1268 and Thr1424). Detailed characteristics of the human EYS protein domains are presented in Figure S3.
Identification of EYS Exons
The EGFL11 gene is made up of 12 exons and encodes a protein with several EGF-like domains. Sequence analysis of the annotated EGFL11 gene in one of the two affected siblings did not reveal any causative sequence variants. Centromeric to EGFL11, several other genes encoding EGF-like domains were predicted, including NT_007299.37, ENST00000237253, NT_007299.35, NT_007299.34, and NT_007299.33. To test the hypothesis that exons of these gene-prediction tracks were part of a longer isoform of the EGFL11 gene, extensive long-range RT-PCR experiments were performed, combined with 5′- and 3′- RACE experiments using human retina cDNA as a template. Interestingly, these analyses showed that in addition to a transcript corresponding to the annotated EGFL11 gene, a second transcript was present, containing several exons of these gene predictions. This larger transcript started at the previously annotated EGFL11 gene and extended up to the final exon of NT_007299.33 (Figure 1A). In total, this extended variant of EGFL11 spans almost 2 Mb of genomic DNA and contains 44 exons, of which several had not previously been predicted by gene-prediction programs. The transcript contains 10,475 nucleotides, including the 3′ untranslated region and poly-A tail. A detailed overview of the identification and characterization of the transcript is presented in Figure S1.
The protein encoded by this transcript is made up of 3165 amino acids and is predicted to contain a signal peptide for secretion into the extracellular environment. In addition, the protein harbors 28 EGF-like and 5 laminin A G-like domains (Figure 1B).
Subsequently, BLAST analyses were performed for the identification of potential orthologs of this human protein in lower species. These analyses led us to discover that the second part of this protein is homologous to Drosophila eyes shut (eys), also known as spacemaker, a protein essential for photoreceptor development and morphology in the insect eye.27,28 The domain organization of eys is comparable to that of the human eys homolog protein, with 14 EGF-like and four laminin A G-like domains positioned in a similar order (Figure 1B). Initially, the Drosophila eys protein was described as a proteoglycan related to agrin and perlecan.27 However, with the sequence of the Drosophila protein used as input in a BLAST search for human orthologs, eys homolog protein, rather than agrin and perlecan, was found to be the closest relative. In addition, the signaling molecules Notch-1 and -2 and the Crumbs-1 and -2 homolog proteins were identified as relatives of Drosophila eys (data not shown). These analyses show that the gene identified in this study is the true ortholog of Drosophila eys. Therefore, we propose to name the human gene eyes shut homolog (EYS).
Tissue Distribution of EYS mRNA
For study of the tissue distribution of human EYS, RT-PCR analysis was performed on cDNA from various tissues, including retina. In total, five primer pairs were used, distributed along the transcript. All five primer pairs that were used showed either specific or enriched expression of EYS in retina, although for one primer pair (exons 41–44), weak PCR products were also observed in the other tissues (Figure 2). This primer pair also amplified two fragments, and sequence analysis showed that the fragments represent alternatively spliced mRNA products of the EYS gene, either lacking or containing exon 42 (63 bp). Together, these results show that this gene is abundantly expressed in retina and support the hypothesis that the encoded protein plays an important role in vision.
Figure 2.
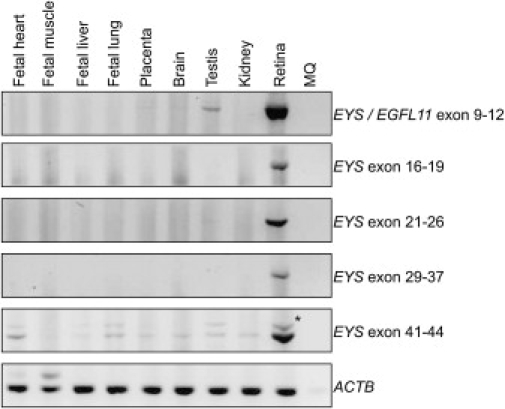
Tissue Distribution of EYS
RT-PCR analysis was performed on total RNA from the various tissues. The expression of EYS was determined with the use of several primer pairs distributed along the transcript (see Table S4). The weak PCR product detected with primers from exon 41 to exon 44 is indicated by an asterisk and represents a transcript resulting from alternative splicing. ACTB (lower panel) was used as a control.
