

Abstract
Linkage studies have implicated 10q22-q23 as a schizophrenia (SZ) susceptibility locus in Ashkenazi Jewish (AJ) and Han Chinese from Taiwan populations. To further explore our previous linkage signal in the AJ population (NPL score: 4.27, empirical p = 2 × 10−5), we performed a peakwide association fine mapping study by using 1414 SNPs across ∼12.5 Mb in 10q22-q23. We genotyped 1515 AJ individuals, including 285 parent-child trios, 173 unrelated cases, and 487 unrelated controls. We analyzed the binary diagnostic phenotype of SZ and 9 heritable quantitative traits derived from a principal components factor analysis of 73 items from our consensus diagnostic ratings and direct assessment interviews. Although no marker withstood multiple test correction for association with the binary SZ phenotype, we found strong evidence of association by using the “delusion” factor as the quantitative trait at three SNPs (rs10883866, rs10748842, and rs6584400) located in a 13 kb interval in intron 1 of Neuregulin 3 (NRG3). Our best p value from family-based association analysis was 7.26 × 10−7. We replicated this association in the collection of 173 unrelated AJ cases (p = 1.55 × 10−2), with a combined p value of 2.30 × 10−7. After performing 10,000 permutations of each of the phenotypes, we estimated the empirical study-wide significance across all 9 factors (90,000 permutations) to be p = 2.7 × 10−3. NRG3 is primarily expressed in the central nervous system and is one of three paralogs of NRG1, a gene strongly implicated in SZ. These biological properties together with our linkage and association results strongly support NRG3 as a gene involved in SZ.
Introduction
Schizophrenia and schizoaffective disorder (MIM #181500), which are hereafter referred to as “SZ,” constitute a common and debilitating disorder affecting ∼1% of the population worldwide.1 Although the causes of SZ are still unknown, multiple lines of evidence, including twin, adoption, and family studies, suggest a strong genetic component,2–4 most likely with extensive genetic heterogeneity.5 More than 20 genome-wide linkage scans and numerous candidate gene/region-based association studies6–10 have been performed to identify susceptibility genes for SZ. Associations between SZ and a few candidate genes (e.g., NRG1 [MIM ∗142445], DTNBP1 [MIM ∗607145], and DISC1 [MIM ∗605210]) have been replicated in various samples, but, aside from a translocation disrupting DISC1,11 no specific genetic alteration has convincingly been demonstrated as a causative variant.
Despite the availability of criteria for reliable diagnosis, SZ is a clinically heterogeneous disorder.12–14 The extent to which the phenotypic variability of SZ reflects underlying genetic heterogeneity is uncertain. With the goal of characterizing a more homogeneous phenotype(s), investigators have attempted to delineate SZ subgroups according to distinct clinical features, endophenotypes, and/or statistically derived measures.14 Accordingly, many genetic studies have been performed with SZ symptom dimensions, subtypes, or endophenotypes as the disease variable(s).15–21
Our previous genome-wide linkage analysis of SZ in the Ashkenazi Jewish (AJ) population was the first report of a significant linkage peak (NPL score = 4.27, p = 2 × 10−5) on chromosome 10q22-q23.22 Subsequently, an independent linkage study based on 1234 affected members in 606 Han Chinese families from Taiwan replicated this linkage signal.23 Also, microdeletions in this region have been associated with neuropsychiatric abnormalities.24 At least two biological candidates, GRID1 (Glutamate receptor, ionotropic, delta 1, [MIM ∗610659]) and NRG3 (MIM ∗605533), located under our linkage peak have been implicated by association studies. Recent candidate gene-based studies in AJ and Han Chinese populations reported association between SZ and GRID1,25,26 whereas in a northern European sample, Benzel et al. found nominal association signals for SZ with NRG3 (as well as NRG1 and NRG2 [MIM ∗603818]) that were not of sufficient magnitude to withstand multiple testing correction.27
To survey the 10q22-q23 interval for the source of our linkage signal, we conducted a peakwide fine mapping association study by genotyping 1414 SNPs across the 12.5 Mb region in our AJ patients, parents, and unrelated controls. The phenotypes for analysis included the binary disease status (affected, not affected) and nine quantitative traits or factors, derived from a principal components factor analysis of 73 items from our consensus diagnostic ratings and direct assessment interviews.28 Although no signal achieved significance for SZ disease status, we find strong evidence that three nearby SNPs in intron 1 of NRG3 are associated with the “delusion” factor of SZ. In addition, several SNPs in other parts of NRG3 show suggestive association signals with other SZ factors.
Subjects and Methods
Ascertainment of Study Subjects
We recruited SZ individuals and controls of Ashkenazi descent in North America via advertisements in newspapers and Jewish newsletters, talks to community organizations, letters to leaders of the Jewish community, letters and talks to service providers, and a study website hosted by the Johns Hopkins Epidemiology-Genetics Program in Psychiatry.22 We determined the ethnicity of all four grandparents of cases and controls on the basis of geographic origin and excluded those with non-Ashkenazi grandparents from our analysis. We considered individuals with grandparents from Spain, Netherlands, Turkey, or Greece as likely being of Sephardic origin and also excluded any with self-reported Sephardic heritage. Controls were also excluded if they screened positive for a history of psychosis, mania, psychiatric hospitalization, depression, or suicide attempt. Cases were recruited if they met criteria for a diagnosis of SZ according to the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV).29 Case-parent trios were eligible for inclusion in these analyses if the probands met the diagnostic criteria and both parents were available for DNA collection. All recruitment methods and protocols for collection of clinical data and blood samples were approved by the Johns Hopkins institutional review board and informed consent was obtained from all individuals.
Diagnostic Instruments and Procedures
All cases were examined in person by a clinical psychologist. Trio parents were not examined but were asked for a blood sample. Most of the subjects were seen in their homes. Examiners used the Diagnostic Interview for Genetic Studies (DIGS, version 2.0; revised for DSM-IV), a semistructured interview that elicits information about lifetime history of psychiatric symptoms and behaviors. On average, the subjects had been affected for 20.9 ± 10.8 years at the time of the examination (minimum of 1, maximum of 63). Thus, the item scores represent a compilation of all features over an average of two decades. The subjects were asked to sign release forms allowing us to receive copies of their psychiatric records. Interviews were tape-recorded for quality control purposes and for review by members of a consensus diagnosis committee. Detailed clinical methods are available from our prior publication.22
Factor Analysis and the Use of Factors as Quantitative Traits for Association Study
To refine our phenotypic characterization of individuals with SZ, we took advantage of the detailed clinical information from our standardized and comprehensive interviews. We performed a principle components factor analysis on 73 items and derived 9 factors best summarizing the clinical symptom dimensions. The procedures and results are described in detail elsewhere.28 In brief, principal components factor analysis was conducted in a sample of 1199 SZ subjects (including all the cases in this AJ collection and other cases in an outbred northern European population) with 73 dichotomous signs and symptoms (Table 1) from consensus diagnostic ratings, DIGS, and the Premorbid Adjustment Scale.30 Information on these signs and symptoms was not collected in our controls. Parallel analyses and Velicer's minimum average partial correlation test provided statistical support for a solution of from 8 to 15 factors. After testing this range, we obtained a reasonable solution of 9 factors (designated prodromal, negative, delusion, affective, scholastic, child/adolescent sociability, disorganization, disability/impairment, and hallucination) (Table 1). We transformed the factor scores to achieve normality with either generalized modulus power transformations31 or natural log transformation28 and used the transformed factor scores as quantitative traits for family-based and unrelated case-only association analyses.
Table 1.
Nine Factors
| Factor Name | Main Phenotype Characteristics for Each Factor |
|---|---|
| Prodromal | prodromal role impairment; prodromal lack of interest, energy; prodromal social isolation/withdrawal; prodromal impaired hygiene; prodromal odd beliefs/magical thinking; prodromal peculiar behavior; prodromal unusual perceptions; premorbid personality disorder; gradual onset of disorder (greater than 3 months) |
| Negative | alogia; flat affect; negative formal thought disorder; apathy; anhedonia; poor adult sociality; failure to establish adult independence |
| Delusion | thought insertion; delusions of influence; somatic hallucinations; thought withdrawal; thought broadcasting; thought echo; primary delusions; olfactory hallucinations; primary delusional perception; delusions of reference; delusions of guilt; nihilistic delusions |
| Affective | affective symptoms early in course; major depression criterion A; mania criterion A; self-damaging acts; affective syndrome (in psychotic illness) |
| Scholastic | poor high school adaptation; poor high school performance; none college; poor elementary school performance; learning disability/hyperactivity; therapeutic school attendance; school deterioration |
| Child/Adolescent Sociability | poor childhood sociability; poor childhood peer relations; poor adolescent sociability; poor adolescent peer relations; poor elementary school adaptation |
| Disorganization | incoherence/loose associations; bizarre behavior; inappropriate affect; self-neglect; poor insight; grandiose delusions; committed assault; persecutory delusions; akathisia; Parkinsonian signs; does not meet criterion A for generalized anxiety disorder; does not meet criterion A for obsessive-compulsive disorder; does not meet criterion A for panic disorder |
| Disability/Impairment | history of occupational deterioration; occupational disability at time of diagnosis; history of occupational impairment; lives alone instead of with family/friends; severe psychotic disorder; tardive dyskinesia; inconsistent, rare, or conflicted intimate relationships; nonremitting course |
| Hallucination | visual hallucinations; auditory hallucinations; tactile hallucinations; catatonic behavior; seizures; abnormal birth or early development; affective syndrome not prominent, or postdates psychosis, or at least 1 week of psychosis occurred in absence of affective symptoms |
Demographic and Clinical Characteristics
We genotyped a total of 1515 individuals, including 458 SZ probands, 570 parents of schizophrenics, and 487 unrelated controls (Figure 1). With SZ disease status as the binary phenotype, we analyzed this data set as 285 independent parent-child trios for family-based study and as 173 cases and 487 controls for an independent case-control study to test for replication of the primary signal in our family-based analysis. We also report an analysis of all 458 cases and 487 controls with the caveat that 285 of the cases overlap with the family-based analysis so that these two results are not independent. When we used the nine factors as quantitative traits, we did not include the controls because factor scores are available only for cases. In that scenario, our data set was analyzed as 285 parent-child trios for the family-based study and 173 unrelated SZ cases that serve as a replication case-only data set.
