

Abstract
Aneuploidy, a chromosomal numerical abnormality in the conceptus or fetus, occurs in at least 5% of all pregnancies and is the leading cause of early pregnancy loss in humans. Accumulating evidence now suggests that the correct segregation of chromosomes is affected by events occurring in prophase during meiosis I. These events include homologous chromosome pairing, sister-chromatid cohesion, and meiotic recombination. In our current study, we show that mutations in SYCP3, a gene encoding an essential component of the synaptonemal complex that is central to the interaction of homologous chromosomes, are associated with recurrent pregnancy loss. Two out of 26 women with recurrent pregnancy loss of unknown cause were found to carry independent heterozygous nucleotide alterations in this gene, neither of which was present among a group of 150 fertile women. Analysis of transcripts from minigenes harboring each of these two mutations revealed that both affected normal splicing, possibly resulting in the production of C-terminally mutated proteins. The mutant proteins were found to interact with their wild-type counterpart in vitro and inhibit the normal fiber formation of the SYCP3 protein when coexpressed in a heterologous system. These data suggest that these mutations are likely to generate an aberrant synaptonemal complex in a dominant-negative manner and contribute to abnormal chromosomal behavior that might lead to recurrent miscarriage. Combined with the fact that similar mutations have been previously identified in two males with azoospermia, our current data suggest that sexual dimorphism in response to meiotic disruption occurs even in humans.
Introduction
Aneuploidy, trisomy or monosomy in the conceptus or fetus, occurs in at least 5% of all pregnancies and is a common reproductive problem in humans.1 Some fetuses with aneuploidy survive to term but suffer from disorders associated with congenital anomalies and also mental retardation, such as Down syndrome ([MIM 190685]). However, most aneuploid conceptuses die in utero, resulting in early pregnancy loss. In addition, whereas approximately 15% of all clinically recognized pregnancies are lost between the 6th and 10th week, more than 50% of these cases are due to numerical cytogenetic errors.2,3 Significantly, although it is the leading cause of early pregnancy loss, the precise underlying mechanism of aneuploidy remains poorly understood.
There is an emerging consensus that a woman with a previous aneuploid conception has an increased risk of recurrence of the same or a different aneuploidy.4,5 Indeed, a subset of women are susceptible to repeat miscarriages, referred to as recurrent pregnancy loss (RPL). RPL is, in fact, a serious reproductive problem affecting approximately 5% of couples trying to conceive.3 RPL has been historically attributed to genetic, structural, infective, endocrine, immunological, or unexplained causes.2 Although a currently prevailing hypothesis is that RPL might be a polygenic disorder, associated with both genetic and environmental determinants, it is also possible that this condition develops as a single-gene disorder.
Aneuploidy occurs as a consequence of the nondisjunction of homologous or sister chromosomes during meiosis I or II. A considerable body of evidence has recently been accumulated in a number of reports in relation to how the meiotic machinery is involved in nondisjunction. There is an emerging consensus that the events that occur during prophase in meiosis I are essential for the proper segregation of homologous or sister chromosomes. These include synapsis between homologous chromosomes and cohesion between sister chromosomes, as well as the location and frequency of meiotic recombination.6-8 It has also been logically hypothesized that genetic defects in these meiotic events induce a greater susceptibility to nondisjunction and the generation of an aneuploid conceptus.9 Given that mice deficient for these genes exclusively manifest reproduction failure without the appearance of any extragonadal symptoms, it is also not unreasonable to hypothesize that human infertility and RPL are caused by mutations in such meiotic genes.
The synaptonemal complex comprises a tripartite protein structure that promotes the connection of homologous chromosomes during prophase in meiosis I. SYCP3 ([MIM 604759]) is an essential component of the axial or lateral element of the synaptonemal complex (Figure 1A).10,11 Male mice that are deficient in SYCP3 are sterile as a result of the onset of meiotic arrest.12 This is consistent with the fact that a mutation in SYCP3 was identified in two human patients with azoospermia ([MIM 270960]).13 Notably, the phenotype of mice harboring a SYCP3 deficiency differs between males and females. Whereas male mice are completely infertile due to meiotic arrest, female mice are subfertile, with a severely reduced oocyte pool. Although two thirds of offspring are healthy, one third is affected by aneuploidy and succumbs during development in utero.14 Hence, SYCP3 is a strong candidate gene for human RPL. In our current study, we screened for mutations in the SYCP3 gene among a cohort of women who suffer from RPL.
Figure 1.