Mutation Analysis
After this transcript was identified, the SNP data were reanalyzed in an attempt to identify RP patients who carry homozygous regions (threshold was set at > 200 consecutive homozygous SNP calls) encompassing this new gene. On the basis of these data, ten arRP patients, including one of the affected individuals of the family described above (Figure 3A, family A), were selected for further mutation analysis of this gene. Sequence analysis of all 44 exons and flanking intronic sequences of the human EYS gene revealed a homozygous mutation, c.9468T → A, in the last exon (Figure 3B), present in the proband of family A. At the protein level, this mutation results in premature termination of the encoded protein at position 3156 (p.Tyr3156X). The mutation was confirmed to be homozygously present in his affected sibling and either absent or heterozygously present in four unaffected siblings (Figure 3A, family A).
Figure 3.
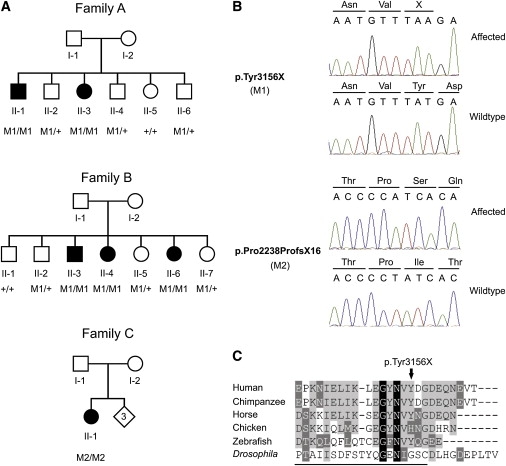
Mutation Analysis of EYS in RP Patients
(A) Pedigrees of three families with individuals affected with RP. Below the individuals, genotypes are presented for either the p.Tyr3156X change (M1, families A and B) or the p.Pro2238ProfsX16 change (M2, family C) detected to segregate with the RP. M1/M1 and M2/M2 represent homozygous mutants; M1/+ indicates heterozygous carriers, whereas +/+ indicates individuals carrying two wild-type alleles.
(B) Upper panel: partial sequence of the EYS gene showing the nonsense c.9486T → A change, in an affected individual (family A, II-1) and an unaffected sibling (family A, II-5). The mutation replaces a tyrosine residue by a termination codon (p.Tyr3156X). Preceding amino acids are indicated above the sequence trace. Lower panel: partial sequence of the EYS gene showing the c.6714 delT change, in an affected individual (family C, II-1) and a control individual. The mutation results in a frame shift and, finally, in premature termination of the protein (p.Pro2238ProfsX16). Amino acids are indicated above the sequence trace.
(C) Sequence comparison of the 25 most C-terminal amino acids of the human EYS protein and several vertebrate and invertebrate orthologs. Residues identical in all sequences are white on a black background, whereas similar amino acids are white on a gray background. Residues that are present in at least three of the six proteins are indicated in black on a light gray background. Residues constituting the most C-terminal laminin A G-like domain in the Drosophila protein are underlined. Accession numbers of the protein sequences used for sequence comparison are as follows: chimpanzee, XM_527426.2 (RefSeq); horse, XM_001918159.1 (RefSeq); chicken, XM_426198.2 (RefSeq); zebrafish, BX005106.5 (EMBL); Drosophila, ABH07112.1 (GenBank).
For detecting whether this mutation occurs more frequently in arRP patients, allele-specific PCR was conducted on another group of 131 unrelated probands affected with RP, resulting in the identification of a second proband carrying this mutation. The mutation was also homozygously present in her affected siblings, but not in her four unaffected family members (Figure 3A, family B). Microsatellite and SNP analysis in the region within and surrounding the EYS gene revealed that p.Tyr3156X was present at the same haplotype in both families, suggesting a founder effect. The p.Tyr3156X mutation was excluded from 552 alleles of ethnically matched control individuals. The nonsense mutation described here results in the absence of the ten C-terminal amino acids of the human EYS protein. Sequence comparison of the C-terminal amino acids of the human EYS protein and various vertebrate and invertebrate orthologs revealed that some of the amino acids that are absent in the human mutant EYS protein are well conserved, even up to zebrafish, and thus may be crucial for proper function of the EYS protein in vertebrates (Figure 3C).
