

Summary
The spondylo-meta-epiphyseal dysplasia [SMED] short limb-hand type [SMED-SL] is a rare autosomal-recessive disease, first reported by Borochowitz et al. in 1993.1 Since then, 14 affected patients have been reported.2–5 We diagnosed 6 patients from 5 different consanguineous Arab Muslim families from the Jerusalem area with SMED-SL. Additionally, we studied two patients from Algerian and Pakistani ancestry and the parents of the first Jewish patients reported.1 Using a homozygosity mapping strategy, we located a candidate region on chromosome 1q23 spanning 2.4 Mb. The position of the Discoidin Domain Receptor 2 (DDR2) gene within the candidate region and the similarity of the ddr2 knockout mouse to the SMED patients' phenotype prompted us to study this gene6. We identified three missense mutations c.2254 C > T [R752C], c. 2177 T > G [I726R], c.2138C > T [T713I] and one splice site mutation [IVS17+1g > a] in the conserved sequence encoding the tyrosine kinase domain of the DDR2 gene. The results of this study will permit an accurate early prenatal diagnosis and carrier screening for families at risk.
Main Text
The spondylo-epiphyseal-metaphyseal dysplasia [SMED] are a group of heterogeneous disorders all defined by the combination of vertebral, epiphyseal, and metaphyseal involvement.7 In 1993, Borochowitz et al. reported on a new autosomal-recessive SMED with short limb-hand type (SMED, short limb-hand type [MIM 271665]).1 Later, Langer et al. emphasized the importance of the chondral calcifications in the diagnosis of this dysplasia and proposed to call it SMED, short limb-abnormal calcification type (SMED-SL).2 Here, we report on eight patients from seven different consanguineous families with SMED-SL: six patients from five Arab Muslim families from the Jerusalem area, one Algerian patient, and one Pakistani patient. The diagnostic clinical and radiological criteria included disproportionate, short stature; platyspondyly; abnormal epiphyses and metaphyses; shortening of the lower and upper limbs; short, broad fingers; and premature calcifications. This report brings the number of patients to 20 and the number of consanguineous families to 13 out of the 17 families reported. The postnatal clinical features of the six “Jerusalem” patients are summarized and compared to data in the literature (Table 1). All the patients presented with typical short hands and broad, puffy fingers (Figure 1). Follow up of the patients permitted us to confirm that this dysplasia is progressive with respect to the severity of the bowing of the lower limbs and to the appearance of the calcifications. Indeed, three of the patients were wheelchair bound from age 11. The chondral calcifications were documented only after 8 years of age and were extremely prominent in the 16-year-old patient, who showed calcifications of the larynx, trachea, and costal cartilages and the choroids plexus (Figure 2). The same patient was severely affected with dense calcifications over the left femoral head pubis and ischium (Figure 3).
Table 1.
Summary of the Clinical Findings of the Six Jerusalem Patients Compared to Findings in the Literature
| Literature | Present Report | Total | |
|---|---|---|---|
| Number of Patients | (14 Patients) | (6 Patients) | (20 Patients) |
| Short broad hands | 14 | 6 | 20 |
| Short puffy fingers | 2 | 6 | 8 |
| Shortening of lower and upper limbs | 14 | 6 | 20 |
| Bowing of legs | 1 | 6 | 7 |
| Pectus excavatum | 5 | 1 | 6 |
| Narrowed chest | 8 | 6 | 14 |
| Respiratory disease | 8 | 3 | 11 |
| Large open fontanel | 5 | 3 | 8 |
| Large broad face | 7 | 6 | 13 |
| Saddle, short, flat nose | 11 | 6 | 17 |
| Wide nostrils | 11 | 6 | 17 |
| Long philtrum | 10 | 6 | 16 |
| Hypertelorism telecanthus | 11 | 6 | 17 |
| Abnormal teeth | 1 | 2 | 3 |
| Short neck | 1 | 6 | 7 |
| Neck hyperextension | 2 | 2 | 4 |
| Peculiar voice | 3 | 1 | 4 |
| Hypotonia | 5 | 6 | 11 |
| Delayed motor development | 9 | 6 | 15 |
| Delayed mental development | 4 | 0 | 4 |
| Platyspondyly | 14 | 6 | 20 |
| Broad and short long bones | 14 | 6 | 20 |
| Broad and short round bones | 14 | 6 | 20 |
| C1-C2 instability | 7 | 4 | 11 |
| Chondral calcifications | 3 | 4 | 7 |
| Rib abnormalities | 14 | 6 | 20 |
| Abnormal metaphyses and epiphyses | 14 | 6 | 20 |
Figure 1.
