

Abstract
Alzheimer's disease (AD) is a genetically complex and heterogeneous disorder. To date four genes have been established to either cause early-onset autosomal-dominant AD (APP, PSEN1, and PSEN21–4) or to increase susceptibility for late-onset AD (APOE5). However, the heritability of late-onset AD is as high as 80%,6 and much of the phenotypic variance remains unexplained to date. We performed a genome-wide association (GWA) analysis using 484,522 single-nucleotide polymorphisms (SNPs) on a large (1,376 samples from 410 families) sample of AD families of self-reported European descent. We identified five SNPs showing either significant or marginally significant genome-wide association with a multivariate phenotype combining affection status and onset age. One of these signals (p = 5.7 × 10−14) was elicited by SNP rs4420638 and probably reflects APOE-ɛ4, which maps 11 kb proximal (r2 = 0.78). The other four signals were tested in three additional independent AD family samples composed of nearly 2700 individuals from almost 900 families. Two of these SNPs showed significant association in the replication samples (combined p values 0.007 and 0.00002). The SNP (rs11159647, on chromosome 14q31) with the strongest association signal also showed evidence of association with the same allele in GWA data generated in an independent sample of ∼1,400 AD cases and controls (p = 0.04). Although the precise identity of the underlying locus(i) remains elusive, our study provides compelling evidence for the existence of at least one previously undescribed AD gene that, like APOE-ɛ4, primarily acts as a modifier of onset age.
Main Text
In contrast to early-onset autosomal-dominant Alzheimer's disease (AD [MIM 104300]), late-onset AD usually shows less obvious or no apparent familial aggregation (“sporadic AD”). Risk for late-onset AD is probably influenced by an array of common risk alleles distributed across different genes affecting a variety of biochemical pathways affecting both the etiology and pathogenesis of AD. Although the identity and total number of these genes remain elusive, recent estimates suggest that together they have a large impact on disease predisposition in the general population.6 In the attempt to identify the remaining AD susceptibility genes, a large body of evidence has accrued over the past 30 years, represented by well over 1000 publications genetically implicating or excluding potential risk factors, the vast majority of which were tested as functional and/or positional candidate genes.7 However, with the exception of the ɛ4-allele of APOE (MIM 107741), these efforts have mostly led to inconsistent findings, although some polymorphisms show significant but modest (∼1.25) summary odds ratios by meta-analysis (for an up-to-date overview of AD genetic association studies see the “AlzGene” database7). Recently, three genome-wide association analyses, all using case-control designs, have been published for AD.8–10 All three studies detected highly significant association at the APOE locus. In addition, they reported the discovery of a number of additional putative AD variants of small effect size, which await independent replication by other groups.7 Here, we set out to identify additional AD genes by performing whole-genome association analysis using 500,668 SNPs on the GeneChip Human Mapping 500K Array Set (Affymetrix, Santa Clara, CA, USA) in four well-characterized samples of AD families.
All data sets tested in this project were originally collected for the study of genetic factors in AD with family-based methods (see Table S1 available online for a detailed summary of sample characteristics). All studies were approved by the institutional review boards of the appropriate institutions, and all subjects gave informed consent for their participation. With the exception of the CAG sample (see below), the majority of pedigrees analyzed here were nuclear families ascertained on the basis of multiple affecteds, generally lacking parental genotypes. In addition to containing at least one affected relative pair, many pedigrees also had DNA available from additional affected or unaffected individuals. These were mostly siblings, and only a minority of additional subjects stemmed from more extended branches (most of these are part of the NIMH sample). The diagnosis of “definite,” “probable,” or “possible” AD was made according to NINCDS/ADRDA11 criteria in all samples. Age of onset for all AD cases was determined by a clinician on the basis of an interview with a knowledgeable informant and review of any available records.