Figure 1.
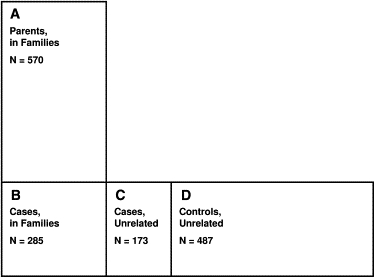
AJ Individuals Enrolled in This Study
For analyses with SZ as a binary phenotype, the subjects for various scenarios are: “Trios,” (a)+(b); “CC-all” (case-control analysis with all SZ individuals), (b)+(c)+(d); “Combined” (all samples), (a)+(b)+(c)+(d). For analyses with factors as quantitative traits, the subjects for various scenarios are: “Trios,” (a)+(b); “Cases-all” (case-only analysis with all SZ individuals), (b)+(c); “Cases-independent” (case-only analysis with independent SZ individuals), (c); “Combined” (all SZ individuals and trios parents), (a)+(b)+(c).
Of the 285 trio probands, the average age at onset was 19.5 (±4.36) years, 74.3% of them were male, and 19.7% of them were diagnosed as having schizoaffective disorder. Of the 173 unrelated cases, the average age at onset was 21.0 (±6.46) years, 60.7% were male, and 21.4% were diagnosed as having schizoaffective disorder. Of all the 487 AJ controls, 45.6% were male.
SNP Selection
By using publicly available information from dbSNP build 124, NCBI genome build 35.1, and the HapMap project32,33 public release #16c.1, we selected 1536 SNPs across the 95% confidence interval of our previous linkage peak at 10q22-q23, which was a ∼12.5 Mb region (chr10:79,550,189-92,037,551). We selected the SNPs in an iterative set of interactions with Illumina (Illumina Corp, CA). An Illumina algorithm that predicted performance on their genotyping platform was used to evaluate the SNPs. Initially, we considered all dbSNPs in the interval (9911 at the time) and obtained their Illumina scores. We discarded those with low scores (<0.4, 628 SNPs) and then selected SNPs based on the following criteria: (1) known minor allele frequency (MAF) > 0.05; (2) submission to dbSNP from more than one source; (3) validation reported; (4) uniform distribution across the region; (5) a >60 basepairs interval from neighboring SNPs; and (6) a priority given to those with a score >0.8. To ensure an unbiased coverage of the whole region, we did not consider gene location in our initial SNP selection. We then added nonsynonymous coding SNPs and then SNPs to fill coverage gaps. There are two segmental duplications in our region (approximately at chr10:80,900,000-81,670,000 and chr10:88,880,000-89,170,000) (also see Figure 2) in which we did not achieve good SNP coverage. Aside from these two segmental duplications, the mean distance between two consecutive SNPs is 7.85 (±5.41) kb and the median distance between two consecutive SNPs is 7.75 kb. The genotyped SNPs and their location are provided in Table S1 available online.
Figure 2.
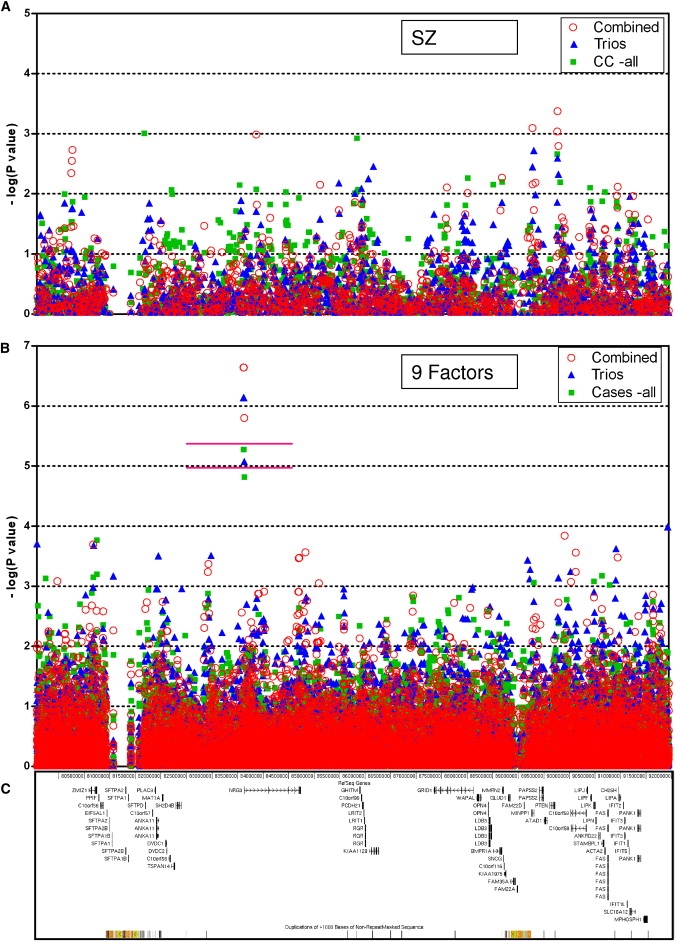
Results of Association Analyses and Genes in the ∼12.5 Mb Region at Chromosome 10q22-q23
The horizontal axis is the sequence position (chr10:79,550,189-92,037,551) according to NCBI genome build 35.1.
(A) Results with SZ as a binary phenotype. Values along the vertical axis are the negative logarithm (based 10) of the p values. CC-all, case-control study of all cases and all controls. Subjects analyzed in various scenarios are described in Figure 1.
(B) Results with the nine factors as the quantitative traits. For clarity, we did not assign different colors to different factors. The upper horizontal pink line marks the study-wide significance cutoff value for Bonferroni correction of all 1414 SNPs and all 9 factors (p = 3.93 × 10−6). The lower horizontal pink line marks the cutoff value for Bonferroni correction of 532 independent SNPs and all 9 factors (p = 1.04 × 10−5).
(C) Genes and segmental duplications in this region, according to UCSC genome browser with the NCBI genome build 35.1 data.
Genotyping and Data Quality Control
Genotyping was performed with the Integrated BeadArray System (Illumina Corp) at the Johns Hopkins Center for Inherited Disease Research (CIDR). We supplied CIDR with 96-well barcoded microtiter plates containing genomic DNA samples (including 80 control samples from Centre d'Etude du Polymorphisme Humain [CEPH]34 and 40 blind duplicates for quality control) at a concentration of 125 ng/μL. Among the 1536 SNPs, 1498 had good quality and were released for analysis. Of these 1498 SNPs, genotyping quality was excellent with an accuracy rate of 99.99% based on blind duplications, a genotype missing rate of 0.09%, and a Mendelian consistency rate of 99.99%. After removing SNPs that were not polymorphic in our sample, there remained for association analysis a total of 1414 SNPs distributed over our 12.5 Mb region.
Statistical Analyses
We use UNPHASED v3.0.9 for family-based, case-control, and quantitative trait association analyses.35,36 The software is a versatile application that can deal with binary phenotypes and quantitative traits and can perform both allelic and genotypic tests. Furthermore, in addition to dealing with familial and unrelated samples separately, UNPHASED can perform a combined analysis on a collection including both familial and unrelated samples, with results presented in this report as the “combined p value.” We consider the allelic test of 285 trios to be our primary statistical analysis, and the set of unrelated 173 cases to serve as an independent sample for replication. When appropriate, we also report the results of the analysis from “all cases” (285 familial plus 173 unrelated, totaling 458) and “all samples” (all 458 cases plus 570 parents and 487 unrelated controls) (see Figure 1). For the strongest association signals detected in the allelic test, we also performed and report the genotypic test. When SZ was used as the binary phenotype, the analyses were a classical transmission disequilibrium test (TDT),37 a case-control study, and a combined analysis. When factors were used as the quantitative traits, the unrelated controls were not included for analysis because factor scores are available only in SZ cases. Therefore, the results reported for our factor analyses are from the quantitative TDT, case-only quantitative traits association studies, and combined analyses.
For our strongest association signal, we also conducted tests with the FBAT38 and QTDT39 software packages to confirm the validity of our results. With FBAT v1.7.3, we performed 1 × 108 Monte Carlo samplings to calculate an empirical p value. With QTDT v2.6.0, we used the default orthogonal model described by Abecasis et al.39
We used PedCheck40 v1.0 to detect Mendelian inconsistencies and HaploView41 v4.0 to calculate linkage disequilibrium and to generate graphical representations of the marker-to-marker disequilibrium and underlying haplotype block structure.
Permutation and Correction for Multiple Testing
We performed 10,000 permutations with UNPHASED for the analysis of each phenotype and used the results to make the quantile-quantile plots (QQ plots) (Figure 3) and to estimate the study-wide significance level of our results.
Figure 3.