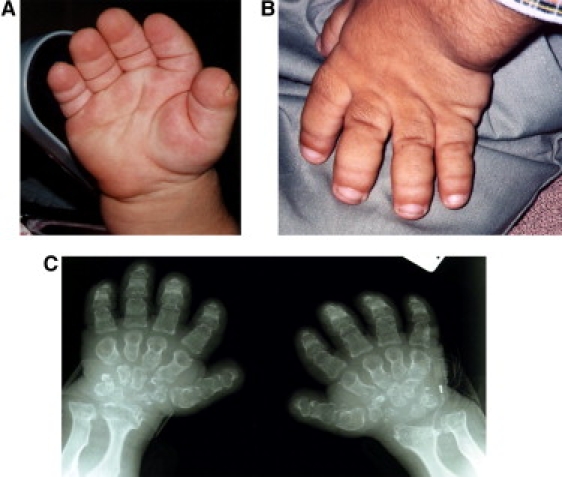
Clinical Picture and Roentgen of the Hands
(A and B) Picture of the hands of 2 patients with typical broad, puffy fingers and short, broad nails.
(C) Hand radiography of patient A shows irregular widened distal radius and ulna. The metacarpals are short and have a narrow neck with meta-epiphyseal widening and distal pointing.
Figure 2.
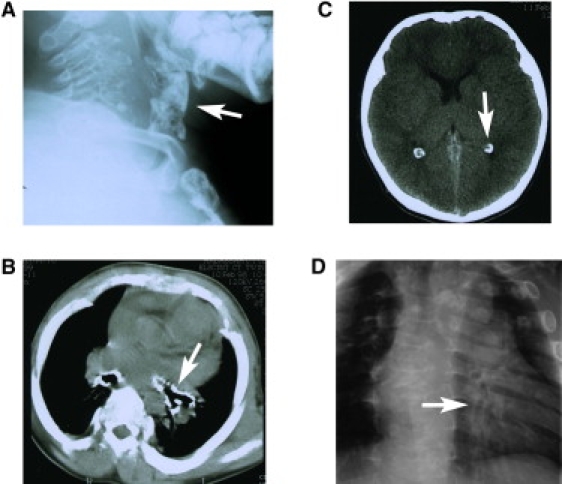
Roentgen of the Cervical Spine, Chest, and Computerized Tomography of the Head and Chest of a 16-Year-Old Patient
Lateral cervical radiography (A) and chest CT (B) showing heavily calcified larynx and tracheal rings (white arrows). (C) Brain CT of the 16-year-old patient demonstrates choroid plexus calcifications not seen normally at this age (white arrow). (D) Chest radiography demonstrates widened ribs and a heavily calcified distal bronchial tree (white arrow).
Figure 3.
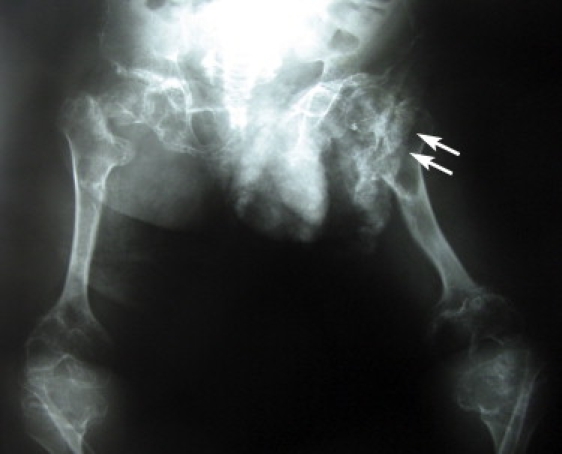
Antero-Posterior Radiography of the Pelvis, Femurs, and Knees of the 16-Year-Old Patient
Dense calcifications are projected over the left femoral head pubis and ischium (two arrows). The right acetabulum is flattened and irregular and shows a varus deformity of the femoral neck. The femurs and tibia metaphyses are widened.