Sequence analysis in the other nine patients whose DNA was homozygous at the region harboring the EYS gene revealed a second mutation, namely a homozygous 1-bp deletion in exon 33 (c.6714 delT; Figure 3B). At the protein level, this mutation is predicted to result in a frame shift and premature termination (p.Pro2238ProfsX16) of EYS. Given that premature truncation is predicted to occur within the second laminin A G-like domain, the mutant protein will lack six EGF-like and three laminin A G-like domains (Figure 1B).
Clinical Characteristics
Clinical examination of the affected individuals of families A, B, and C showed that all patients, with the exception of patient II-3 of family A, displayed characteristic RP abnormalities, including night blindness as the initial symptom, retinal bone-spicule pigmentations and attenuated retinal vessels (Figures 4A and 4B), constriction of the visual fields, and a nonrecordable ERG or ERG responses in a rod-cone pattern. A posterior subcapsular cataract could be observed in patient II-1 of family A (age 53) and in patient II-1 of family C (age 39). Patient II-3 of family A also demonstrated a photoreceptor dystrophy, but in this patient, the cones were more severly affected than were the rods (cone-rod pattern; Figure 4C). This is also reflected by the central scotomas on the kinetic visual field. Fundus abnormalities included central abnormalities at the level of the RPE and moderate attenuation of the retinal vessels.
Figure 4.
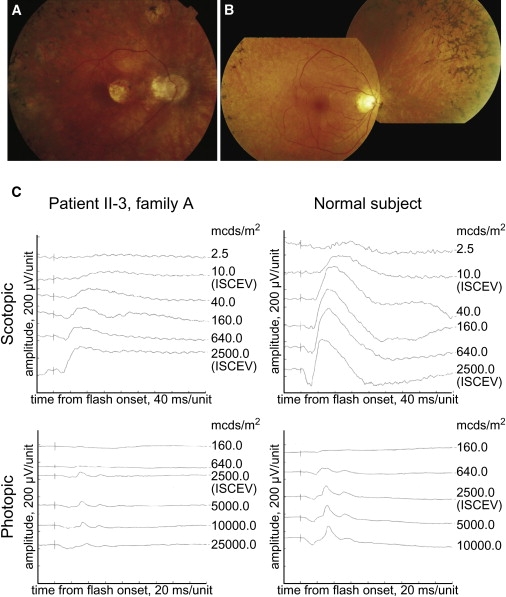
Clinical Characteristics of RP Patients with a Homozygous p.Tyr3156X Mutation in EYS
(A) Fundus photograph of the right eye of patient II-1 of family A, showing mild pallor of the optic disc, a peripapillary crescent, attenuated retinal vessels, and bone-spicule pigmentations. An area of sharply demarcated chorioretinal atrophy is located nasal to the fovea, with similar atrophic lesions along the vascular arcades, conflating to diffuse atrophy in the midperiphery.
(B) Fundus photograph of the posterior pole and nasal peripheral retina of the right eye of patient II-6 of family B, showing mild pallor of the optic disc, severely attenuated vessels, pronounced atrophic changes in the (mid) periphery that spare the posterior pole, and extensive bone spicules in the peripheral retina.
(C) Scotopic and photopic ERG of the right eye of patient II-3 of family A and a normal subject. Scotopic mixed response (ISCEV measurement; 2500 mcds/m2) had a b-wave amplitude of 274 μV (normal > 195 μV, mean 424 μV). The b-wave amplitude of the photopic response (ISCEV measurement; 2500 mcds/m2) was 58.8 μV (normal > 69 μV, mean 79 μV).
The clinical characteristics of the patients in families A, B, and C are summarized in Table 1.
Table 1.