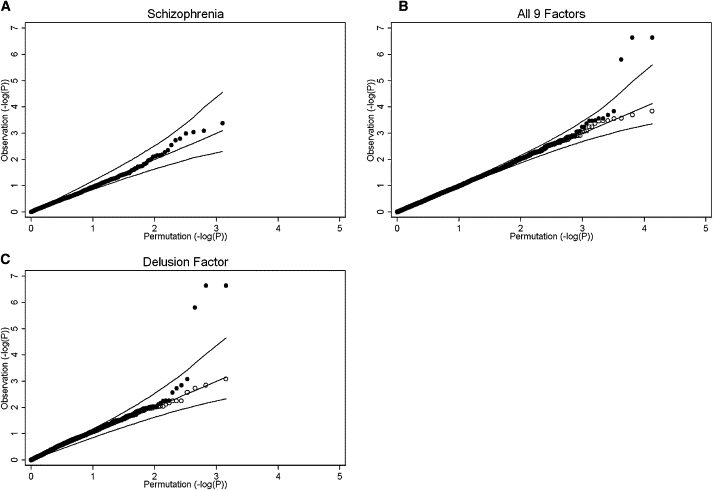
Representative Quantile-Quantile Plots Based on 10,000 Permutations
The values plotted are the negative logarithm (based 10) of p values, either of the observed results (vertical axis) or the permutation results (horizontal axis) of the corresponding phenotypes. The three lines in each plot represent the 97.5, 50, and 2.5 percentile values of the permutation. The area between the upper line and lower line represents the 95% confidence interval of the corresponding permutation. The solid circles are the observed values plotted against the permutation values.
(A) The quantile-quantile (QQ) plot with SZ as a binary phenotype. Our results show no obvious inflation of type I error.
(B) The QQ plot with data from all nine factors analyzed together. There are three observations above the 97.5 percentile of the study-wide permutation data. When those three observations are removed (the result shown in open circles), the distribution shows no evidence of type I error inflation.
(C) The QQ plot with the “delusion” factor as a quantitative trait. The three observations with highly significant values are the same as those of (B); removal of those three observations (open circles) yields a distribution that fits the permutation data quite well.
We applied two additional strategies for multiple test correction. For the first, we assumed that the genotypes of the 1414 SNPs were independent and used the Bonferroni method to correct for 1414 tests (“all” SNPs). For the second, we took into account the linkage disequilibrium between nearby SNPs to calculate the number of independent SNPs (“independent” SNPs).42,43 In brief, we considered SNP genotypes within the same LD block (the criteria used by Gabriel et al.44) as a single independent test, and we counted genotypes for SNPs outside of LD blocks individually. These calculations reduced the number of “independent” tests for Bonferroni correction to 532. With these strategies, the cut-off p value for claiming statistical significance is either 3.54 × 10−5 (= 0.05/1414, “all” tests) or 9.40 × 10−5 (= 0.05/532, “independent” tests). The study-wide cut-off p values for the analysis of the nine factors were calculated at 3.93 × 10−6 for “all” tests and 1.04 × 10−5 for “independent” tests.
Investigation of “Failed” SNPs and Detection of Copy-Number Variations
Among the 1536 SNPs, 59 were flagged for low quality or Mendelian inconsistency and were not used in the filtered data set in our analysis. We looked at the detailed genotyping results for these SNPs individually to determine whether their apparent poor performance could be explained by other than technical reasons. We considered Mendelian inconsistencies in certain pedigrees as a possible indication of CNV, such as a deletion, at the corresponding regions. For those SNPs, we then looked at the “Log R Ratio” readings of Illumina genotyping data, which represented the level of fluorescence probing intensity and provided further evidence of copy-number difference of the genomic DNA. If necessary, we used quantitative PCR to confirm CNVs and conducted long-range PCR and sequencing to demonstrate the boundaries. Several unrelated individuals were sequenced for each confirmed CNV to compare the junction points.
Sequencing
We sequenced known exons and several conserved noncoding regions of two genes (GRID1 and NRG3) in amplified products of the genomic DNA from six AJ normal controls and 15 unrelated AJ SZ individuals from the original linkage families.22 We also sequenced presumptive break point fragments to confirm deletions. Sequencing of each PCR product was conducted from both directions with a capillary DNA sequencer (ABI 3100 Genetic Analyzer, Applied Biosystems, CA) according to the company-recommended protocol.
Results
Association Analyses with SZ as a Discrete Phenotype
The results of the allelic test for all 1414 SNPs in family-based association analysis, case-control analysis, and combined (all samples together) association study with SZ affected status as the discrete phenotype are shown in Figure 2A. The plots comparing the observed versus the expected at random distribution of p values (QQ plots) show that the distribution of the majority of p values fits the prediction of our permutation results, suggesting no gross inflation of type I errors (Figure 3). SNPs with the 20 smallest p values are listed in Table 2 and results of all SNPs are in Table S1. None of these SNPs are statistically significant according to either stringent or LD-based Bonferroni corrections.
Table 2.
20 SNPs with the Smallest p Values with SZ as the Discrete Phenotype
| SNP Name | Position (Build 35) | Gene/Locus |
Trios |
Case-Control |
Combined |
|||
|---|---|---|---|---|---|---|---|---|
| p Value | Odds Ratio | p Value | Odds Ratio | p Value | Odds Ratio | |||
| rs7913285 | 80228807 | 8.34 × 10−2 | 1.24 (0.98–1.57) | 2.99 × 10−2 | 1.24 (1.02–1.49) | 4.54 × 10−3 | 1.28 (1.08–1.51) | |
| rs1013005 | 80239045 | 1.76 × 10−2 | 1.34 (1.06–1.69) | 2.93 × 10−2 | 1.23 (1.03–1.47) | 2.82 × 10−3 | 1.29 (1.09–1.52) | |
| rs10762810 | 80250777 | 1.73 × 10−2 | 1.35 (1.06–1.72) | 1.35 × 10−2 | 1.27 (1.06–1.52) | 1.86 × 10−3 | 1.31 (1.11–0.15) | |
| rs1080293 | 83886240 | NRG3 | 1.96 × 10−2 | 1.44 (1.07–1.93) | 8.43 × 10−3 | 1.36 (1.08–1.70) | 1.04 × 10−3 | 1.41 (1.16–1.73) |
| rs1649967 | 83897704 | NRG3 | 1.04 × 10−1 | 1.25 (0.96–1.61) | 4.78 × 10−2 | 1.22 (1.00–1.48) | 1.52 × 10−2 | 1.25 (1.05–1.49) |
| rs11592457 | 85145754 | 2.62 × 10−2 | 2.18 (1.05–4.55) | 1.48 × 10−1 | 1.41 (0.88–2.24) | 7.09 × 10−3 | 1.79 (1.15–2.78) | |
| rs4244944 | 85834794 | 9.84 × 10−3 | 1.35 (1.08–1.68) | 9.77 × 10−3 | 1.27 (1.06–1.53) | 1.38 × 10−2 | 1.23 (1.04–1.44) | |
| rs4244946 | 85963245 | PCDH21 | 8.11 × 10−4 | 1.39 (1.08–1.78) | 7.58 × 10−2 | 1.19 (0.98–1.45) | 1.47 × 10−2 | 1.24 (1.04–1.48) |
| rs2222546 | 87652091 | GRID1 | 4.30 × 10−1 | 1.10 (0.86–1.41) | 1.53 × 10−2 | 1.26 (1.04–1.52) | 7.86 × 10−3 | 1.25 (1.06–1.48) |
| rs12098567 | 88063754 | GRID1 | 1.23 × 10−1 | 2.00 (0.82–4.86) | 5.42 × 10−3 | 2.71 (1.30–5.65) | 9.71 × 10−3 | 2.13 (1.22–3.71) |
| rs1745903 | 88742186 | KIAA1975 | 1.39 × 10−2 | 1.34 (1.07–1.69) | 6.33 × 10−3 | 1.29 (1.07–1.56) | 5.40 × 10−3 | 1.26 (1.08–1.49) |
| rs12771728 | 89345293 | 3.56 × 10−3 | 1.49 (1.12–1.97) | 9.57 × 10−2 | 1.18 (0.97–1.44) | 8.08 × 10−4 | 1.36 (1.14–1.63) | |
| rs12778872 | 89346913 | 2.75 × 10−2 | 1.46 (1.02–2.07) | 2.03 × 10−1 | 1.17 (0.92–1.50) | 6.96 × 10−3 | 1.36 (1.08–1.72) | |
| rs791888 | 89402555 | 1.04 × 10−2 | 1.38 (1.08–1.77) | 1.73 × 10−1 | 1.14 (0.95–1.37) | 6.54 × 10−3 | 1.26 (1.07–1.50) | |
| rs9630112 | 89834555 | 1.38 × 10−2 | 1.34 (1.06–1.69) | 2.18 × 10−3 | 1.33 (1.11–1.59) | 9.19 × 10−4 | 1.32 (1.12–1.55) | |
| rs4933466 | 89839499 | 2.54 × 10−3 | 1.43 (1.13–1.82) | 3.49 × 10−2 | 1.22 (1.02–1.46) | 4.23 × 10−4 | 1.35 (1.14–1.59) | |
| rs11202656 | 89856629 | 4.73 × 10−3 | 1.45 (1.11–1.90) | 1.90 × 10−1 | 1.15 (0.94–1.41) | 1.61 × 10−3 | 1.35 (1.12–1.63) | |
| rs7922193 | 91011625 | 8.61 × 10−2 | 1.22 (0.97–1.52) | 1.37 × 10−2 | 1.26 (1.05–1.50) | 1.08 × 10−2 | 1.23 (1.05–1.44) | |
| rs10430719 | 91028961 | 3.29 × 10−2 | 1.40 (1.01–1.95) | 3.69 × 10−2 | 1.29 (1.01–1.63) | 7.69 × 10−3 | 1.33 (1.07–1.66) | |
| rs3824603 | 91333587 | PANK1 | 3.09 × 10−2 | 1.35 (1.02–1.79) | 4.73 × 10−2 | 1.24 (1.00–1.55) | 1.10 × 10−2 | 1.29 (1.06–1.56) |
Association Analyses with Nine Factors as Quantitative Traits
The results of our association analyses with factors as the quantitative trait phenotypes are shown in Figure 2B. For visual clarity, we overlay the results of nine factors together without using different symbols for each factor. The comprehensive results for each individual factor can be found in Table S2 and a description of the 20 SNPs with the smallest p values across all nine factors is provided in Table 3. We observed no gross inflation of type I errors according to the comparison to the permutation results (Figure 3).