NIMH Families
This sample was collected as part of the National Institute of Mental Health Genetics Initiative Study12 and comprised a total of 1528 subjects from 457 families. Only families in which all affected family members showed an onset age ≥50 years, and in which DNA was available from at least two affected family members, were included in these analyses, i.e., 1439 individuals from 436 families. Of these, 1376 individuals from 410 families were of self-reported European ancestry and used for the initial 500K screening. Fifty-eight individuals from 24 families were of African descent and were included in the follow-up analyses.
NIA and NCRAD Families
Both of these data sets were obtained from the National Repository of Research on Alzheimer's Disease (NCRAD), and ascertainment and collection details can be found at the NCRAD website. For this study, we used families of self-reported European ancestry with DNA available from at least two first-degree relatives (concordant or discordant) and in which all individuals affected with AD showed onset ages ≥50 years. For the NIA collection, this comprised 1040 samples from 329 pedigrees, and for NCRAD, this comprised 1108 samples from 331 pedigrees.
CAG Families
Samples in this data set were recruited under the auspices of the “Consortium on Alzheimer's Genetics” (see Bertram et al.13 for more details). Probands were included only if they had at least one unaffected living sibling willing to participate in this study. As for the other replication samples, only families of self-reported European ancestry and with onset ages ≥50 years were included here, i.e., 483 individuals from 215 sibships.
We genotyped 500,668 SNPs of the GeneChip Human Mapping 500K Array Set in 1505 individuals comprising the publicly available NIMH AD genetics population, using two chips (Nsp and Sty) that each assayed approximately 250,000 SNPs per sample. Modifications to the manufacturer's protocol (see below and Supplemental Data) increased the quality and quantity of data obtained from each chip assay. Genotyping was carried out according to the manufacturer's protocol except for the following modifications: Restriction enzyme digestion, ligation, PCR, and purification were completed in 96-well plates containing 92 samples and four blanks. The PCR normalization step was performed with a Biomek F/X robot. After normalization, PCR products were divided into four separate 96-well plates each containing only 23 samples. Both the fragmentation and labeling steps were performed on 23 samples at a time, while a constant temperature with cold blocks was maintained. For the hybridization step, samples were denatured in hybridization cocktail for 10 min at 99°C and 2 min at 49°C with an MJ Tetrad, then transferred immediately to an external heating block kept at a constant temperature of 49°C. Prior to sample injection, the 500K arrays were warmed in hybridization ovens at 49°C for at least 10 min. During sample injection the arrays were maintained at 40°C–49°C and were immediately returned to the hybridization oven for 19 to 27 hr of incubation at 49°C. Posthybridization wash, staining, and detection were performed in accordance with the manufacturer's protocol. The average call rates increased from 87.5% to 97.3% and from 94.8% to 96.1% for Nsp and Sty arrays, respectively. Out of the entire sample, data from only eight arrays on a total of five DNA samples failed to exceed a 93% call-rate threshold necessary to be included in the analyses. Genotype calls were based on the Bayesian Robust Linear Model with Mahalanobis Distance algorithm (BRLMM14) for which we developed an improved protocol that led to greater call rates without affecting accuracy or reproducibility of the data and that was used here (see below). Overall, our protocol achieved an average SNP genotype call rate of 98.95%. SNPs with genotype call rates below 90% (5758 markers [1.1%]) were excluded. In addition, we excluded all SNPs located on the X chromosome (10,388 markers [2.1%], resulting in 484,522 markers used in the whole-genome association analyses. SNPs on the X chromosome were excluded because there is currently no method available for association testing of these markers in family-based settings.
We accepted only genotype calls passing a stringent quality-control threshold in which 93% of the SNPs on a 250K array yielded a genotype (with the DM algorithm at a confidence threshold of 0.33). Of the 3010 500K GeneChips necessary to complete genotyping of our family-based sample set, only eight chips failed to meet or exceed the 93% call-rate threshold. The DM algorithm calculates genotypes for one sample at a time, relying on assumptions about the behavior of SNP allele signals. However, an alternative genotype-calling algorithm was recently developed by Rabbee and Speed termed Bayesian Robust Linear Modeling using Mahalanobis Distance (BRLMM14). The BRLMM method simultaneously analyzes data from multiple chips, by calling genotypes via multiple-sample cluster analysis. BRLMM accounts for probe effects on variation in allele signal intensity of individual SNPs in making genotype calls. This new algorithm is an improvement over DM in terms of overall call rates, accuracy, and detection of heterozygous genotypes. Both Affymetrix and The Broad Institute (MIT/Harvard) have shown improved efficacy of genotyping calling by using BRLMM on their data sets.