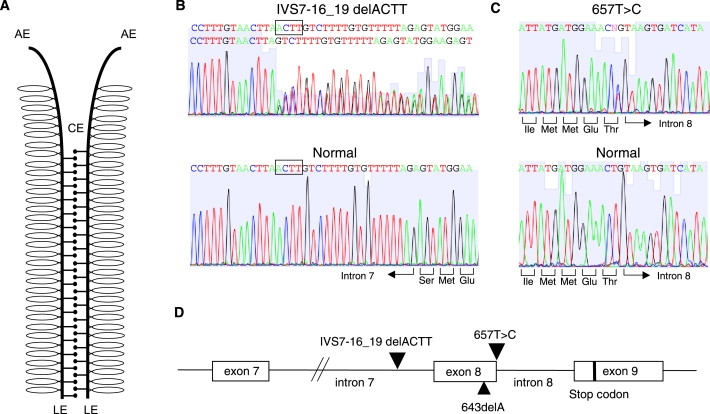
Characterization of Two SYCP3 Gene Mutations Identified in Women with RPL
(A) Schematic representation of the synaptonemal complex. Abbreviations are as follows: AE, axial element; LE, lateral element; CE, central element. SYCP3 is an essential component of the AE or LE.
(B) The c.IVS7–16_19delACTT mutation. The upper panel indicates the sequence found in case 46, whereas the lower panel indicates that of a control subject. The deleted nucleotides are boxed.
(C) The c.657T→C mutation. The upper panel indicates the sequence found in case 14, whereas the lower panel indicates that of a control subject. The identified heterozygous mutation is indicated by “N.”
(D) Localization of the two SYCP3 mutations identified in this study. Boxes indicate exons and black triangles indicate locations of the mutations. c.643delA is the mutation that was previously identified in azoospermia patients.13
Subjects and Methods
Samples
A total of 26 Japanese women with a history of RPL (more than three consecutive pregnancy losses) of unknown cause were enrolled in this study. None of these individuals has a family history of birth defects. RPL cases of known cause, such as hormonal, structural, immunological, and coagulation disorders, were excluded. All of the women were examined by ultrasonography or hysterosalpingography for detection of anatomic abnormalities of the genital tract, and all had blood drawn for testing for immunological risk factors, including natural killer cell activity, anti-nuclear antibodies, and phospholipid antibodies, such as lupus anti-coagulant. Screening for thrombophilia, with the measurement of plasma levels of antithrombin and proteins C and S, was performed. Blood tests for hyperthyroidism, diabetes mellitus, hyperprolactinemia, and infections, such as chlamydia, were also evaluated. Cytogenetic analyses demonstrated a normal karyotype in all of these individuals, as well as in their partners, but were not performed in all of the abortuses. Peripheral-blood samples were drawn at the clinic after appropriate informed consent was obtained. A total of 150 blood samples from Japanese women with at least one child and no history of infertility or miscarriage were used as controls. The study was approved by the Ethical Review Board for Human Genome Studies at Fujita Health University.
DNA Analysis
Genomic DNA was extracted from peripheral-blood samples with PureGene (Gentra). Oligonucleotide primers were designed to amplify each coding sequence, as well as the exon-intron boundaries of the human SYCP3 gene, which contains 9 exons (GenBank accession no. NM_153694). PCR was performed under standard conditions. After the removal of unincorporated primers and excess dNTPs by exonuclease I and shrimp alkaline phosphatase (ExoSAP-IT, USB), the amplified fragments were directly sequenced with the use of the same primer sets.
RNA Analysis
The SYCP3 genomic region encompassing exon 8 and a part of introns 7 and 8 was amplified for both wild-type (WT) and mutant alleles. The resulting PCR products were then subcloned into the intronic region of a mammalian expression vector (Exontrap, MoBiTec). These plasmids were then transfected into HEK293 or HeLa cells with Lipofectamine 2000 (Invitrogen). Cells were harvested at 48 hr after transfection, and total RNA extracts were isolated with RNeasy (QIAGEN). After treatment with DNase I, first-strand cDNAs were synthesized with the use of an oligo(dT) primer and Superscript III (Invitrogen). RT-PCR was then performed with primers designed with vector sequences. PCR products were resolved in 2% agarose gels and evaluated with ImageJ software.