Clinical Characteristics of Patients with Mutations in EYS
| ID | Age∗ (yrs) | Sex | Age at Onset (yrs) | Initial Symptom | Visual Acuity |
Ophthalmoscopy | Goldmann Kinetic Perimetry | ERG |
Phenotype | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | Rod | Cone | ||||||||
| Family A | |||||||||||
| II-1 | 61 | M | 12 | Night blindness and photophobia | 20/100 | 20/40 | Pallor of the optic disc, attenuated retinal vessels, bone spicules in midperiphery, and atrophic lesion in the posterior pole. | Constricted visual fields (50–80°). Marked decrease of the central visual field (I-4e not observed). | NR | NR | RP |
| II-3 | 57 | F | 28 | Night blindness and photophobia | 20/50 | 20/80 | Mild attenuation of the retinal vessels and RPE alterations in the central macula. | Large central scotomas without constriction of the peripheral visual field | ↓ | ↓↓ | CRD |
| Family B | |||||||||||
| II-3 | 53 | F | 45 | Night blindness | 20/20 | 20/20 | Normal aspect of the optic disc, attenuated retinal vessels, and bone spicules in the midperiphery. | Constricted visual fields (50–60°) | NR | NR | RP |
| II-4 | 52 | F | 46 | Night blindness | 20/25 | 20/20 | Normal aspect of the optic disc, attenuated retinal vessels, and bone spicules in the nasal fundus. | NP | ↓↓ | ↓ | RP |
| II-6 | 49 | F | 42 | Night blindness | 20/20 | 20/20 | Mild pallor of the optic disc, severely attenuated vessels, and bone spicules throughout the entire retina. | Severely constricted visual fields, with some residual visual field temporally | NR | NR | RP |
| Family C | |||||||||||
| II-2 | 48 | F | 13 | Night blindness | 20/50 | 20/40 | Pallor of the optic disc, attenuated vessels, RPE alterations in the central macula, and extensive bone-spicule pigmentation throughout the entire retina. | Severely constricted visual fields (<5°) | NR | NR | RP |
Abbrevations are as follows: M, male; F, female; RP, retinitis pigmentosa; CRD, cone-rod dystrophy; RPE, retinal pigment epithelium; NP, not performed; ERG, electroretinogram; ↓, decreased; ↓↓, severely decreased; NR, nonrecordable.
Current age.
Discussion
In the present study, we describe an extended transcript of the EGFL11 gene, containing 33 as-yet-uncharacterized exons downstream of the previously annotated gene. The resulting transcript is more than 10 kb in size and is abundantly expressed in human retina. The protein encoded by this gene is predicted to contain 28 EGF-like and five laminin A G-like domains. Interestingly, the second part of the protein was found to be homologous to the Drosophila eys protein. Therefore, we name the corresponding human gene eyes shut homolog (EYS). In two unrelated families, the same homozygous nonsense mutation (p.Tyr3156X) was identified as segregating with arRP, whereas in an isolated patient, a homozygous frame-shift mutation was identified (p.Pro2238ProfsX16).
The patients of all three families display typical signs of RP, with night blindness, fundus abnormalities (including bone-spicule pigmentations and narrowing of the retinal vessels), constriction of the visual fields, and evidence of cone- and rod-photoreceptor abnormalities on the ERG. Although all affected individuals share a similar molecular defect, there are, nevertheless, differences in the ensuing photoreceptor dystrophy. Although the affected individual of family C is the youngest of all patients described in this study, she is the only one who is legally blind, a result of her severely constricted visual fields. The phenotype of family A, on the other hand, shows a more prominent involvement of cone degeneration compared to the other families. This is reflected by the moderate to severe impairment of the visual acuity (see Table 1), the cone and mixed rod-cone responses of the ERG, and the photophobia as an early symptom. In patient II-3, especially, the ERG shows relatively more impairment of the cone-photoreceptor system compared to the rod-photoreceptor system. The visual fields in this patient are not constricted, as in the other patients, but show bilateral central scotomas, also indicative of cone-rod dystrophy (CRD). Her elder brother (II-1) also shows central fundus lesions, but his ERG (at age 60) no longer shows either cone or rod activity. We do not know whether a cone-rod pattern of deterioration was present in the earlier stages of his disease. Family B, finally, has a relatively late onset of a classic form of RP, with preservation of central vision. In this regard, the phenotype in this family is relatively mild compared to many forms of arRP. In two patients, cataracts were observed at a relatively young age. The development of cataracts, however, is often seen in patients affected by RP at an early age and is not exclusively present in patients with RP due to EYS mutations. It appears that in this type of photoreceptor dystrophy, like in many forms of inherited retinal diseases, other modifying factors besides the genetic defect in EYS exert their influence on the phenotypic outcome and explain the intra- and interfamilial variability.
The frame-shift mutation identified in the isolated RP patient (family C) results in the absence of 927 amino acids that altogether form six EGF-like and three laminin A G-like domains. As a result of the absence of these functionally important domains, the truncated protein will probably have little or no residual function. Alternatively, the mRNA is degraded via a mechanism called nonsense-mediated decay (NMD).29 Premature termination of the EYS protein due to the nonsense mutation in families A and B results only in the absence of the ten C-terminal amino acids that apparently fulfil a crucial function. Although the C-terminal amino acids of Drosophila eys are not highly conserved compared to the human protein, several residues of this segment are conserved in vertebrate species, including zebrafish. These results may indicate that during evolution of the vertebrate eye, the C-terminal part of EYS became essential for proper functioning of the entire protein.