We compared the DM and BRLMM genotype-calling algorithms with respect to call rates, accuracy, and concordance on our 500K data set. Initial analysis of our data with BRLMM increased the number of heterozygous genotypes (Figure S1), as well as the total number of genotypes called (data not shown). Accuracy of called genotypes was assessed via inheritance-error checks on replicate data with varying DM-derived call rates, collected for a family trio (mother, father, and child) contained in our data set. Inheritance errors consistently decreased with increasing initial DM call rate (Table S2), and even fewer inheritance errors were observed when BRLMM analysis was applied. Overall, genotype calls made by DM and BRLMM were in close agreement with one another, and the concordance increased with data from chips having higher initial DM call rates, indicating that higher call-rate data is more reliable (Figure S2A). Although BRLMM was able to make calls on a significant number of SNPs that were previously not called with DM, we did observe the reverse scenario as well, albeit with a much lower number of SNPs (Figure S2B).
Because the BRLMM algorithm processes chips in batches and uses a clustering algorithm to make genotype calls, we determined whether batch size and/or batch composition had an effect on call rate. Experiments were carried out with batch sizes of 50 and 100 chip-data CEL files. We tested samples with moderate (93%), good (95%), and excellent (98%) chip call rates1 and processed them in varying batch environments. Single test samples (e.g., 93% initial DM call rate) were grouped with samples that had (1) like or similar call rates (e.g., 93%), (2) mixed call rates (samples with call rates ranging between 93% and 99%), or (3) unlike or dissimilar call rates (e.g., 98%). CEL data files were analyzed in “like,” “mixed,” and “unlike” environments with the BRLMM algorithm. The “mixed” environment was designed to emulate the batch composition one would obtain if processing batches were built randomly. Contrary to our expectations, we found that batch context does make a difference in genotype calling efficacy. Specifically, we observed that call rates for the test samples were substantially improved when processed with data files of similar call rates (Figure S3). The most dramatic results were observed with samples at the lower end of the range of DM call rates tested, those with “moderate” call rates (93%). For example, when a sample with a moderate DM call rate was called in a batch environment composed of moderate DM call-rate samples, chip call rates were boosted substantially. Chip call rates for these samples were consistently superior in the “like” environment rather than the “unlike,” boosting call rates on average 2.3% ± 0.9%. In addition, the “like” environment consistently outperformed the “mixed” environment, boosting call rates on average 0.5% ± 0.4%.