For expression in meiotic cells, the vector promoter was replaced with the mouse Sycp1 promoter (−722/+102).15,16 The resultant plasmids were then injected into the rete testis, because this produced less damage to testicular tissue than injections into seminiferous tubules or direct injection into the parenchyma of the testis. We used a 4-wk-old DDY mouse without elongated spermatids or mature sperm, which might inhibit successful transfection into spermatocytes. The plasmids were introduced into the meiotic cells by in vivo electroporation with a CUY21 electroporator (BEX).17 The electroporation settings involved eight pulses of 30 V, with a 50 ms pulse length and a 950 ms interval. Testicular tissue was resected at 24 hr after transfection, and RT-PCR was performed with similar primers. The protocols adopted for the use of laboratory mice were approved by the Animal Care and Use Committee at Fujita Health University.
Immunoprecipitations
We cloned cDNAs encoding the WT and C-terminally truncated mutant human SYCP3 protein into the pET9 (Novagen) and pT7-FLAG (Sigma) vectors to obtain N-terminal T7- or FLAG-tagged proteins, respectively. These recombinant proteins were subsequently expressed in Rosetta DE3 competent cells (Novagen). FLAG-tagged WT proteins were first bound to anti-FLAG affinity gel (Sigma) for 30 min and then incubated with each T7-tagged WT or mutant protein for 1 hr. After washing, the proteins were eluted with 0.1 M citric acid pH2.2, followed by neutralization with 2M Tris base pH10.4. Western blotting was performed with FLAG antibodies (M2, Sigma) or T7 antibodies (Novagen) and evaluated with ImageJ software.
Expression in Mammalian Cell Lines
cDNAs encoding the WT mouse SYCP3 protein and its C-terminal truncated mutants were cloned into the pFLAG-CMV2 (Sigma) and pCMV-Tag3 (Stratagene) vectors for expression of N-terminal FLAG- or myc-tagged protein, respectively. These expression vectors were then either solely transfected or cotransfected into COS-7 cells. At 24 hr after transfection, the expressed proteins were detected with FLAG-M2 or myc antibodies (A-14, Santa Cruz Biotechnology). Alexa 488-conjugated donkey anti-mouse IgG and Alexa 594-conjugated donkey anti-rabbit IgG (Molecular Probes) were used as secondary antibodies for detection by fluorescence microscopy.
Results
We identified heterozygous point mutations within the SYCP3 gene in two of the women with RPL in our study cohort. One mutation was found in case 46 (39 years old, three miscarriages between six and ten weeks of gestation, no liveborns) and identified as c.IVS7–16_19delACTT (Figures 1B and 1D). This mutation is located at the putative branch site of intron 7 and possibly affects the normal splicing of the intron, inducing skipping of the next exon or partial skipping via a cryptic splice site in the exon. The other SYCP3 mutation was found in case 14 (29 years old, three miscarriages between six and ten weeks of gestation, no liveborns) and identified as c.657T→C in exon 8 (Figure 1C). Although this second mutation does not affect the encoded amino acid at this position (Thr), it is located at the extreme end of the exon and thus might also affect normal splicing, inducing skipping of the exon or partial intron inclusion via a cryptic splice site in the intron (Figure 1D). These changes do not appear in the NCBI database for single-nucleotide polymorphisms (SNPs). Neither of these nucleotide substitutions was found among the 150 control fertile women that we tested.
To examine how the c.IVS7–16_19delACTT and c.657T→C mutations might affect splicing events during SYCP3 transcription, we constructed expression vectors encoding minigenes with or without these mutations. When the expression vector harboring c.IVS7–16_19delACTT was introduced into human cultured somatic cell lines, RT-PCR analysis revealed that a significant fraction of the SYCP3 transcripts were shorter than WT and did not include exon 8 (Figure 2A). This skipping of exon 8 gives rise to an in-frame deletion resulting in a C-terminal truncation. We did not observe a similar abnormal-splicing phenomenon for the c.657T→C mutant in this experiment.
Figure 2.