Platyspondyly of the cervical spine with widened disc spaces was already detected at the age of 1 year and was a major finding in all the patients. Four patients presented with atlanto axial instability already from age 3, and three of them required neurosurgery decompression at ages 10, 11, and 16, respectively.
The rarity of the disease, the common geographic origin, and the consanguinity of these families prompted us to use a homozygosity mapping strategy as a first step for gene identification. Although homozygosity mapping has been shown to be a powerful method for localizing genes causing autosomal-recessive diseases in consanguineous families, this strategy has potential pitfalls because disease mutations can arise on nonhomozygous haplotypes, although this is quite rare.8 Blood samples of the patients and family members were obtained after informed consent was obtained from the institutional review board (0127-08-HMO) and the patients. DNA was extracted from whole blood, fibroblasts, or lymphoblastoid lines by standard methods. We analyzed the DNA of three unrelated patients of the Jerusalem families by using an Affymetrix human mapping 250K (NspI) array with an average distance of 12 Kb between the SNPs (Figure 4). We manually detected homozygous regions by looking for shared homozygous regions larger than 5 cM.9 Among the homozygous intervals shared by the three patients studied, the largest one was 2.4 Mb on chromosome 1q23 (it was bordered by markers rs12059277 [159186252] and rs10799915 [161622219]). This critical region contains approximately 50 different genes, including the gene coding for Discoidin Domain Receptor 2 (DDR2 [MIM 191311]), which was a strong candidate because of the similarity between the phenotype of the ddr2 knock-out mouse and that of the patients. The ddr2−/− mouse generated by Labrador et al. in 2001 developed a skeletal phenotype characterized by shortening of long bones, irregular growth of flat bones, and a short snout.6 The reduced chondrocyte proliferation and bone growth found in the ddr2−/− mouse was compatible with the previously reported extremely abnormal cartilage documented at the costochondral junction of one SMED-SL patient.1,5,6 The DDR2 gene is a member of the receptor tyrosine kinase [RTK] family and is involved in signal transduction.10,11 The DDR2 protein consists of an extracellular discoidin domain and a cytosolic tyrosine kinase domain (TK).10,11 This cell-surface receptor tyrosine kinase is activated by the binding of its discoidin domain to fibrillar collagen and is mainly expressed in mesenchymal cells12,13. We tested for mutations at the cDNA level. We extracted RNA from fibroblasts or lymphoblastoid lines (1.106 cells) by using the high pure RNA isolation kit (Roche) according to the manufacturer's instructions. The DDR2 transcript (accession number NM_006182.2. GI:62420883) was divided into 5 overlapping fragments (Table S1 in the Supplemental Data available online). First-strand cDNA was synthesized with Superscript II (Invitrogen), then amplified, purified (QIA quick gel extraction kit) and sequenced with the BigDye terminator system on an ABI prism 3700 sequencer. Sequence analysis of the patients' cDNA permitted us to identify three missense mutations, all localized in exon 17 of the DDR2 gene. The mutation c.2254 C > T [R752C] was found in the six patients from the Jerusalem families; the mutations c. 2177 T > G [I726R] and c.2138C > T [T713I] were identified in the Algerian and in the Pakistani patients, respectively. Study of the consanguineous parents of the Jewish patients revealed abnormal short transcripts. At the genomic level, both parents were carriers of a splice mutation IVS17+1g > a. To test and screen for the different mutations, we used the following methods. The mutation c.2254 C > T [R752C] abolishes a restriction site for MspA1I. To identify the c. 2177 T > G [I726R] mutation, we created a restriction site for SacI in the mutated sequence. For mutations c.2138C > T [T713I] and IVS17+1g > a, we created restriction sites for Tsp45I and BstNI, respectively, in the normal sequence. In all the cases studied, parents were heterozygous for the respective mutations, and the eight affected patients were homozygous. In order to rule out a rare polymorphic variant, we tested a panel of 200 chromosomes of matched healthy, unrelated individuals for each of the three missense mutations, and no carriers were found. We performed haplotype analysis of the family members carrying the mutation c.2254 C > T [R752C] by selecting microsatellite markers spanning the DDR2 region [D1S2675- D1S2768–D1S2844] from the UCSC database. PCR products were analyzed on the ABI 3100 with Genescan software. The chromosomes carrying the R752C mutation share a common haplotype, suggesting a founder mutation (Figure 4).