Chips from the “good” (95%) and “excellent” (98%) call-rate1 classes showed a similar pattern to the “moderate” chips when analyzed in the three different batch environments with BRLMM; however, this trend was not absolute. The majority of the cases tested showed improved call rates when analyzed in “like” environments as compared to “mixed” or “unlike” environments, with a few exceptions (Figure S3). To better understand this “batch-effect” phenomenon, we investigated the properties of probe signals for several SNPs in which a genotype was called in the “like” environment and not called in the “unlike” environment. For the SNPs of interest, the BRLMM-derived allele signal for of all SNPs in the cluster of samples was transformed into Cluster-Center-Stretch space (see the Affymetrix website and Figure S4). This example illustrates that both the allele contrast and the signal strength can shift markedly with different input data sets. Given this phenomenon, it is clear that the contents of a data set can influence the genotype-calling behavior for some outlying samples. Indeed, it is evident in this example that the uncalled genotype (blue triangle, Figure S4B) fell well outside of the expected cluster (pink triangles) and was thus not called. Examination of several such examples showed two trends that help explain the ineffectiveness of high DM call-rate batches to call lower DM call-rate raw data: (1) The genotype clusters for high DM call-rate data are typically tighter than those from lower call-rate data. (2) The signal strength (y axis) axis is generally higher for high call-rate data. The differences observed underscore the need to process CEL-data files in batches with similar or mixed call rates, in order to create genotype-calling clusters that are valid for all samples in the batch. These experiments suggest that call-rate outcome with BRLMM can vary, depending on the batch environment chosen for analysis, and that careful attention to the batch selection can improve the number of genotypes called by greater than 2%. On the basis of the analysis above, we developed a “workflow” for maximizing SNP call rates while maintaining high accuracy and reproducibility of our data. We used DM as an initial quality measure for individual chips and then reanalyzed the raw data with the BRLMM algorithm in appropriately defined clusters. With this method, the average chip call rates across the entire sample set improved to 98.95% with BRLMM (Figure S5) with over half of the chips yielding genotype calls for greater than 99% of the SNPs assayed (Figure S5).
In addition to the chip-based genotypes, we also genotyped all four SNPs implied by the GWA analyses as well as APOE SNPs rs429358 and rs7412 in all four samples with high-efficiency fluorescence polarization (HEFP) detection of a single-base extension assay.15 The HEFP procedures were essentially identical to those previously described.13 Primer sequences and thermocycling conditions are available on request. Neither genotyping method showed evidence for Mendelian errors for these four SNPs, although our power to detect such inconsistencies is low because of the lack of parental genotypes (see above) and relatively small family size.
Association analyses were performed with PBAT (v3.6), an extension16 of the family-based association test (FBAT) program.17 To maximize statistical power, we tested AD affection status and age of onset jointly, using the multivariate extension of the FBAT-approach, FBAT-GEE.18 To minimize the multiple testing problem, we applied the weighted Bonferroni-testing strategy by Ionita-Laza et al.19 which is an extension of the VanSteen algorithm.20 On the basis of the between-family information that is statistically independent from the FBAT-statistic,20 the testing strategy evaluates the evidence for association at a population level and then estimates the conditional power of the FBAT-GEE statistic for each marker in the first step. The FBAT-GEE statistic contains affection status and time to onset as phenotypes, coded as Wilcoxon statistic. The choice of which statistic to use in the association test is determined on the basis of the highest conditional-power estimate for each coding. In the second step of the testing strategy, FBAT statistics are computed for all markers. Because none of the traits here were quantitative, the conditional power was estimated on the basis of the nonparametric extension of the conditional mean model approach to dichotomous traits and time-to-onset variables proposed by Jiang et al.21 Their significance is assessed on the basis of individually adjusted alpha levels that maintain the overall type 1 error and that are weighted on the basis of the conditional-power estimate for the corresponding marker according to their conditional-power estimates. The computation of the weights is described in detail in Ionita-Laza et al.19 Here, the approach was applied with the following tuning parameters: The size of the first partition was five and the parameter was set to two. When the weighted Bonferroni-approach was applied to the 809,208 p values of the FBAT-GEE statistic for AD affection status and time to onset, four SNPs not related to APOE ɛ4 reached genome-wide significance (thresholds for genome-wide significance are p ≤ 5 × 10−3 for markers rs11159647, rs179943, and rs2049161, and p ≤ 4.88 × 10−6 for rs3826656). Affection status was coded