Table 3.
20 SNPs with the Smallest p Values with Each of the Nine Factors as Quantitative Traits
| SNP Name | Position (Build 35) | Gene/Locus |
Trios |
Cases |
Combined |
Factor Responsible for the p Value | |||
|---|---|---|---|---|---|---|---|---|---|
| p Value | Effect Sizea | p Value | Effect Sizea | p Value | Effect Sizea | ||||
| rs2019080 | 79951742 | 3.92 × 10−2 | 0.25 (0.00–0.50) | 1.23 × 10−2 | 0.17 (0.03–0.31) | 8.24 × 10−4 | 0.28 (0.11–0.44) | delusion | |
| rs1574190 | 80662387 | RAI17 | 1.04 × 10−3 | 0.43 (0.14–0.72) | 7.02 × 10−4 | 0.24 (0.10–0.39) | 2.02 × 10−4 | 0.33 (0.14–0.51) | prodromal |
| rs4319456 | 82927110 | ∗ | 1.11 × 10−2 | 0.50 (0.09–0.92) | 1.31 × 10−3 | 0.35 (0.13–0.57) | 5.79 × 10−4 | 0.46 (0.20–0.71) | disorganization |
| rs7899151 | 82938902 | ∗ | 1.62 × 10−3 | 0.67 (0.22–1.12) | 2.01 × 10−3 | 0.34 (0.10–0.57) | 4.27 × 10−4 | 0.48 (0.20–0.76) | disorganization |
| rs10883866 | 83633619 | NRG3 | 7.26 × 10−7 | 0.97 (0.49–1.45) | 5.27 × 10−6 | 0.51 (0.28–0.74) | 2.30 × 10−7 | 0.67 (0.37–0.97) | delusion |
| rs10748842 | 83639719 | NRG3 | 7.26 × 10−7 | 0.97 (0.49–1.45) | 5.27 × 10−6 | 0.51 (0.28–0.74) | 2.30 × 10−7 | 0.67 (0.37–0.97) | delusion |
| rs6584400 | 83646506 | NRG3 | 8.40 × 10−6 | 0.75 (0.36–1.14) | 1.51 × 10−5 | 0.44 (0.24–0.64) | 1.59 × 10−6 | 0.55 (0.30–0.81) | delusion |
| rs17099528 | 83921863 | NRG3 | 5.72 × 10−3 | 67.2 (−4.8–139) | 2.94 × 10−3 | 1.90 (0.46–3.33) | 1.34 × 10−3 | 2.53 (0.85–4.22) | disorganization |
| rs951204 | 84038745 | NRG3 | 5.46 × 10−3 | 0.47 (0.12–0.83) | 1.60 × 10−2 | 0.23 (0.03–0.43) | 1.24 × 10−3 | 0.37 (0.13–0.61) | disorganization |
| rs12416489 | 84717603 | NRG3 | 4.73 × 10−2 | 0.22 (0.00–0.44) | 3.78 × 10−2 | 0.14 (0.00–0.27) | 1.24 × 10−3 | 0.25 (0.09–0.41) | scholastic |
| rs11196700 | 84724720 | NRG3 | 6.01 × 10−3 | 0.32 (0.09–0.54) | 1.24 × 10−2 | 0.17 (0.03–0.31) | 3.42 × 10−4 | 0.29 (0.12–0.46) | scholastic |
| rs3818306 | 84728393 | NRG3 | 4.73 × 10−2 | 0.22 (0.00–0.44) | 3.78 × 10−2 | 0.14 (0.00–0.27) | 1.24 × 10−3 | 0.25 (0.09–0.41) | scholastic |
| rs8421 | 84736601 | NRG3b | 6.01 × 10−3 | 0.32 (0.09–0.54) | 1.20 × 10−2 | 0.17 (0.03–0.31) | 3.36 × 10−4 | 0.29 (0.12–0.45) | scholastic |
| rs1339844 | 84856649 | NRG3b | 3.76 × 10−3 | 0.36 (0.09–0.64) | 3.15 × 10−2 | 0.16 (0.01–0.30) | 2.74 × 10−4 | 0.32 (0.14–0.49) | scholastic |
| rs10886221 | 85123936 | ∗ | 1.12 × 10−1 | 0.22 (−0.06–0.50) | 7.48 × 10−2 | 0.13 (−0.02–0.29) | 8.94 × 10−4 | 0.30 (0.12–0.47) | hallucination |
| rs12220927 | 89980527 | 6.04 × 10−2 | 0.28 (0.01–0.56) | 8.00 × 10−3 | 0.23 (0.07–0.38) | 1.45 × 10−4 | 0.40 (0.19–0.60) | disability/impairment | |
| rs4934396 | 90108138 | C10orf59 | 1.09 × 10−2 | 0.61 (0.05–1.18) | 6.64 × 10−3 | 0.33 (0.07–0.59) | 8.50 × 10−4 | 0.50 (0.18–0.82) | disorganization |
| rs11202748 | 90199318 | C10orf59 | 5.50 × 10−3 | 0.42 (0.10–0.74) | 2.24 × 10−2 | 0.19 (0.02–0.36) | 2.76 × 10−4 | 0.37 (0.16–0.58) | disorganization |
| rs10509546 | 90201633 | C10orf59 | 4.25 × 10−3 | 0.53 (0.14–0.92) | 2.13 × 10−2 | 0.23 (0.03–0.43) | 5.77 × 10−4 | 0.42 (0.17–0.67) | disorganization |
| rs10430719 | 91028961 | 4.75 × 10−2 | 0.32 (0.00–0.63) | 2.27 × 10−2 | 0.19 (0.01–0.37) | 3.33 × 10−4 | 0.37 (0.14–0.60) | prodromal | |
AddVal values from the UNPHASED output results. For quantitative traits, this value shows the estimated additive genetic value for this risk allele compared to the nonrisk allele, assuming a normally distributed trait and small deviations from the mean.
3′ downstream of NRG3, which is within the LD block containing the last 5 exons of NRG3.
Located in the gene deserts flanking NRG3.
We identified a strong association signal by using the “delusion” factor as the phenotype. Our primary analysis (allelic test in 285 trios) with UNPHASED gave the smallest p values at three consecutive SNPs located in a 13 kb interval (p = 7.26 × 10−7 at rs10883866 and rs10748842; p = 8.40 × 10−6 at rs6584400). We examined the genotyping quality of these three SNPs by using Illumina's BeadStudio software and found the results reliable (GenTrain scores: 0.94, 0.83, and 0.83, respectively; and none of the three SNPs violates Hardy-Weinberg equilibrium). Two of these SNPs (rs10883866 and rs10748842) are only 6.1 kb apart and in complete linkage disequilibrium (r2 = 1) and thus have the same p values. Hereafter, unless otherwise specified, we report the p value(s) for rs10883866 as the representative for this association signal. The result is study-wide significant, even under the most stringent Bonferroni correction (the study-wide cut-off p value = 3.93 × 10−6, for 9 quantitative phenotypes and 1414 SNPs). We performed analyses by QTDT (p = 3.0 × 10−7) and FBAT (p = 6.7 × 10−7) to ensure that the results are robust to the use of different analytical software packages. Furthermore, association analysis with our 173 independent cases replicated the signal (p = 1.55 × 10−2) and pointed to the same susceptibility allele. The combined p value (with 285 trios and 173 independent cases together) was 2.30 × 10−7. Further analysis with the genotypic test also showed significant results (family-based analysis, p = 2.63 × 10−5; 173 independent cases, p = 9.96 × 10−3; and combined samples, p = 3.86 × 10−6). The alleles associated with higher scores of the delusion factor are the minor alleles for all three SNPs. Detailed results are summarized in Table 4.
Table 4.
SNPs Strongly Associated with the Delusion Factor
|
Allelic Test (p Values) |
Genotypic Test (p Values) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
285 Trios |
173 Cases, Independent |
458 Cases, All |
1028 Persons, Combined |
285 Trios |
173 Cases, Independent |
458 Cases, All |
1028 Persons, Combined |
||||||
| SNP Name | Allele | Risk Allele | Freq. of Risk Allele | UNPHASED | FBAT | QTDT | |||||||
| rs10883866 | C/G | G | 0.11 | 7.26 × 10−7 | 6.7 × 10−7 | 3.0 × 10−7 | 1.55 × 10−2 | 5.27 × 10−6 | 2.30 × 10−7 | 2.63 × 10−5 | 9.96 × 10−3 | 8.36 × 10−5 | 3.86 × 10−6 |
| rs10748842 | T/C | C | 0.11 | 7.26 × 10−7 | 6.7 × 10−7 | 3.0 × 10−7 | 1.55 × 10−2 | 5.27 × 10−6 | 2.30 × 10−7 | 2.63 × 10−5 | 9.96 × 10−3 | 8.36 × 10−5 | 3.86 × 10−6 |
| rs6584400 | G/A | A | 0.13 | 8.40 × 10−6 | 1.8 × 10−5 | 3.0 × 10−6 | 1.97 × 10−2 | 1.51 × 10−5 | 1.59 × 10−6 | 2.29 × 10−4 | 5.28 × 10−2 | 1.85 × 10−4 | 2.36 × 10−5 |
In addition to theoretical approaches, study-wide significance level can be empirically estimated with permutation data. We performed 10,000 permutations for each of the 9 factors, and only in 27 permutations observed p values smaller than 2.30 × 10−7. Accordingly, we estimate the empirical study-wide significance level for 9 quantitative traits as p = 2.7 × 10−3.