Figure 4.
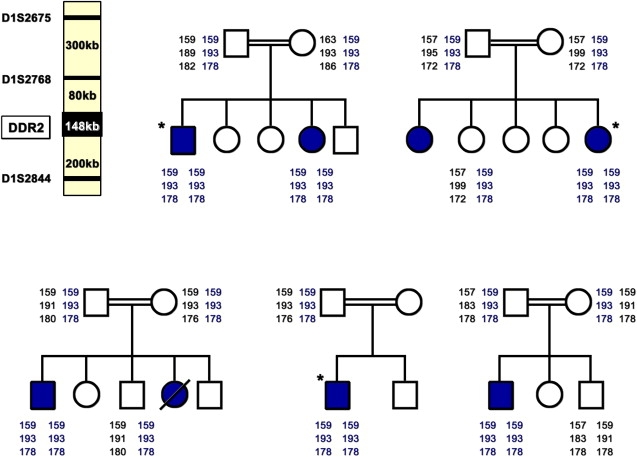
Partial Pedigrees of the Five Arab Muslim Jerusalem Families and Haplotype Analysis Using Three Microsatellite Markers, D1S2675, D1S2768, and D1S2844, in the Critical Region on Chromosome 1q23, Showing a Common Haplotype
The microsatellite markers are shown in the upper left panel. An asterisk indicates the three patients studied by SNP analysis.
Analysis of the parents' cDNA carrying the IVS17+1g > a revealed three different transcripts, which were sequenced (Figure 5A). The shortest transcript c is the result of out of frame skipping of exon 17 and includes a premature termination codon 196 bp downstream. Transcript b represents partial out-of-frame skipping of exon 17 (128 bp) as a result of an alternative donor site recognized by the spliceosome at c.2156 (GTAAGA), and transcript a corresponds to the normal allele. Importantly, the skipped exon is the same exon 17 that contains the three missense mutations identified here.
Figure 5.
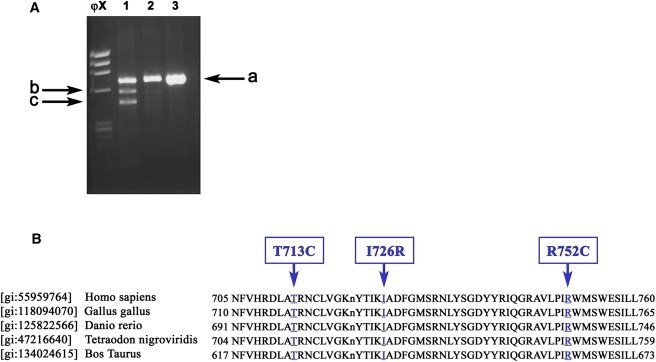
RT-PCR of the DDR2 c.DNA and Partial Alignment of the Human DDR2 Tyrosine Kinase Domain with Sequences from Four Different Species
(A) RT-PCR of the DDR2 c.DNA (fragment E). Panel 1: Heterozygous parent for IVS17+1g > a (Lymphoblastoid line). Panels 2 and 3: Normal control (lymphoblastoid line and fibroblasts, respectively). Sequencing of band c showed skipping of exon 17, band b showed partial skipping of exon 17, and band a represents the normal allele.