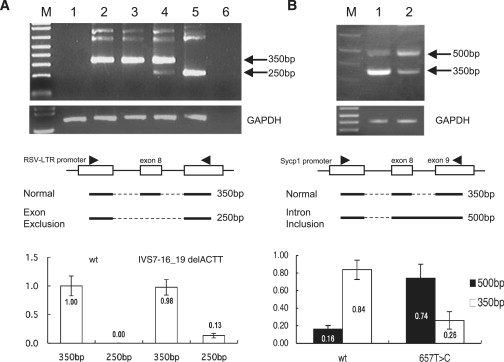
Effects of the Identified Mutations upon Normal Splicing
(A) RT-PCR analysis of SYCP3 minigenes transfected into HEK293 cells. Lane M, size markers; lane 1, untransfected parental cells; lane 2, wild-type (WT) SYCP3; lane 3, c.657T→C mutant; lane 4, c.IVS7–16_19delACTT mutant; lane 5, vector without exon 8; lane 6, no template-DNA control. The sizes of the PCR products are indicated on the right of the panels. The origin of each PCR product is indicated in the middle panels. Boxes indicate exons within the expression vector, whereas triangles depict the regions homologous to the PCR primers. Bold horizontal lines indicate the transcribed regions. The lower panel indicates semiquantification of the PCR products for WT and for the c.IVS7–16_19delACTT mutant. The given data are the mean values of three independent experiments. Vertical bars indicate the standard deviation. Similar results were observed when these constructs were transfected into HeLa cells.
(B) RT-PCR analysis of SYCP3 minigenes under the control of the Sycp1 promoter transfected into mouse testicular cells. Lane M, size markers; lane 1, WT SYCP3; lane 2, c.657T→C mutant. The lower panel shows the semiquantification of the PCR products for WT and for the c.657T→C mutant. Open boxes indicate the normal transcript, whereas closed boxes indicate an abnormal transcript with an intron retention. Ratio of intensity corresponding to these two PCR products was significantly different between WT and the c.657T→C mutant. Vertical bars indicate the standard deviation (Student's t test, p < 0.000005).
To further examine the effects of the c.657T→C mutation upon SYCP3 splicing, a minigene harboring this mutation was introduced directly into mouse testes by in vivo electroporation. When we first examined the transfection efficiency with a GFP-expression vector, the plasmids were found to be transfected with a far higher efficiency in somatic cells but less so in meiotic cells. We then utilized the Sycp1 promoter to restrict this expression to meiotic cells. We speculated that because this base substitution occurred at the last nucleotide of exon 8, an overlarge transcript bearing intron 8 might be generated as a result of a read through of the splicing junction. Although the minigene construct for the WT SYCP3 gene produced small amounts of such transcripts in this experiment, the c.657T→C mutant-expression vector generated much more aberrant transcripts (Figure 2B). The ratio of unspliced/spliced transcripts was significantly higher for the c.657T→C mutant than for the WT. In contrast, the abnormal transcript detected for the c.IVS7–16_19delACTT mutation in the previous assay using somatic cells was not observed in this assay (data not shown).
Because both of the women with RPL who harbored SYCP3 mutations were heterozygous for the identified substitutions and no other mutations were evident on the WT allele, we looked for possible effects of interactions between the mutant and WT SYCP3 proteins. On the basis of the assumptions that the mutated SYCP3 allele in case 46 (c.IVS7–16_19delACTT) produces a C-terminally truncated SYCP3 protein as a result of the exclusion of exon 8 and that that found in case 14 (c.657T→C) produces a C-terminal mutant extended protein as a result of the inclusion of intron 8 (Figure 3A), we constructed expression vectors that produced these mutant products and the WT SYCP3 protein. We first analyzed the in vitro interaction between recombinant proteins produced using an E. coli-expression system. WT SYCP3 proteins that were differentially tagged at the N terminus were found to coimmunoprecipitate each other (Figures 3B and 3C). The two mutant SYCP3 proteins also interacted with their WT counterpart, although this affinity was weaker than that between the WT proteins. The c.643delA frameshift mutation previously found in azoospermia patients has been predicted to also produce a C-terminally mutated SYCP3 protein,13 and this was found to be the case with the mutants identified in this study.
Figure 3.
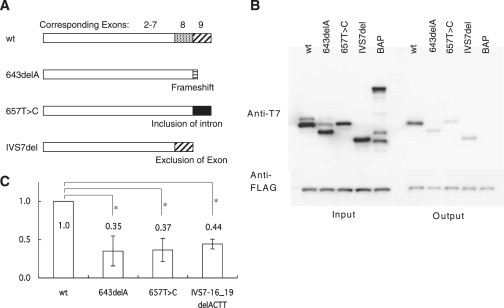
Interactions between Wild-Type and Mutant SYCP3 Proteins
(A) Schematic diagram showing the structure of the wild-type (WT) and putative mutant SYCP3 proteins. Dotted and hatched boxes indicate peptide regions encoded by exons 8 and 9, respectively. The c.IVS7–16_19 delACTT mutant is predicted to produce a C-terminally truncated protein via the exclusion of exon 8. The c.657T→C mutant is predicted to produce a C-terminally mutated protein resulting from the inclusion of intron 8 (black box). The c.643delA allele is predicted to produce C-terminally truncated protein via a frameshift.