The EYS gene is located on chromosome 6q12 and resides within the 15 Mb RP25 locus.3,4 Recently, a ∼100 kb clone from a tiling-path array located within the RP25 interval was found to be deleted in all affected members of a Spanish family linked to RP25,8 suggesting that genes residing within this deletion might be underlying RP in families linked to RP25. On the basis of the array-CGH data, the total length of the deletion was predicted to be 100–200 kb in size, spanning EYS exons 14–19. Altogether, these data support our conclusion that EYS is the gene responsible for RP in families that link to the RP25 locus. The prevalence of EYS mutations remains to be established, because we have thus far only fully analyzed the presence of mutations in the 44 exons and flanking intronic sequences of EYS in ten patients with arRP.
The human EYS protein is composed of 3165 amino acids and has a number of remarkable features. The first part of the protein corresponds to the previously annotated EGFL11 protein, which seems not to be present in Drosophila. The second part of human EYS is the ortholog of the Drosophila eys protein. Whereas the domain organization is similar between human and Drosophila eys, other features are not conserved. In both proteins, a less conserved region of the protein, located between the first series of EGF-like domains and the C-terminal end (with both EGF-like and laminin A G-like domains), is present. In Drosophila eys, multiple sites for the attachment of glycosaminoglycan side chains are predicted in this region, and indeed, the protein is heavily glycosylated in the insect eye.27 The consensus for such an attachment is composed of a serine residue directly followed by a glycine residue, with either a second serine-glycine tandem or a series of acidic amino acids in close proximity.30 Several of these serine-glycine clusters are found in Drosophila eys, but, remarkably, they are not conserved in the human ortholog. Apparently, extensive glycosylation is not required for proper functioning of the human eys homolog protein in the retina.
Drosophila eys is an extracellular matrix protein that occupies the interrhabdomeral space.27,28 The generation of the interrhabdomeral space has been a critical event in the transition of compound eyes from a closed to an open system. In insects with a “fused rhabdomer” configuration, such as bees, photoreceptors 1–6 within one ommatidium behave as one photosensitive system and collect light from the same area. In contrast, in insects with an open system, such as flies, photoreceptors 1–6 detect light from different areas in the visual field, because they are isolated from each other. Consequently, flies have an improved angular sensitivity, allowing the detection of smaller moving objects. Both mutant Drosophila lines spam and prominin (prom) showed a failure of interrhabdomeral space separation.28 Drosophila prom, a pentaspan transmembrane protein, is present throughout rhabdomer biogenesis and, at the time of eclosion, is selectively localized to the stalk membrane and the tips of the rhabdomer microvilli. There is evidence that spam binds to prom in orchestrating the open-rhabdomer configuration. Interestingly, Drosophila crumbs, a single-span transmembrane protein consisting of 30 extracellular EGF-like and four laminin A G-like domains, is also expressed at the stalk membrane.
What function can be attributed to Eys, Prom, and Crumbs in mammalian photoreceptors? In the mouse embryonic eye, Prom1 is located between the progenitors of the photoreceptor and retinal pigment epithelium (RPE) cells, whereas in adult murine retina, Prom1 was found at the microvilli of the RPE cells and in the rod outer segment (ROS) layer, with a high concentration in the plasma-membrane evaginations.31 Mutations in human PROM1 are associated with arRP31,32 or macular degeneration.33
Crumbs homolog 1 (Crb1) is expressed in mouse Müller cells, at the outer limiting membrane opposing photoreceptor cell inner segments, the functional equivalent of the Drosophila photoreceptor stalk.34 Loss-of-function mutations in human CRB1 result in Leber congenital amaurosis or arRP.35–37
We hypothesize three different functions for human EYS. First, through an interaction with PROM1, EYS could be involved in ROS disc morphogenesis. Second, EYS might interact directly or indirectly with the extracellular domain of CRB1 or its homolog CRB2 and in this way form a critical component of Müller cell–photoreceptor cell and photoreceptor cell–photoreceptor cell interactions. Third, reminiscent of the function of its Drosophila ortholog, EYS might be sequestered in the extracellular matrix, also known as the subretinal space, between the (developing) photoreceptors and RPE.