(B) Partial alignment of the human DDR2 tyrosine kinase domain with sequences from four different species. The mutated residues T713C, I726R, and R752C, highlighted and indicated by arrows, are found in SMED patients and are localized in the active site of the DDR2 tyrosine kinase domain.
During recent years, the DDR2 gene has been extensively studied.10,11The crucial event in DDR2 cellular signaling after ligand binding is the phosphorylation of the tyrosines in the activation loop of the intracellular Tyrosine Kinase [TK] domain.14 Phosphorylation of the tyrosine residues in the activation loop causes change in the three-dimensional structure of the activation loop and stimulates tyrosine kinase activity by providing ATP and peptide substrate access to the active-site pocket.15,16 DDR2 has a similar pattern of activation as well as sequence homology to other RTKs15 and is especially homologous to the tyrosine kinase domain of the insulin receptor (IR [MIM 14760]). This homology enables us to speculate on the effect of the DDR2 mutations reported here.14 Phosphorylation of the three tyrosine residues [Tyr 1158, 1161, and 1162] within the activation loop of the Insulin Receptor Tyrosine Kinase [IRTK] were shown to be essential for IRTK autophosphorylation and tyrosine kinase activity.16,17 These residues correspond to Tyr 736, Tyr 740, and Tyr 741 of the DDR2 protein.14 Notably, the three missense mutations reported here are localized in the active site of DDR2 in the vicinity of the activation loop near these tyrosine residues. By analogy with IRTK's three-dimensional conformation, threonine 713 of the DDR2 protein should be localized in the vicinity of the active aspartate 710 (corresponding to aspartate 1132 in the IRTK protein). Indeed, crystallographic studies of IRTK demonstrate that aspartate 1132 and Tyr 1162 [corresponding to Tyr 740 of DDR2] form a specific hydrogen bond within the active site of IRTK to stabilize the activation loop into an inhibitory conformation.16 We suggest that the T713I mutation inhibits the opening of the activation loop and thus leads to abnormal switching on or switching off of DDR2. DDR2-Ile 726 corresponds to the IRTK gene's Ile 1148, which localizes near D1150, which is involved in the ATP binding. This suggests that the mutation I726R might affect the ATP binding affinity of DDR2. The IRTK arginine 1174, which belongs to one of the IRTK substrate binding pockets16, corresponds to DDR2 arginine 752; We propose that the R752C mutation affects the substrate specificity of DDR2 and thus influences the downstream signaling pathways.14
All the missense mutations identified in this work localized in the active site of the DDR2 tyrosine kinase domain, which is highly conserved among different species (Figure 5B), whereas the splice mutation causes deletion and frameshift in this same conserved sequence.
In summary, we report for the first time on four different mutations in the DDR2 gene in the patients affected with the rare autosomal-recessive spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type syndrome [SMED-SL]. Future studies will permit elucidation of the downstream events mediated by DDR2. Families at risk can now benefit from carrier identification and early prenatal diagnosis, including preimplantation diagnosis.