The three SNPs with the strongest association with the delusion factor span a 13 kb region in intron 1 of the NRG3 gene. LD block structure around NRG3 derived from our AJ samples does not differ from results of the CEU samples in the HapMap project (Figure 4). The three associated SNPs reside in a ∼160 kb LD block containing the promoter, exon 1, and part of intron 1 of NRG3 (Figure 4).
Figure 4.
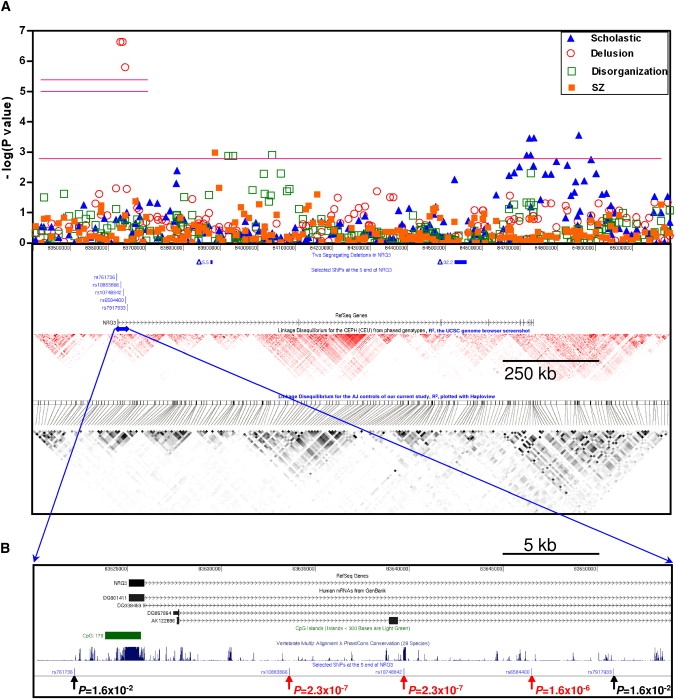
Results of Association Analyses and Selected Genomic Features in and around NRG3
(A) Association analysis results (combined samples) with SZ as the binary phenotype and three factors (scholastic, delusion, and disorganization) as quantitative traits. As in Figure 2, the two short upper horizontal pink lines represent the study-wide significance cut-off p values (either for all SNPs, higher, or for independent SNPs, lower) for all nine factors. The long horizontal pink line (p = 1.34 × 10−3) marks the 20th smallest p value from all 1414 SNPs of the ∼12.5 Mb region with any of the nine factors as the phenotype. We also plot the two segregating deletions (solid rectangles) and the three SNPs (and two flanking SNPs) with the most significant associations. We also provide the NRG3 gene and the LD block structure (based on r2 values) for the CEPH (CEU) from the HapMap project and the LD block structure for the AJ population from 487 controls of our current study to show the relative positions of these association results.
(B) A closer look at the 5′ region of NRG3. The p values of the same three SNPs (and two flanking SNPs) are indicated.
We also observed an excess of association signals in NRG3 overall. In addition to the three SNPs associated with the delusion factor, there are seven other SNPs either in NRG3 or its immediate flanking sequences that ranked in the top 20 best p value list (Table 3). Moreover, three other top 20 SNPs are located 388–697 kb outside of NRG3 in the two flanking gene deserts (Table 3). These 10 additional SNPs are not in LD with the three SNPs associated with the delusion factor (data not shown) and showed association with the scholastic, disorganization, and hallucination factors but not with delusion factor. Thus, taken together, 13 of the top 20 SNPs are located in or around NRG3 and show evidence for association with 4 of the SZ phenotypic factors.
Identification of Two Small Segregating Deletions in NRG3
As part of our fine mapping experiment, we detected Mendelian inconsistencies in the genotypes of 19 of the total 1536 SNPs genotyped. We considered that some of these might represent copy-number variations and therefore focused on regions with consecutive SNPs with Mendelian inconsistency. We found two regions that fit these criteria, both within NRG3. The genotyping plots and intensity data in those families exhibited reduced fluorescence intensity in some family members, compatible with the presence of segregating deletions. We performed quantitative PCR (data not shown) for one of the two regions to confirm and precisely locate the putative deletion. Long-range PCR followed by cloning and sequencing of the PCR products confirmed and defined the deletion break points in both regions. The more 5′ break point is a ∼5.5 kb deletion (hereafter referred to as Δ5.5) (starting at chr10:83,873,517 and ending at chr10:83,879,043) with an allele frequency of 2.62% in cases and 2.57% in controls. The more 3′ break point is a ∼32.2 kb deletion (hereafter referred to as Δ32.2) (starting at chr10:84,524,457 and ending at chr10:84,556,666) with an allele frequency of 0.88% in cases and 0.72% in controls. We sequenced amplified break point fragments from 12 unrelated individuals with Δ5.5 and 2 unrelated individuals with Δ32.2, and found the break point for each deletion to be identical in all samples. In our small sample, we did not find association between these deletions and SZ or any of the nine factors (data not shown), but our power to detect association signal for genetic variants with these frequencies is minimal.
Both of these inherited microdeletions are in NRG3; deletion Δ5.5 is in intron 1 and deletion Δ32.2 is in intron 3 (Figure 4). Deletion Δ32.2 includes an alternative noncoding first exon of NRG3, which is annotated in a human mRNA clone (AK098823)45 and confirmed by our RT-PCR experiment (data not shown).
Discussion
We conducted a peak-wide association fine mapping of a 12.5 Mb region on 10q22-q23 in the AJ population, a region identified in our previous genome-wide linkage analysis of SZ in the AJ.22 In addition to the binary phenotype of SZ, we tested nine quantitative trait phenotypes derived from principal components factor analysis. By using 285 parent-child trios, we found that a ∼13 kb region containing three SNPs in the 5′ region of NRG3 showed strong evidence of association with the “delusion” factor at a study-wide significance level. We replicated this association signal by using 173 unrelated SZ individuals. We also found multiple weaker association signals in other segments of NRG3 as well as two small deletions, one of which removes an alternative, first noncoding exon of NRG3.
Challenges for Identifying SZ Susceptibility Genes
Genetic heterogeneity, including both allelic and locus heterogeneity, is one of several challenges for discovery of genes and alleles contributing to human disease phenotypes. To minimize heterogeneity, we conducted our study only in the AJ population, where founder effects have been reported for both monogenic diseases and complex traits,46–50 and LD structure has shown this group to be somewhat more homogeneous than the outbred northern European population.51 A second challenge is phenotypic misclassification. To deal with this, we established a rigorous assessment protocol that included tape-recorded interviews performed by skilled diagnosticians and reviewed independently by two members of a consensus diagnosis committee.22 A third challenge involves the complexity of the SZ phenotypes; for example, these include positive and negative symptoms likely reflecting different central nervous system (CNS) dysfunctions that are not adequately described by a binary variable (SZ, yes/no).13,14 To account for this complexity, we incorporated more extensive phenotypic information (73 items from the consensus ratings and direct assessment interviews) and used principal components analysis to identify nine independent factors to serve as quantitative phenotypes in our analyses.
SZ as the Discrete Phenotype
When SZ was used as the discrete phenotype, several SNPs had nominal p values reaching the 0.05 significance level, but none reached our multiple test correction thresholds. The association signal in intron 2 of GRID1 previously reported by our group in a candidate gene-based study25 persisted in this current analysis at about the same level of significance (p = ∼0.01). This was not sufficiently strong to be reliably detected in our current analyses. Intriguingly, GRID1 recently has also been implicated as a SZ susceptibility gene in a case-control study (260 cases and 307 controls) in a Northern Han Chinese sample, with the most significant nominal p value of 1.1 × 10−5 (rs11591408, near exon 2).26 We did not genotype rs11591408 in this study and there is no information about the linkage disequilibrium or allele frequency of this SNP in the public databases. To summarize, by using SZ as a discrete phenotype, we found some nominally significant associations but none strong enough to withstand rigorous correction for multiple testing. We list our best results in Table 2 for future testing in independent samples.
Assuming validity of our linkage result, there are multiple reasons for failing to find an association signal for genuine susceptibility variants, including multiple causative genes in the same linkage interval; multiple (rare) responsible alleles in a single gene;52 interactions between genes/alleles; insufficient power; inability of regular SNP genotypes to represent the information of causative genetic variants (such as CNVs or inversions); inadequate marker coverage; genetic heterogeneity; and phenotypic heterogeneity. Despite the lack of significant association with the diagnosis of SZ, our results are useful for comparison with other studies and will allow for unbiased meta-analyses.
Nine Factors as the Quantitative Trait Phenotypes
It has long been argued that schizophrenia is not a nosological monolith but rather a heterogeneous collection of related phenotypes.12,14 The importance of careful delineation of phenotypes of diseases with genetic heterogeneity has been well addressed.53,54 To this end, we tested nine symptom factors as the quantitative phenotypes for our association study, with the expectation that these factors would provide more homogeneous and meaningful phenotypes for genetic analysis. In practice, this approach comes at the price of increasing the number of hypotheses tested.