In conclusion, we have identified the human ortholog of Drosophila eyes shut, a 3165-aa extracellular protein that is encoded by one of the largest human genes described thus far. The 2 Mb size of this gene, which we have coined eyes shut homolog (EYS), is a little short of that of the dystrophin gene mutated in X-linked Duchenne and Becker muscular dystrophies, which spans 2.2 Mb.38,39 EYS is mutated in six patients of three families with arRP and, on the basis of previous linkage studies, is probably an important cause of inherited retinal blindness. On the basis of the function of its Drosophila counterpart (eys) and interactor (prom), it probably serves an important function in photoreceptor morphogenesis.
Supplemental Data
Supplemental Data include three figures and five tables and can be found with this paper online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi/
NetOGlyc 3.1 Server, http://www.cbs.dtu.dk/services/NetOGlyc/
UCSC Genome Browser build hg18, March 2006, http://www.genome.ucsc.edu
Unigene, http://www.ncbi.nlm.nih.gov/unigene
Accession Numbers
For our human cDNA encoding the human ortholog of Drosophila eys, an accession number was requested at the EMBL Nucleotide Sequence Database and was provided; namely, FM209056. After a request to the Human Gene Nomenclature Committee was submitted, the human gene is now officially named eyes shut homolog, abbreviated EYS.
Note Added in Proof
Note added in proof: Very recently, Abd El-Aziz and coworkers described the identification of EYS as a novel gene mutated in patients with retinitis pigmentosa. This study supports our data that EYS is essential for retinal function and shows that mutations in this gene are causative for RP in populations of various ethnicity.
Reference: Abd El-Aziz, M.M., Barragan, I., O'Driscoll, C.A., Goodstadt, L., Prigmore, E., Borrego, S., Mena, M., Pieras, J.I., El-Ahsry, M.F., Abu Safieh, L., Shah, A., Cheetham, M.E., Carter, N.P., Chakarova, C., Ponting, C.P., Bhattacharya, S.S., and Antinolo, G. (2008). EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet, published online October 5, 2008.
Acknowledgments
The authors thank C. Beumer, S. van Beersum, D. Cremers, I. Lopez, S. van der Velde-Visser, and E. van Wijk for excellent technical assistance and J. Hehir-Kwa for data analysis. We also thank all of the participating RP patients and their families. This study was financially supported by: (1) the Dutch Organisation for Scientific Research (grant 916.56.160 to A.I.d.H.), (2) the Foundation Fighting Blindness USA (grant BR-GE-0606-0349-RAD to A.I.d.H.), (3) Stichting Wetenschappelijk Onderzoek Oogziekenhuis Rotterdam (grant 2005-13 to L.I.v.d.B., A.I.d.H., and F.P.M.C.), (4) Fonds de la recherché en santé Quebec (to R.K.K), (5) the Foundation Fighting Blindness-Canada (to R.K.K and F.P.M.C), (6) Toronto Dominion financial group (to R.K.K), (7) Algemene Nederlandse Vereniging ter Voorkoming van Blindheid (to F.P.M.C.), (8) Landelijke Stichting voor Blinden en Slechtzienden (to F.P.M.C.), (9) Rotterdamse Vereniging Blindenbelangen (to F.P.M.C.), (10) Stichting Blindenhulp (to F.P.M.C.), and (11) Stichting Ondersteuning Oogheelkunde 's-Gravenhage (to F.P.M.C.).