Acknowledgments
We are grateful to the families for their participation in this study; we wish to thank Professor Orly Elpeleg for her help with computational SNP analysis and Dr. Mira Korner of the National Center of Genomic Technologies at the Hebrew University of Jerusalem for the SNP analysis. We thank Dr. Zeev Paroush for his fruitful comments and support. This research was supported by a grant provided by the Z. family.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Ensembl genome browser, http://ensembl.org
University of California, Santa Cruz (UCSC) genome bioinformatics, http://genome.ucsc.edu/
GDB database, http://gdb.org
References
- 1.Borochowitz Z., Langer L.O., Jr., Gruber H.E., Lachman R., Katznelson M.B., Rimoin D.L. Spondylo-meta-epiphyseal dysplasia (SMED), short limb-hand type: A congenital familial skeletal dysplasia with distinctive features and histopathology. Am. J. Med. Genet. 1993;45:320–326. doi: 10.1002/ajmg.1320450308. [DOI] [PubMed] [Google Scholar]
- 2.Langer L.O., Jr., Wolfson B.J., Scott C.I., Jr., Reid C.S., Schidlow D.V., Millar E.A., Borns P.F., Lubicky J.P., Carpenter B.L. Further delineation of spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type, with emphasis on diagnostic features. Am. J. Med. Genet. 1993;45:488–500. doi: 10.1002/ajmg.1320450419. [DOI] [PubMed] [Google Scholar]
- 3.Al-Gazali L.I., Bakalinova D., Sztriha L. Spondylo-meta-epiphyseal dysplasia, short limb, abnormal calcification type. Clin. Dysmorphol. 1996;5:197–206. [PubMed] [Google Scholar]
- 4.Fano V., Lejarraga H., Barreiro C. Spondylo-meta-epiphyseal dysplasia, short limbs, abnormal calcification type: A new case with severe neurological involvement. Pediatr. Radiol. 2001;31:19–22. doi: 10.1007/s002470000359. [DOI] [PubMed] [Google Scholar]
- 5.Tüysüz B., Gazioğlu N., Ungür S., Aji D.Y., Türkmen S. The time of onset of abnormal calcification in spondylometaepiphyseal dysplasia, short limb-abnormal calcification type. Pediatr. Radiol. 2009;39:84–89. doi: 10.1007/s00247-008-1036-1. [DOI] [PubMed] [Google Scholar]
- 6.Labrador J.P., Azcoitia V., Tuckermann J., Lin C., Olaso E., Mañes S., Brückner K., Goergen J.L., Lemke G., Yancopoulos G. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001;2:446–452. doi: 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cormier-Daire V. Spondylo-epi-metaphyseal dysplasia. Best Pract. Res. Clin. Rheumatol. 2008;22:33–44. doi: 10.1016/j.berh.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Frishberg Y., Ben-Neriah Z., Suvanto M., Rinat C., Männikkö M., Feinstein S., Becker-Cohen R., Jalanko H., Zlotogora J., Kestilä M. Misleading findings of homozygosity mapping resulting from three novel mutations in NPHS1 encoding nephrin in a highly inbred community. Genet. Med. 2007;9:180–184. doi: 10.1097/gim.0b013e318031c7de. [DOI] [PubMed] [Google Scholar]
- 9.Woods C.G., Cox J., Springell K., Hampshire D.J., Mohamed M.D., McKibbin M., Stern R., Raymond F.L., Sandford R., Malik Sharif S. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leitinger B. Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. J Biol Chem. 2003;278:16761–16769. doi: 10.1074/jbc.M301370200. [DOI] [PubMed] [Google Scholar]
- 11.Vogel W. Discoidin Domain receptors: Structural relations and functional implications. FASEB J. 1999;13(Suppl.):S77–S82. doi: 10.1096/fasebj.13.9001.s77. [DOI] [PubMed] [Google Scholar]
- 12.Leitinger B., Kwan A.P. The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. 2006;25:355–364. doi: 10.1016/j.matbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Ichikawa O., Osawa M., Nishida N., Goshima N., Nomura N., Shimada I. Structural basis of the collagen-binding mode of discoidin domain receptor 2. EMBO J. 2007;26:4168–4176. doi: 10.1038/sj.emboj.7601833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang K., Kim J.H., Kim H.J., Park I.S., Kim I.Y., Yang B.S. Tyrosine 740 phosphorylation of discoidin domain receptor 2 by Src stimulates intramolecular autophosphorylation and Shc signaling complex formation. J. Biol. Chem. 2005;280:39058–39066. doi: 10.1074/jbc.M506921200. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard S.R., Till J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Till J.H., Ablooglu A.J., Frankel M., Bishop S.M., Kohanski R.A., Hubbard S.R. Crystallographic and solution studies of an activation loop mutant of the insulin receptor tyrosine kinase: Insights into kinase mechanism. J. Biol. Chem. 2001;276:10049–10055. doi: 10.1074/jbc.M010161200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.