With this approach, we identified a strong association at the 5′ region of NRG3 by using the “delusion” factor as the quantitative trait phenotype. The primary analysis (allelic test in 285 parent-child trios) yielded a significant p value (p = 7.26 × 10−7) that can sustain the most stringent study-wide Bonferroni correction for 9 quantitative phenotypes and 1414 SNPs. Most importantly, this association signal was replicated in our sample of 173 independent, unrelated SZ individuals of AJ descent. Analyses with combined family samples and independent cases yielded an even stronger signal (p = 2.30 × 10−7). Genotypic tests provided similar results (Table 4). We estimated the study-wide significance level to be p = 2.7 × 10−3 after 10,000 permutations.
We found several additional association signals in and around NRG3 (Figure 4). Including the 5′ and 3′ LD blocks extending from both ends, NRG3 spans a ∼1.5 Mb genomic region, or about 12% of the 12.5 Mb region we covered in our fine mapping study (13.1%, if we exclude the ∼1.06 Mb duplicated regions poorly covered by the SNPs we genotyped). Stimulated by our results implicating NRG3, we asked whether any other SNPs among our 20 most significant signals across all factors supported involvement of NRG3. The list of the 20 SNPs with the smallest p values in our factor analysis includes a total of 10 (50%) SNPs in NRG3. In addition to the 3 SNPs associated with the delusion factor, there are 7 SNPs that demonstrate suggestive evidence of association with 2 other factors (scholastic and disorganization). There are 3 more SNPs on the top 20 list located in the gene deserts flanking NRG3 (Table 3). The SNPs responsible for these different factor associations are not in LD with each of the others (data not shown). Although the association signals for these additional 10 SNPs fail to withstand multiple test correction, in aggregate this result is consistent with multiple causative variants in different regions of the NRG3 gene. Allelic heterogeneity is the rule in rare disorders and has also been observed in various common traits.55–59 The highly significant result for the delusion factor together with the multiple nominal associations with other factors strongly suggests that, in our population, multiple alleles of NRG3 play an important role in SZ. The biological basis of this effect could be the result of multiple alleles influencing the risk for SZ, each with some distinct genotype-phenotype correlation, or it could be that NRG3 variants influence the phenotypic features manifested by individuals who have SZ because of a constellation of other genetic and environmental variables. Determining between these or other possibilities will require identification of the causative variants, elucidation of their functional consequences on NRG3 or other genes, and a better understanding of the biological functions of NRG3 and its multiple spliceforms.
We obtained a 9 factor solution based on 73 items (Table 1) of the consensus ratings and direct assessment interviews. This number of factors is larger than identified in other analyses of SZ in part because we scored a larger number of items including those that may reflect developmental problems and premorbid features. Each factor has a measurable heritability in our population ranging from 0.27 to 0.61, and each factor is independent of the other eight.28 Many of the factors, such as the delusion, negative, disorganization, hallucination, and affective, have been derived in similar forms in other studies of SZ.60–62 It is noteworthy that the “delusion” factor which has the most prominent association signal in our analysis is roughly equivalent to the classic Schneiderian “first-rank” symptoms,63 or “nuclear syndrome” factors in other studies, where sib-pair correlations have been demonstrated.64,65 These first-rank symptoms have been given particular weight for making diagnosis of SZ and might be expected to have etiological significance.64,65
Identification of NRG3 Microdeletions
CNVs, including small deletions, are plausible disease-causing variants that are easily overlooked but can be identified by careful analysis of SNP genotyping data. Taking advantage of the inheritance information in our family sample, genotype information, and intensity variation of SNP calls, we discovered, confirmed, and precisely delineated two novel microdeletions in NRG3. Although neither of the deletions we identified showed association with SZ or any of the nine factors, our power to detect association signal for genetic variants of such low frequencies was limited. Intriguingly, the Δ5.5 deletion is in intron 1 where we find an association signal, and the Δ32.2 deletion in intron 3 removes an alternative, first noncoding exon of NRG3. In future studies, it will be important to investigate the potential impact of these CNVs on the expression and function of NRG3.
Analogy of the Association Signal for NRG1 and NRG3
Associations of variants in NRG1 with SZ have been replicated by several groups.66,67 NRG3 is a paralog of NRG1 with an expression pattern that is more limited to the CNS68 and, like NRG1, encodes a single-pass membrane protein with an N-terminal EGF domain on the extracellular side of the plasma membrane. When we designed our SNP fine mapping project as an unbiased approach, we selected SNPs to evenly cover the whole ∼12.5 Mb region without taking into account the location of known genes (see Subjects and Methods). It is intriguing that our best association signal mapped to a LD block at the 5′ end of NRG3, analogous to the results implicating NRG1.66
NRG1 spans a ∼1.25 Mb genomic region, with a large first intron and a ∼300 kb LD block containing the promoter, exon 1, and part of the intron 1. The initial study implicating NRG1 as a SZ susceptibility gene showed association to a risk haplotype located at the 5′ end of the gene.66 Several studies have replicated the association signal, and almost all of the published risk SNPs/haplotypes in northern Europeans were mapped to the 5′ end of NRG1.67 Recently, an alternative NRG1 first exon, 479 base-pairs upstream of the standard exon 1 and also contained in the 5′ LD block, was identified and shown to be transcribed only in the CNS. Increased levels of this CNS-specific NRG1 transcript in human postmortem hippocampus were shown to be associated with the SNP risk allele for SZ.69–71 How these susceptibility SNPs/haplotypes at the 5′ end of NRG1 lead to functional changes in NRG1 isoforms is currently under vigorous investigation.
The NRG3 gene spans a ∼1.2 Mb genomic region and, like NRG1, has a large first intron and a large 5′ LD block (∼160 kb) containing the promoter, exon 1, and part of the intron 1. The association signal we report here is at the 5′ end of intron 1 within 10 kb of exon 1. An alternative transcription start site, located between the original exon 1 and the association signal we report here, has been identified in human fetal brain and produces a neural-specific NRG3 transcript capable of affecting oligodendrocyte survival in vitro.72 Additionally, there are several potentially functional elements in and around the region of our best association signal that could harbor susceptibility variants affecting NRG3 expression or function, including the proximal promoter, exon 1, alternative transcription start site(s), possible alternative exons, and conserved noncoding elements. We sequenced all the exons and axon-intron junction regions in 15 unrelated AJ SZ probands from our original linkage pedigrees22 and found no sequence variants that could obviously alter NRG3 function (data not shown). It will be important to conduct more comprehensive sequencing of NRG3 in the future. Studies to clarify the transcription profile and function of NRG3 are also warranted.
Although little has been published regarding NRG3, its function shares some common pathways with NRG1 in the central nervous system. The full-length human NRG3 protein, 720 amino acids in length, has an extracellular EGF-like domain (31% identity with the NRG1 EGF-like domain) that has been shown to be functional.68,72 The only known binding receptor for NRG3 is erbB4, although NRG1 binds to both erbB3 and erbB4.73,74 SZ-related traits were observed in various genetically modified mice with mutation of Nrg1 or erbB4 (but not erbB3).75 A recent association analysis reported suggestive evidence of interaction between NRG1 and NRG3 on their effect of SZ susceptibility.27 During the preparation of this manuscript, a genome-wide association study of SZ in an Israeli AJ sample was published.76 Although the focus of this report was on an association of a variant in RELN limited to females with SZ, we note that the 206th best SNP (the top 0.04%) out of 510,552 genotyped in the overall sample and the 11th best (the top 0.002%) in the male sample was rs17559044. This SNP is located in intron 1 of NRG3, 228.9 kb 3′ from our strongest association signal for the delusion factor and 23.7 kb 5′ from our best association signal for the diagnosis of SZ in NRG3. We could find no evidence for an effect of sex on our signal. While this manuscript was being revised, Wang et al.77 reported association of SZ in a Han Chinese population in Taiwan with two SNPs (rs1937970 and rs677221) located in intron 2 of NRG3, approximately 0.6 Mb telomeric of our peak signal. These two SNPs were genotyped in our study and did not show significant association in our samples (Tables S1 and S2).
In summary, we performed SNP fine mapping of SZ and related phenotypes at our previous linkage peak. With the “delusion” factor as quantitative trait phenotype, we found a strong association signal at the 5′ end of NRG3 when analyzing 285 parent-child trios, and we subsequently replicated the signal with 173 independent SZ individuals. Our results suggest that NRG3 influences the SZ risk and presentation in multiple ways, the clarification of which requires further investigation. The analogy with NRG1 results and the location of possible NRG3 functional elements in and around the implicated risk region support our conclusion that alleles of NRG3 play a role in the etiology and/or phenotypic manifestation of SZ. Further efforts to replicate the association result with other independent samples, to sequence the 5′ end region with reasonable coverage, to clarify the transcription profile, and to study the function of NRG3 are warranted.
Supplemental Data
Supplemental Data include two tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CEPH, http://www.cephb.fr
HapMap, http://www.hapmap.org
Johns Hopkins Epidemiology-Genetics Program in Psychiatry, http://www.hopkinsmedicine.org/epigen
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
The UNPHASED software, http://www.mrc-bsu.cam.ac.uk/personal/frank/software/unphased/
Acknowledgments
We are indebted to all the patients and controls for participation in this research. Genotyping services were provided by the Johns Hopkins Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number NO1-HG-65403. This study was funded by National Institutes of Mental Health grant RO1MH057314 and RO1MH068406, and by the Wasie Foundation (A.E.P.). This project also was funded in part by the National Alliance for Research on Schizophrenia and Depression (D.V. and D.A.). None of the authors have any conflict of interest concerning this manuscript. We also thank Sandy Muscelli for assistance with the manuscript.