References
- 1.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Daiger S.P., Shankar S.P., Schindler A.B., Sullivan L.S., Bowne S.J., King T.M., Daw E.W., Stone E.M., Heckenlively J.R. Genetic factors modifying clinical expression of autosomal dominant RP. Adv. Exp. Med. Biol. 2006;572:3–8. doi: 10.1007/0-387-32442-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barragan I., Abd El-Aziz M.M., Borrego S., El-Ashry M.F., O'Driscoll C., Bhattacharya S.S., Antinolo G. Linkage validation of RP25 using the 10K genechip array and further refinement of the locus by new linked families. Ann. Hum. Genet. 2008;72:454–462. doi: 10.1111/j.1469-1809.2008.00448.x. [DOI] [PubMed] [Google Scholar]
- 4.Ruiz A., Borrego S., Marcos I., Antinolo G. A major locus for autosomal recessive retinitis pigmentosa on 6q, determined by homozygosity mapping of chromosomal regions that contain gamma-aminobutyric acid-receptor clusters. Am. J. Hum. Genet. 1998;62:1452–1459. doi: 10.1086/301866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abd El-Aziz M.M., El-Ashry M.F., Barragan I., Marcos I., Borrego S., Antinolo G., Bhattacharya S.S. Molecular genetic analysis of two functional candidate genes in the autosomal recessive retinitis pigmentosa, RP25, locus. Curr. Eye Res. 2005;30:1081–1087. doi: 10.1080/02713680500351039. [DOI] [PubMed] [Google Scholar]
- 6.Abd El-Aziz M.M., Patel R.J., El-Ashry M.F., Barragan I., Marcos I., Borrego S., Antinolo G., Bhattacharya S.S. Exclusion of four candidate genes, KHDRBS2, PTP4A1, KIAA1411 and OGFRL1, as causative of autosomal recessive retinitis pigmentosa. Ophthalmic Res. 2006;38:19–23. doi: 10.1159/000088493. [DOI] [PubMed] [Google Scholar]
- 7.Abd El-Aziz M.M., El-Ashry M.F., Chan W.M., Chong K.L., Barragan I., Antinolo G., Pang C.P., Bhattacharya S.S. A novel genetic study of Chinese families with autosomal recessive retinitis pigmentosa. Ann. Hum. Genet. 2007;71:281–294. doi: 10.1111/j.1469-1809.2006.00333.x. [DOI] [PubMed] [Google Scholar]
- 8.Abd El-Aziz M.M., Barragan I., O'Driscoll C., Borrego S., Abu-Safieh L., Pieras J.I., El-Ashry M.F., Prigmore E., Carter N., Antinolo G. Large-scale molecular analysis of a 34 Mb interval on chromosome 6q: major refinement of the RP25 interval. Ann. Hum. Genet. 2008;72:463–477. doi: 10.1111/j.1469-1809.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barragan I., Marcos I., Borrego S., Antinolo G. Mutation screening of three candidate genes, ELOVL5, SMAP1 and GLULD1 in autosomal recessive retinitis pigmentosa. Int. J. Mol. Med. 2005;16:1163–1167. [PubMed] [Google Scholar]
- 10.Barragan I., Marcos I., Borrego S., Antinolo G. Molecular analysis of RIM1 in autosomal recessive Retinitis pigmentosa. Ophthalmic Res. 2005;37:89–93. doi: 10.1159/000084250. [DOI] [PubMed] [Google Scholar]
- 11.Barragan I., Borrego S., Abd El-Aziz M.M., El-Ashry M.F., Abu-Safieh L., Bhattacharya S.S., Antinolo G. Genetic analysis of FAM46A in Spanish families with autosomal recessive retinitis pigmentosa: characterisation of novel VNTRs. Ann. Hum. Genet. 2008;72:26–34. doi: 10.1111/j.1469-1809.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- 12.Li Y., Marcos I., Borrego S., Yu Z., Zhang K., Antinolo G. Evaluation of the ELOVL4 gene in families with retinitis pigmentosa linked to the RP25 locus. J. Med. Genet. 2001;38:478–480. doi: 10.1136/jmg.38.7.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcos I., Ruiz A., Blaschak C.J., Borrego S., Cutting G.R., Antinolo G. Mutation analysis of GABRR1 and GABRR2 in autosomal recessive retinitis pigmentosa. J. Med. Genet. 2000;37:E5. doi: 10.1136/jmg.37.6.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcos I., Galan J.J., Borrego S., Antinolo G. Cloning, characterization, and chromosome mapping of the human GlcAT-S gene. J. Hum. Genet. 2002;47:677–680. doi: 10.1007/s100380200103. [DOI] [PubMed] [Google Scholar]
- 15.Marcos I., Borrego S., Antinolo G. Molecular cloning and characterization of human RAB23, a member of the group of Rab GTPases. Int. J. Mol. Med. 2003;12:983–987. [PubMed] [Google Scholar]