with an offset of 0.10 (approximate prevalence of AD among individuals over 65 years). Sensitivity analyses using offsets ranging from 0.05 to 0.2 did not change the results appreciably (data not shown). The age of onset variable was constructed with the Wilcoxon approach.21 Although SNPs on the X chromosome and those with low call rates or poor reproducibility across duplicated genotypes were excluded from the analyses (16,146 SNPs), we retained SNPs that deviated from HWE, which affected a total of ∼85,000 SNPs at p = 0.01, and ∼56,000 at p = 0.001. Approximately half of the HWE-deviating SNPs showed low minor allele frequencies (≤0.10). Inclusion of HWE-deviating SNPs was based on the assumption that most departures from HWE in this context are caused by miscalling heterozygous genotypes. Under these circumstances, dominant and recessive models, which treat the heterozygous genotype and one of the homozygous genotypes as one category, will provide test results that are fairly robust against such genotyping errors. This was the case for marker rs326656, which significantly deviated from HWE (p = 1 × 10−23) in the 500K data set, but not in the families of the follow-up samples (all p values > 0.05), which were genotyped with the HEFP technology (see above). Regenotyping of this SNP by HEFP in the NIMH families resolved the HWE deviation (p = 0.6), decreasing the statistical significance to p = 0.04 in the FBAT-GEE analyses. The FBAT-GEE test statistics were only calculated for SNPs for which the number of informative families was at least 20 (i.e. 404,604 out of the 484,522 SNPs with available genotypes). The 404,604 SNPs were tested under additive and dominant transmission models. Accordingly, all nominal p values were adjusted conservatively for 2 × 404,604 = 809,208 comparisons, with the weighted Bonferroni method by Ionita-Laza et al.13 We calculated p values on the combined samples by using the method described by Fisher22 taking into account the direction of the transmissions in each individual sample. Pairwise LD estimations were performed in Haploview (v3.32) on the 500K SNP chip genotype data in self-reported European NIMH families (using the regenotyped data for rs4777936) as well as on genotype data available on the International HapMap Consortium website (public release #22 based on NCBI build 36 [dbSNP b126]).
In the first stage of our project, we screened 1376 individuals from 410 families of self-reported European descent from the National Institute of Mental Health (NIMH) Genetics Initiative Study sample, the largest uniformly ascertained and evaluated AD family sample to date.12,23 We optimized methods for both the genotyping assay and genotype-calling algorithm that led to increased quality and quantity of the data (see above). After removal of all 10,388 X chromosome markers, as well as 5,758 SNPs that did not pass genotype quality assessment or showed a minor allele frequency (MAF) that resulted in less than 20 informative families, a total of 404,604 (80.8%) SNPs were used for the whole-genome screening. Statistical analyses were performed in PBAT with affection status and age of onset as a multivariate phenotype in the FBAT-GEE statistic for which p values were adjusted on the basis of the weighted Bonferroni-testing strategy by Ionita-Laza et al.19 The Q-Q plot displaying observed versus expected p values shows that the overall alpha level is maintained, despite a slight departure from the expected values for the smallest p values (Figure 1). After correction for the number of tests performed, four markers not related to APOE ɛ4 attained genome-wide significance at an overall alpha level of 5%. The first marker, rs4420638 (p = 5.7 × 10−14), is located 340 bp 3′ of APOC1 on chromosome 19q13 and very likely reflects the well-established effects of the APOE ɛ4-allele (rs429358), which maps 11 kb proximal (r2 between both SNPs = 0.78) and shows highly significant association in the NIMH families as well as the three follow-up data sets (see below and Schjeide et al.24). The other markers are rs11159647 (p = 0.001; located in predicted gene NT_026437.1360 on chromosome 14q31.2), rs179943 (p = 0.002; in ATXN1 [MIM 601556] on chromosome 6p22.3), rs3826656 (p = 4 × 10−6; located in predicted gene NT_011109.848 on 19q13.33), and rs2049161 (p = 0.002; in cDNA BC040718 on 18p11.31). None of these markers were previously described as modifiers of AD risk or onset age. Interestingly, with the exception of rs2049161, all SNPs are located either in or close to previously described early- and late-onset AD linkage regions.7,23 Analyses using affection status and age of onset as separate phenotypes revealed that SNP rs11159647 on chromosome 14q31.2 was primarily associated with age of onset ([two-tailed] p = 0.006, median reduction in onset age 1.1 years; odds ratio [OR] ∼1.4; Figure 2A), whereas the remaining markers only showed association in the analyses using affection status (ORs ranging from ∼1.1 to 1.3). All markers showed their strongest signals in an additive transmission model, with the exception of SNP rs3826656 on chromosome 19q13.33, for which dominant transmission of the minor allele yielded the strongest association. None of the four markers showed evidence of association in NIMH families of African American descent, possibly because of lower power in that this subset only consists of 24 families (data not shown).