(B) Interactions between various SYCP3 proteins. After immunoprecipitation with FLAG antibodies, coprecipitated proteins were detected with T7 antibodies. Lane 1, WT SYCP3; lane 2, c.643delA mutant; lane 3, c.657T→C mutant; lane 4, c.IVS7–16_19delACTT mutant; lane 5, bacterial alkaline phosphatase (BAP) negative control.
(C) Semiquantification of the western blot. The height of the boxes represents the mean of value relative to that of WT obtained from three independent experiments. Vertical bars indicate the standard deviation. Asterisk indicates p < 0.005 (Student's t test).
The fiber-forming properties of the self-assembling SYCP3 protein can be analyzed in cultured somatic cells. Therefore, to analyze the effects of the c.IVS7–16_19delACTT and c.657T→C mutations upon this fiber-formation process, we expressed the corresponding mutant and WT SYCP3 proteins in COS-7 cells. The WT SYCP3 protein, as expected, generated a thick fiber in a loop-like structure in the nucleus regardless of the presence of any epitope tags (Figure 4A). In contrast, the intron-inclusion mutant (c.657T→C) did not form any fiber structure and the exon-exclusion mutant (c.IVS7–16_19delACTT) formed only short rod-like fibers. Notably, when these two mutants were coexpressed with the WT in these cells, we could not detect the typical loop-like fibers characteristic of WT SYCP3 and we observed only several short fibers composed of both WT and mutant proteins (Figure 4B and Figure 5). This suggests that normal SYCP3-fiber formation is inhibited by the presence of C-terminally mutated protein. These findings also suggest that the two identified heterozygous mutations are likely to form aberrant lateral elements in the synaptonemal complex in a dominant-negative manner, which would probably lead to abnormal chromosomal behavior in meiosis I during oogenesis.
Figure 4.
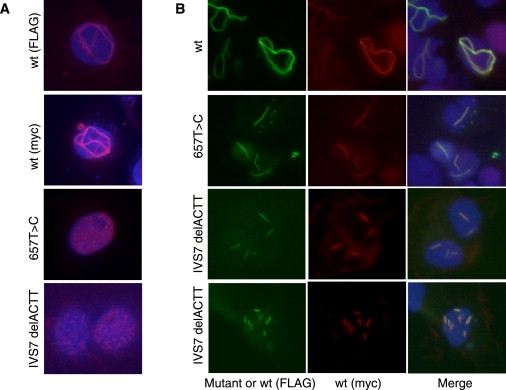
Characterization of SYCP3 Fiber Formation
(A) Microscopic observation of SYCP3 fibers produced in COS-7 cells. N-terminal FLAG- or myc-tagged wild-type (WT) SYCP3 proteins form a loop-like fiber structure, whereas the currently identified mutants appear to form short rod-like structures.
(B) Effects of the coexpression of FLAG-tagged WT and mutant SYCP3 proteins (green) upon the fiber formation of myc-tagged WT SYCP3 protein (red). WT SYCP3 forms typical loop-like structures even in the presence of differently tagged WT SYCP3 (upper panel). However, coexpressed mutant SYCP3 proteins (c.657T→C and c.IVS7delACTT) colocalize with their WT counterparts and disrupt the complex fiber conformations to yield shorter products. The levels of inhibition appear dependent on the amount of mutant protein (lower panels).
Figure 5.
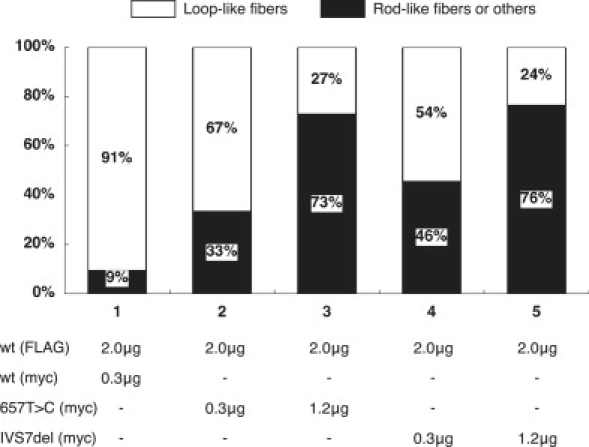
Dose-Dependent Inhibition of SYCP3 Fiber Formation by Mutant Proteins
Plasmids encoding FLAG-tagged wild-type (WT) SYCP3 were mixed with various amounts of plasmids encoding myc-tagged WT or mutant SYCP3, then the mixtures were transfected into COS-7 cells. We observed a total of 100 cells for each experiment and determined the conformation of the SYCP3 fibers for each cell. White bars indicate the percentages of cells with loop-like SYCP3 fibers, whereas black bars indicate the percentages of cells with short rod-like fibers or other structures.