- 16.den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 17.Helbling-Leclerc A., Zhang X., Topaloglu H., Cruaud C., Tesson F., Weissenbach J., Tome F.M., Schwartz K., Fardeau M., Tryggvason K. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- 18.Perrault I., Rozet J.M., Calvas P., Gerber S., Camuzat A., Dollfus H., Chatelin S., Souied E., Ghazi I., Leowski C. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 19.den Hollander A.I., Lopez I., Yzer S., Zonneveld M.N., Janssen I.M., Strom T.M., Hehir-Kwa J.Y., Veltman J.A., Arends M.L., Meitinger T. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest. Ophthalmol. Vis. Sci. 2007;48:5690–5698. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 20.Marmor M.F., Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Doc. Ophthalmol. 1993;85:115–124. doi: 10.1007/BF01371127. [DOI] [PubMed] [Google Scholar]
- 21.Miller S.A., Dykes D.D., Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 23.Letunic I., Copley R.R., Pils B., Pinkert S., Schultz J., Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34(Database issue):D257–D260. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz J., Milpetz F., Bork P., Ponting C.P. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R., Ceric G., Forslund K., Eddy S.R., Sonnhammer E.L. The Pfam protein families database. Nucleic Acids Res. 2008;36(Database issue):D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julenius K., Molgaard A., Gupta R., Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005;15:153–164. doi: 10.1093/glycob/cwh151. [DOI] [PubMed] [Google Scholar]
- 27.Husain N., Pellikka M., Hong H., Klimentova T., Choe K.M., Clandinin T.R., Tepass U. The agrin/perlecan-related protein eyes shut is essential for epithelial lumen formation in the Drosophila retina. Dev. Cell. 2006;11:483–493. doi: 10.1016/j.devcel.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Zelhof A.C., Hardy R.W., Becker A., Zuker C.S. Transforming the architecture of compound eyes. Nature. 2006;443:696–699. doi: 10.1038/nature05128. [DOI] [PubMed] [Google Scholar]
- 29.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 30.Winzen U., Cole G.J., Halfter W. Agrin is a chimeric proteoglycan with the attachment sites for heparan sulfate/chondroitin sulfate located in two multiple serine-glycine clusters. J. Biol. Chem. 2003;278:30106–30114. doi: 10.1074/jbc.M212676200. [DOI] [PubMed] [Google Scholar]
- 31.Maw M.A., Corbeil D., Koch J., Hellwig A., Wilson-Wheeler J.C., Bridges R.J., Kumaramanickavel G., John S., Nancarrow D., Roper K. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet. 2000;9:27–34. doi: 10.1093/hmg/9.1.27. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Q., Zulfiqar F., Xiao X., Riazuddin S.A., Ahmad Z., Caruso R., MacDonald I., Sieving P., Riazuddin S., Hejtmancik J.F. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum. Genet. 2007;122:293–299. doi: 10.1007/s00439-007-0395-2. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z., Chen Y., Lillo C., Chien J., Yu Z., Michaelides M., Klein M., Howes K.A., Li Y., Kaminoh Y. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Invest. 2008;118:2908–2916. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Rossum A.G., Aartsen W.M., Meuleman J., Klooster J., Malysheva A., Versteeg I., Arsanto J.P., Le Bivic A., Wijnholds J. Pals1/Mpp5 is required for correct localization of Crb1 at the subapical region in polarized Muller glia cells. Hum. Mol. Genet. 2006;15:2659–2672. doi: 10.1093/hmg/ddl194. [DOI] [PubMed] [Google Scholar]
- 35.den Hollander A.I., ten Brink J.B., de Kok Y.J.M., van Soest S., van den Born L.I., van Driel M.A., van de Pol D.J., Payne A.M., Bhattacharya S.S., Kellner U. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12) Nat. Genet. 1999;23:217–221. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 36.den Hollander A.I., Heckenlively J.R., van den Born L.I., de Kok Y.J.M., van der Velde-Visser S.D., Kellner U., Jurklies B., van Schooneveld M.J., Blankenagel A., Rohrschneider K. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am. J. Hum. Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lotery A.J., Jacobson S.G., Fishman G.A., Weleber R.G., Fulton A.B., Namperumalsamy P., Heon E., Levin A.V., Grover S., Rosenow J.R. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch. Ophthalmol. 2001;119:415–420. doi: 10.1001/archopht.119.3.415. [DOI] [PubMed] [Google Scholar]
- 38.den Dunnen J.T., Grootscholten P.M., Bakker E., Blonden L.A., Ginjaar H.B., Wapenaar M.C., van Paassen H.M., van Broeckhoven C., Pearson P.L., van Ommen G.J. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am. J. Hum. Genet. 1989;45:835–847. [PMC free article] [PubMed] [Google Scholar]
- 39.Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.