References
- 1.Eaton W.W. Epidemiology of schizophrenia. Epidemiol. Rev. 1985;7:105–126. doi: 10.1093/oxfordjournals.epirev.a036278. [DOI] [PubMed] [Google Scholar]
- 2.McGue M., Gottesman I.I. The genetic epidemiology of schizophrenia and the design of linkage studies. Eur. Arch. Psychiatry Clin. Neurosci. 1991;240:174–181. doi: 10.1007/BF02190760. [DOI] [PubMed] [Google Scholar]
- 3.Tsuang M.T., Gilbertson M.W., Faraone S.V. The genetics of schizophrenia. Current knowledge and future directions. Schizophr. Res. 1991;4:157–171. doi: 10.1016/0920-9964(91)90031-l. [DOI] [PubMed] [Google Scholar]
- 4.Cardno A.G., Gottesman I.I. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am. J. Med. Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- 5.Owen M.J., Craddock N., Jablensky A. The genetic deconstruction of psychosis. Schizophr. Bull. 2007;33:905–911. doi: 10.1093/schbul/sbm053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badner J.A., Gershon E.S. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol. Psychiatry. 2002;7:405–411. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]
- 7.Lewis C.M., Levinson D.F., Wise L.H., DeLisi L.E., Straub R.E., Hovatta I., Williams N.M., Schwab S.G., Pulver A.E., Faraone S.V. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am. J. Hum. Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crow T.J. How and why genetic linkage has not solved the problem of psychosis: review and hypothesis. Am. J. Psychiatry. 2007;164:13–21. doi: 10.1176/ajp.2007.164.1.13. [DOI] [PubMed] [Google Scholar]
- 9.Norton N., Williams H.J., Owen M.J. An update on the genetics of schizophrenia. Curr. Opin. Psychiatry. 2006;19:158–164. doi: 10.1097/01.yco.0000214341.52249.59. [DOI] [PubMed] [Google Scholar]
- 10.Sanders A.R., Duan J., Levinson D.F., Shi J., He D., Hou C., Burrell G.J., Rice J.P., Nertney D.A., Olincy A. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am. J. Psychiatry. 2008;165:497–506. doi: 10.1176/appi.ajp.2007.07101573. [DOI] [PubMed] [Google Scholar]
- 11.St Clair D., Blackwood D., Muir W., Carothers A., Walker M., Spowart G., Gosden C., Evans H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 12.Fanous A.H., Kendler K.S. Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Mol. Psychiatry. 2005;10:6–13. doi: 10.1038/sj.mp.4001571. [DOI] [PubMed] [Google Scholar]
- 13.Craddock N., Owen M.J. The beginning of the end for the Kraepelinian dichotomy. Br. J. Psychiatry. 2005;186:364–366. doi: 10.1192/bjp.186.5.364. [DOI] [PubMed] [Google Scholar]
- 14.Jablensky A. Subtyping schizophrenia: implications for genetic research. Mol. Psychiatry. 2006;11:815–836. doi: 10.1038/sj.mp.4001857. [DOI] [PubMed] [Google Scholar]
- 15.Serretti A., Lilli R., Lorenzi C., Lattuada E., Smeraldi E. DRD4 exon 3 variants associated with delusional symptomatology in major psychoses: a study on 2,011 affected subjects. Am. J. Med. Genet. 2001;105:283–290. doi: 10.1002/ajmg.1321. [DOI] [PubMed] [Google Scholar]
- 16.Wilcox M.A., Faraone S.V., Su J., Van Eerdewegh P., Tsuang M.T. Genome scan of three quantitative traits in schizophrenia pedigrees. Biol. Psychiatry. 2002;52:847–854. doi: 10.1016/s0006-3223(02)01465-8. [DOI] [PubMed] [Google Scholar]
- 17.Fanous A.H., van den Oord E.J., Riley B.P., Aggen S.H., Neale M.C., O'Neill F.A., Walsh D., Kendler K.S. Relationship between a high-risk haplotype in the DTNBP1 (dysbindin) gene and clinical features of schizophrenia. Am. J. Psychiatry. 2005;162:1824–1832. doi: 10.1176/appi.ajp.162.10.1824. [DOI] [PubMed] [Google Scholar]
- 18.Fanous A.H., Neale M.C., Webb B.T., Straub R.E., Amdur R.L., O'Neill F.A., Walsh D., Riley B.P., Kendler K.S. A genome-wide scan for modifier loci in schizophrenia. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144:589–595. doi: 10.1002/ajmg.b.30442. [DOI] [PubMed] [Google Scholar]
- 19.Hall J., Whalley H.C., Job D.E., Baig B.J., McIntosh A.M., Evans K.L., Thomson P.A., Porteous D.J., Cunningham-Owens D.G., Johnstone E.C. A neuregulin 1 variant associated with abnormal cortical function and psychotic symptoms. Nat. Neurosci. 2006;9:1477–1478. doi: 10.1038/nn1795. [DOI] [PubMed] [Google Scholar]
- 20.Freedman R., Adams C.E., Adler L.E., Bickford P.C., Gault J., Harris J.G., Nagamoto H.T., Olincy A., Ross R.G., Stevens K.E. Inhibitory neurophysiological deficit as a phenotype for genetic investigation of schizophrenia. Am. J. Med. Genet. 2000;97:58–64. doi: 10.1002/(sici)1096-8628(200021)97:1<58::aid-ajmg8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 21.Nicodemus K.K., Luna A., Vakkalanka R., Goldberg T., Egan M., Straub R.E., Weinberger D.R. Further evidence for association between ErbB4 and schizophrenia and influence on cognitive intermediate phenotypes in healthy controls. Mol. Psychiatry. 2006;11:1062–1065. doi: 10.1038/sj.mp.4001878. [DOI] [PubMed] [Google Scholar]
- 22.Fallin M.D., Lasseter V.K., Wolyniec P.S., McGrath J.A., Nestadt G., Valle D., Liang K.Y., Pulver A.E. Genomewide linkage scan for schizophrenia susceptibility loci among Ashkenazi Jewish families shows evidence of linkage on chromosome 10q22. Am. J. Hum. Genet. 2003;73:601–611. doi: 10.1086/378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faraone S.V., Hwu H.G., Liu C.M., Chen W.J., Tsuang M.M., Liu S.K., Shieh M.H., Hwang T.J., Ou-Yang W.C., Chen C.Y. Genome scan of Han Chinese schizophrenia families from Taiwan: confirmation of linkage to 10q22.3. Am. J. Psychiatry. 2006;163:1760–1766. doi: 10.1176/ajp.2006.163.10.1760. [DOI] [PubMed] [Google Scholar]
- 24.Balciuniene J., Feng N., Iyadurai K., Hirsch B., Charnas L., Bill B.R., Easterday M.C., Staaf J., Oseth L., Czapansky-Beilman D. Recurrent 10q22-q23 deletions: a genomic disorder on 10q associated with cognitive and behavioral abnormalities. Am. J. Hum. Genet. 2007;80:938–947. doi: 10.1086/513607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fallin M.D., Lasseter V.K., Avramopoulos D., Nicodemus K.K., Wolyniec P.S., McGrath J.A., Steel G., Nestadt G., Liang K.Y., Huganir R.L. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am. J. Hum. Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo S.Z., Huang K., Shi Y.Y., Tang W., Zhou J., Feng G.Y., Zhu S.M., Liu H.J., Chen Y., Sun X.D. A case-control association study between the GRID1 gene and schizophrenia in the Chinese Northern Han population. Schizophr. Res. 2007;93:385–390. doi: 10.1016/j.schres.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Benzel I., Bansal A., Browning B.L., Galwey N.W., Maycox P.R., McGinnis R., Smart D., St Clair D., Yates P., Purvis I. Interactions among genes in the ErbB-Neuregulin signalling network are associated with increased susceptibility to schizophrenia. Behav. Brain Funct. 2007;3:31. doi: 10.1186/1744-9081-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGrath J.A., Avramopoulos D., Lasseter V.K., Wolyniec P.S., Fallin M.D., Liang K.-Y., Nestadt G., Thornquist M.H., Luke J.R., Chen P.-L. Familiality of novel factorial dimensions of schizophrenia. Arch. Gen. Psychiatry. 2009 doi: 10.1001/archgenpsychiatry.2009.56. in press. [DOI] [PubMed] [Google Scholar]
- 29.American Psychiatric Association . Fourth Edition. American Psychiatric Association; Washington, DC: 1994. Diagnostic and Statistical Manual of Mental Disorders. [Google Scholar]
- 30.Cannon-Spoor H.E., Potkin S.G., Wyatt R.J. Measurement of premorbid adjustment in chronic schizophrenia. Schizophr. Bull. 1982;8:470–484. doi: 10.1093/schbul/8.3.470. [DOI] [PubMed] [Google Scholar]
- 31.George V.T., Elston R.C. Generalized modulus power transformations. Commun. Statist. Theory Meth. 1988;17:2933–2952. [Google Scholar]
- 32.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 34.Dausset J., Cann H., Cohen D., Lathrop M., Lalouel J.M., White R. Centre d'etude du polymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics. 1990;6:575–577. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- 35.Dudbridge, F. (2006) UNPHASED user guide. Technical Report 2006/5, MRC Biostatistics Unit, Cambridge, UK.