Discussion
In our current study, we have identified a subset of women who have a history of RPL and harbor subtle nucleotide alterations in their SYCP3 genes. Although cytogenetic studies were not performed on the corresponding aborted fetuses, these identified mutations are likely to be causative for RPL in humans, because this observation is analogous to the phenotype observed previously in female mice with a SYCP3 deficiency.14 The loss of SYCP3 in female mice has two effects: a reduced oocyte pool and embryonic death due to aneuploidy. Our patients did not show the severe oocyte loss, however, possibly because the identified mutations do not completely abrogate SYCP3 function. Additionally, our patients had no liveborns, although two-thirds of the offspring are healthy in SYCP3-deficient female mice. This might be due to differences in checkpoint robustness between two mammalian species. Indeed, mice have rates of aneuploidy that are an order of magnitude lower than those observed in humans.9
Both of the screened mutations showed effects upon normal splicing, resulting in a C-terminally mutated SYCP3 protein under our experimental conditions. Although a minigene harboring the c.657T→C mutation did not show the abnormal transcript in our assay system using somatic cells, we could detect when the minigene was expressed in mouse testis under the control of a meiotic promoter. Recently, the regulation of meiosis-specific splicing by a meiotic promoter has been reported.18 On the basis of a previous report showing that a 5′ splice-site mutation often results in exon skipping or usage of a cryptic splice site, the inclusion of an entire intron as a result of the c.657T→C mutation is unusual.19,20 We reasoned that in this instance, no cryptic site was available, because this intron is short (150 bp). On the other hand, we did not detect abnormal transcripts from the minigene harboring c.IVS7–16_19delACTT in our assay system using mouse testis, possibly because the region mutated in c.IVS7–16_19delACTT is not conserved between mice and humans.
Both of the mutations identified in this study, in addition to the c.643delA mutation previously found in azoospermia patients, are located within the C-terminal region of the SYCP3 protein.13 The SYCP3 C terminus is highly conserved between mice and humans, in terms of not only the protein structure but also genomic organization. Hence, this region, encoding one of two coiled-coil motifs within the conserved Cor1 domain, appears to fulfill an important function in the development of the synaptonemal complex.21 Functional domains of the SYCP3 protein have been examined in detail with the use of multiple-deletion mutants, and the results of these examinations appear to be consistent to our results.22
The two RPL females that were found to harbor the SYCP3 mutation were found to be heterozygous for their respective mutations. Our data also demonstrated that the resulting C-terminally mutated proteins in both cases showed weak but substantial interactions with their WT counterparts. Moreover, their coexpression in cultured mammalian cells shows that the mutants colocalize with WT protein, suggesting that they act in concert to form abnormal SYCP3 fibers. The resultant fibers are short, possibly as a result of the lower binding affinity of the mutants, which might interfere with fiber elongation or render the fibers more fragile. The crucial question then arose as to how these mutant SYCP3 proteins could contribute to nondisjunction during female meiosis I. Given that the SYCP3 protein has already disappeared by the dictyate stage in female mice, it is reasonable to assume that homologous pairing is partly inhibited prior to the dictyate stage.23 Quantification of RT-PCR products suggests that the two patients might produce only a small amount of mutant proteins relative to WT SYCP3. Thus, it is possible that the SYCP3 fibers can still form a synaptonemal complex that is functional enough to repair SPO11-induced double-strand breaks and escape oocyte loss. However, it has been speculated that another function of the synaptonemal complex, assurance and interference in crossover, might be partly impaired, leading to nondisjunction in meiosis I.24 We are now investigating the effects of these mutations upon nondisjunction by introducing an oligonucleotide corresponding to the SYCP3 splicing junction into mouse oocytes using a primary culture system.25
Given that mutations in the SYCP3 gene were first reported in two out of 19 patients with male infertility,13 it is intriguing that similar mutations do not cause infertility but result in RPL in females. However, mice that are deficient in meiotic genes often show different phenotypes between males and females, and it has been speculated that the checkpoint systems that mediate the completion of synapsis in meiotic prophase differ between males and females.26 Indeed, SYCP3-deficient mice show complete meiotic arrest leading to infertility in male, whereas in females this leads to aneuploidy in the oocytes, which resembles human recurrent pregnancy loss.12,14 Similar phenotypic discordance was also been reported in mice with a SMC1B deficiency or in those with hypomorphic mutations in the Rad51c gene.27-29 Our results herein indicate that a similar sexual-dimorphism phenomenon also exists in humans.
In conclusion, our present data suggest that mutations in the SYCP3 gene are a predisposing factor for human nondisjunction. Given that we identified the SYCP3 mutations only in a subset of RPL women, it is possible that the remaining women in this study group might harbor mutations in other meiotic genes. Larger-scale investigations in the future will be needed for a more precise elucidation of the etiology of human reproduction failure.
Web Resources
The URLs for data presented herein are as follows:
ImageJ, http://rsb.info.nih.gov/ij/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
The authors wish to thank M. Yoshida, K. Tsukada, A. Nakamura, and T. Koishi for providing samples and to thank M. Suzuki for technical assistance. This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H.K.).
References
- 1.Hassold T., Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 2.Rai R., Regan L. Recurrent miscarriage. Lancet. 2006;368:601–611. doi: 10.1016/S0140-6736(06)69204-0. [DOI] [PubMed] [Google Scholar]
- 3.Sierra S., Stephenson M. Genetics of recurrent pregnancy loss. Semin. Reprod. Med. 2006;24:17–24. doi: 10.1055/s-2006-931797. [DOI] [PubMed] [Google Scholar]
- 4.Rubio C., Simón C., Vidal F., Rodrigo L., Pehlivan T., Remohí J., Pellicer A. Chromosomal abnormalities and embryo development in recurrent miscarriage couples. Hum. Reprod. 2003;18:182–188. doi: 10.1093/humrep/deg015. [DOI] [PubMed] [Google Scholar]
- 5.Warburton D., Dallaire L., Thangavelu M., Ross L., Levin B., Kline J. Trisomy recurrence: a reconsideration based on North American data. Am. J. Hum. Genet. 2004;75:376–385. doi: 10.1086/423331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherman S.L., Freeman S.B., Allen E.G., Lamb N.E. Risk factors for nondisjunction of trisomy 21. Cytogenet. Genome Res. 2005;111:273–280. doi: 10.1159/000086900. [DOI] [PubMed] [Google Scholar]
- 7.Eichenlaub-Ritter U. Mouse genetic models for aneuploidy induction in germ cells. Cytogenet. Genome Res. 2005;111:392–400. doi: 10.1159/000086917. [DOI] [PubMed] [Google Scholar]
- 8.Martin R.H. Meiotic errors in human oogenesis and spermatogenesis. Reprod. Biomed. Online. 2008;16:523–531. doi: 10.1016/s1472-6483(10)60459-2. [DOI] [PubMed] [Google Scholar]
- 9.Hassold T., Hall H., Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum. Mol. Genet. 2007;16:R203–R208. doi: 10.1093/hmg/ddm243. [DOI] [PubMed] [Google Scholar]
- 10.Dobson M.J., Pearlman R.E., Karaiskakis A., Spyropoulos B., Moens P.B. Synaptonemal complex proteins: occurrence, epitope mapping and chromosome disjunction. J. Cell Sci. 1994;107:2749–2760. doi: 10.1242/jcs.107.10.2749. [DOI] [PubMed] [Google Scholar]
- 11.Costa Y., Cooke H.J. Dissecting the mammalian synaptonemal complex using targeted mutations. Chromosome Res. 2007;15:579–589. doi: 10.1007/s10577-007-1142-1. [DOI] [PubMed] [Google Scholar]
- 12.Yuan L., Liu J.G., Zhao J., Brundell E., Daneholt B., Hoog C. The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol. Cell. 2000;5:73–83. doi: 10.1016/s1097-2765(00)80404-9. [DOI] [PubMed] [Google Scholar]
- 13.Miyamoto T., Hasuike S., Yogev L., Maduro M.R., Ishikawa M., Westphal H., Lamb D.J. Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet. 2003;362:1714–1719. doi: 10.1016/S0140-6736(03)14845-3. [DOI] [PubMed] [Google Scholar]
- 14.Yuan L., Liu J.G., Hoja M.R., Wilbertz J., Nordqvist K., Hoog C. Female germ cell aneuploidy and embryo death in mice lacking the meiosis-specific protein SCP3. Science. 2002;296:1115–1118. doi: 10.1126/science.1070594. [DOI] [PubMed] [Google Scholar]