- 36.Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum. Hered. 2008;66:87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spielman R.S., McGinnis R.E., Ewens W.J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am. J. Hum. Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- 38.Laird N.M., Horvath S., Xu X. Implementing a unified approach to family-based tests of association. Genet. Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 39.Abecasis G.R., Cardon L.R., Cookson W.O. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 42.Chang Y.P., Liu X., Kim J.D., Ikeda M.A., Layton M.R., Weder A.B., Cooper R.S., Kardia S.L., Rao D.C., Hunt S.C. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am. J. Hum. Genet. 2007;80:253–264. doi: 10.1086/510918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicodemus K.K., Liu W., Chase G.A., Tsai Y.Y., Fallin M.D. Comparison of type I error for multiple test corrections in large single-nucleotide polymorphism studies using principal components versus haplotype blocking algorithms. BMC Genet. 2005;6(Suppl 1):S78. doi: 10.1186/1471-2156-6-S1-S78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabriel S.B., Schaffner S.F., Nguyen H., Moore J.M., Roy J., Blumenstiel B., Higgins J., DeFelice M., Lochner A., Faggart M. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 45.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank: update. Nucleic Acids Res. 2004;32:D23–D26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ostrer H. A genetic profile of contemporary Jewish populations. Nat. Rev. Genet. 2001;2:891–898. doi: 10.1038/35098506. [DOI] [PubMed] [Google Scholar]
- 47.Neuhausen S.L., Godwin A.K., Gershoni-Baruch R., Schubert E., Garber J., Stoppa-Lyonnet D., Olah E., Csokay B., Serova O., Lalloo F. Haplotype and phenotype analysis of nine recurrent BRCA2 mutations in 111 families: results of an international study. Am. J. Hum. Genet. 1998;62:1381–1388. doi: 10.1086/301885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rennert H., Bercovich D., Hubert A., Abeliovich D., Rozovsky U., Bar-Shira A., Soloviov S., Schreiber L., Matzkin H., Rennert G. A novel founder mutation in the RNASEL gene, 471delAAAG, is associated with prostate cancer in Ashkenazi Jews. Am. J. Hum. Genet. 2002;71:981–984. doi: 10.1086/342775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stern H.S., Viertelhausen S., Hunter A.G., O'Rourke K., Cappelli M., Perras H., Serfas K., Blumenthall A., Dewar D., Baumann E. APC I1307K increases risk of transition from polyp to colorectal carcinoma in Ashkenazi Jews. Gastroenterology. 2001;120:392–400. doi: 10.1053/gast.2001.21170. [DOI] [PubMed] [Google Scholar]
- 50.Sugimura K., Taylor K.D., Lin Y.C., Hang T., Wang D., Tang Y.M., Fischel-Ghodsian N., Targan S.R., Rotter J.I., Yang H. A novel NOD2/CARD15 haplotype conferring risk for Crohn disease in Ashkenazi Jews. Am. J. Hum. Genet. 2003;72:509–518. doi: 10.1086/367848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Service S., DeYoung J., Karayiorgou M., Roos J.L., Pretorious H., Bedoya G., Ospina J., Ruiz-Linares A., Macedo A., Palha J.A. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat. Genet. 2006;38:556–560. doi: 10.1038/ng1770. [DOI] [PubMed] [Google Scholar]
- 52.McClellan J.M., Susser E., King M.C. Schizophrenia: a common disease caused by multiple rare alleles. Br. J. Psychiatry. 2007;190:194–199. doi: 10.1192/bjp.bp.106.025585. [DOI] [PubMed] [Google Scholar]
- 53.McKusick V.A. On lumpers and splitters, or the nosology of genetic disease. Perspect. Biol. Med. 1969;12:298–312. doi: 10.1353/pbm.1969.0039. [DOI] [PubMed] [Google Scholar]
- 54.Childs B., Finucci J.M., Preston M.S., Pulver A.E. Human behavior genetics. Adv. Hum. Genet. 1976;7:57–97. doi: 10.1007/978-1-4757-0659-8_2. [DOI] [PubMed] [Google Scholar]
- 55.Watson M.S., Cutting G.R., Desnick R.J., Driscoll D.A., Klinger K., Mennuti M., Palomaki G.E., Popovich B.W., Pratt V.M., Rohlfs E.M. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet. Med. 2004;6:387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lakhani V.T., You Y.N., Wells S.A. The multiple endocrine neoplasia syndromes. Annu. Rev. Med. 2007;58:253–265. doi: 10.1146/annurev.med.58.100305.115303. [DOI] [PubMed] [Google Scholar]
- 57.Scriver C.R., Hurtubise M., Konecki D., Phommarinh M., Prevost L., Erlandsen H., Stevens R., Waters P.J., Ryan S., McDonald D. PAHdb 2003: what a locus-specific knowledgebase can do. Hum. Mutat. 2003;21:333–344. doi: 10.1002/humu.10200. [DOI] [PubMed] [Google Scholar]
- 58.Bobadilla J.L., Macek M., Fine J.P., Farrell P.M. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum. Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 59.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 60.Peralta V., Cuesta M.J. How many and which are the psychopathological dimensions in schizophrenia? Issues influencing their ascertainment. Schizophr. Res. 2001;49:269–285. doi: 10.1016/s0920-9964(00)00071-2. [DOI] [PubMed] [Google Scholar]
- 61.Cardno A.G., Jones L.A., Murphy K.C., Sanders R.D., Asherson P., Owen M.J., McGuffin P. Dimensions of psychosis in affected sibling pairs. Schizophr. Bull. 1999;25:841–850. doi: 10.1093/oxfordjournals.schbul.a033423. [DOI] [PubMed] [Google Scholar]
- 62.McGrath J.A., Nestadt G., Liang K.Y., Lasseter V.K., Wolyniec P.S., Fallin M.D., Thornquist M.H., Luke J.R., Pulver A.E. Five latent factors underlying schizophrenia: analysis and relationship to illnesses in relatives. Schizophr. Bull. 2004;30:855–873. doi: 10.1093/oxfordjournals.schbul.a007138. [DOI] [PubMed] [Google Scholar]
- 63.Schneider K. Grune & Stratton; New York: 1959. Clinical Psychopathology (transl. M.W. Hamilton) [Google Scholar]
- 64.Loftus J., Delisi L.E., Crow T.J. Factor structure and familiality of first-rank symptoms in sibling pairs with schizophrenia and schizoaffective disorder. Br. J. Psychiatry. 2000;177:15–19. doi: 10.1192/bjp.177.1.15. [DOI] [PubMed] [Google Scholar]
- 65.Cardno A.G., Sham P.C., Farmer A.E., Murray R.M., McGuffin P. Heritability of Schneider's first-rank symptoms. Br. J. Psychiatry. 2002;180:35–38. doi: 10.1192/bjp.180.1.35. [DOI] [PubMed] [Google Scholar]
- 66.Stefansson H., Sigurdsson E., Steinthorsdottir V., Bjornsdottir S., Sigmundsson T., Ghosh S., Brynjolfsson J., Gunnarsdottir S., Ivarsson O., Chou T.T. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Munafo M.R., Thiselton D.L., Clark T.G., Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol. Psychiatry. 2006;11:539–546. doi: 10.1038/sj.mp.4001817. [DOI] [PubMed] [Google Scholar]
- 68.Zhang D., Sliwkowski M.X., Mark M., Frantz G., Akita R., Sun Y., Hillan K., Crowley C., Brush J., Godowski P.J. Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA. 1997;94:9562–9567. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steinthorsdottir V., Stefansson H., Ghosh S., Birgisdottir B., Bjornsdottir S., Fasquel A.C., Olafsson O., Stefansson K., Gulcher J.R. Multiple novel transcription initiation sites for NRG1. Gene. 2004;342:97–105. doi: 10.1016/j.gene.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 70.Law A.J., Lipska B.K., Weickert C.S., Hyde T.M., Straub R.E., Hashimoto R., Harrison P.J., Kleinman J.E., Weinberger D.R. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc. Natl. Acad. Sci. USA. 2006;103:6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan W., Wang Y., Gold B., Chen J., Dean M., Harrison P.J., Weinberger D.R., Law A.J. Molecular cloning of a brain-specific, developmentally regulated neuregulin 1 (NRG1) isoform and identification of a functional promoter variant associated with schizophrenia. J. Biol. Chem. 2007;282:24343–24351. doi: 10.1074/jbc.M702953200. [DOI] [PubMed] [Google Scholar]
- 72.Carteron C., Ferrer-Montiel A., Cabedo H. Characterization of a neural-specific splicing form of the human neuregulin 3 gene involved in oligodendrocyte survival. J. Cell Sci. 2006;119:898–909. doi: 10.1242/jcs.02799. [DOI] [PubMed] [Google Scholar]
- 73.Hobbs S.S., Coffing S.L., Le A.T., Cameron E.M., Williams E.E., Andrew M., Blommel E.N., Hammer R.P., Chang H., Riese D.J. Neuregulin isoforms exhibit distinct patterns of ErbB family receptor activation. Oncogene. 2002;21:8442–8452. doi: 10.1038/sj.onc.1205960. [DOI] [PubMed] [Google Scholar]
- 74.Falls D.L. Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 75.O'Tuathaigh C.M., Babovic D., O'Meara G., Clifford J.J., Croke D.T., Waddington J.L. Susceptibility genes for schizophrenia: characterisation of mutant mouse models at the level of phenotypic behaviour. Neurosci. Biobehav. Rev. 2007;31:60–78. doi: 10.1016/j.neubiorev.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 76.Shifman S., Johannesson M., Bronstein M., Chen S.X., Collier D.A., Craddock N.J., Kendler K.S., Li T., O'Donovan M., O'Neill F.A. Genome-wide association identifies a common variant in the Reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 2008;4:e28. doi: 10.1371/journal.pgen.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y.C., Chen J.Y., Chen M.L., Chen C.H., Lai I.C., Chen T.T., Hong C.J., Tsai S.J., Liou Y.J. Neuregulin 3 genetic variations and susceptibility to schizophrenia in a Chinese population. Biol. Psychiatry. 2008;64:1093–1096. doi: 10.1016/j.biopsych.2008.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.