- 15.Sage J., Yuan L., Martin L., Mattei M.G., Guénet J.L., Liu J.G., Hoög C., Rassoulzadegan M., Cuzin F. The Sycp1 loci of the mouse genome: successive retropositions of a meiotic gene during the recent evolution of the genus. Genomics. 1997;44:118–126. doi: 10.1006/geno.1997.4832. [DOI] [PubMed] [Google Scholar]
- 16.Vidal F., Sage J., Cuzin F., Rassoulzadegan M. Cre expression in primary spermatocytes: a tool for genetic engineering of the germ line. Mol. Reprod. Dev. 1998;51:274–280. doi: 10.1002/(SICI)1098-2795(199811)51:3<274::AID-MRD6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 17.Shoji M., Chuma S., Yoshida K., Morita T., Nakatsuji N. RNA interference during spermatogenesis in mice. Dev. Biol. 2005;282:524–534. doi: 10.1016/j.ydbio.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 18.Moldón A., Malapeira J., Gabrielli N., Gogol M., Gómez-Escoda B., Ivanova T., Seidel C., Ayté J. Promoter-driven splicing regulation in fission yeast. Nature. 2008;455:997–1000. doi: 10.1038/nature07325. [DOI] [PubMed] [Google Scholar]
- 19.Krawczak M., Reiss J., Cooper D.N. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 20.O'Neill J.P., Rogan P.K., Cariello N., Nicklas J.A. Mutations that alter RNA splicing of the human HPRT gene: a review of the spectrum. Mutat. Res. 1998;411:179–214. doi: 10.1016/s1383-5742(98)00013-1. [DOI] [PubMed] [Google Scholar]
- 21.Yuan L., Pelttari J., Brundell E., Björkroth B., Zhao J., Liu J.G., Brismar H., Daneholt B., Höög C. The synaptonemal complex protein SCP3 can form multistranded, cross-striated fibers in vivo. J. Cell Biol. 1998;142:331–339. doi: 10.1083/jcb.142.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baier A., Alsheimer M., Volff J.N., Benavente R. Synaptonemal complex protein SYCP3 of the rat: evolutionarily conserved domains and the assembly of higher order structures. Sex Dev. 2007;1:161–168. doi: 10.1159/000102105. [DOI] [PubMed] [Google Scholar]
- 23.Hodges C.A., LeMaire-Adkins R., Hunt P.A. Coordinating the segregation of sister chromatids during the first meiotic division: evidence for sexual dimorphism. J. Cell Sci. 2001;114:2417–2426. doi: 10.1242/jcs.114.13.2417. [DOI] [PubMed] [Google Scholar]
- 24.Bishop D.K., Zickler D. Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell. 2004;117:9–15. doi: 10.1016/s0092-8674(04)00297-1. [DOI] [PubMed] [Google Scholar]
- 25.Paredes A., Garcia-Rudaz C., Kerr B., Tapia V., Dissen G.A., Costa M.E., Cornea A., Ojeda S.R. Loss of synaptonemal complex protein-1, a synaptonemal complex protein, contributes to the initiation of follicular assembly in the developing rat ovary. Endocrinology. 2005;146:5267–5277. doi: 10.1210/en.2005-0965. [DOI] [PubMed] [Google Scholar]
- 26.Hunt P.A., Hassold T.J. Sex matters in meiosis. Science. 2002;296:2181–2183. doi: 10.1126/science.1071907. [DOI] [PubMed] [Google Scholar]
- 27.Revenkova E., Eijpe M., Heyting C., Hodges C.A., Hunt P.A., Liebe B., Scherthan H., Jessberger R. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat. Cell Biol. 2004;6:555–562. doi: 10.1038/ncb1135. [DOI] [PubMed] [Google Scholar]
- 28.Hodges C.A., Revenkova E., Jessberger R., Hassold T.J., Hunt P.A. SMC1beta-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat. Genet. 2005;37:1351–1355. doi: 10.1038/ng1672. [DOI] [PubMed] [Google Scholar]
- 29.Kuznetsov S., Pellegrini M., Shuda K., Fernandez-Capetillo O., Liu Y., Martin B.K., Burkett S., Southon E., Pati D., Tessarollo L. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J. Cell Biol. 2007;176:581–592. doi: 10.1083/jcb.200608130. [DOI] [PMC free article] [PubMed] [Google Scholar]