Figure 1.
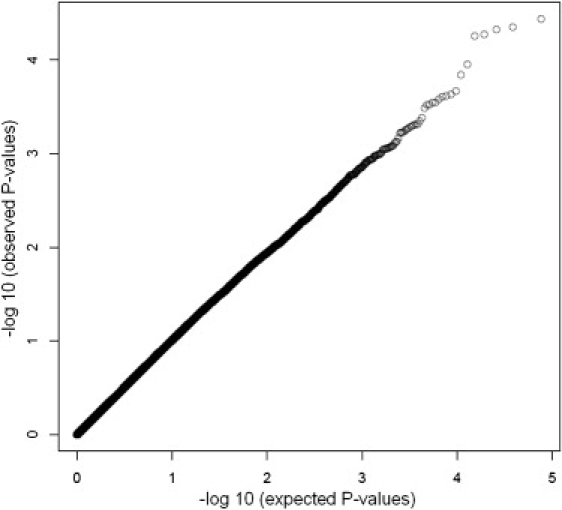
Q-Q Plot of Markers Tested in the GWA Screening Phase
Distribution of FBAT-GEE p values for all 404,604 SNPs on the 500K array with ≥20 informative families as Q-Q plot depicting observed versus expected p values.
Figure 2.
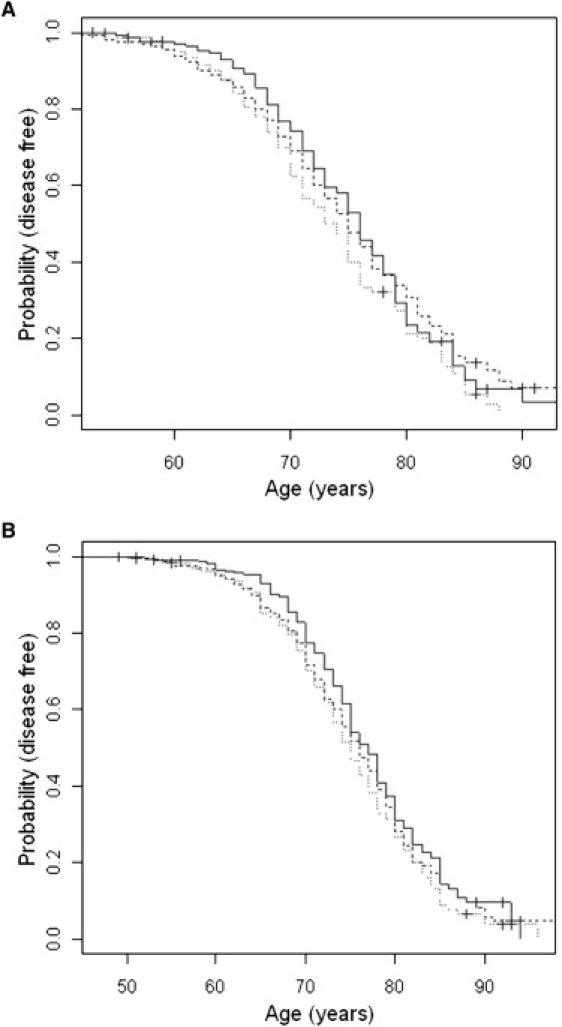
Kaplan-Meier Survival Curves for rs11159647 in 500K Screening Sample and Combined Follow-Up Data Set
Dotted lines represent carriers of the A/A genotype, broken lines are A/G-carriers, solid lines are G/G-carriers. (A) shows the NIMH sample used in the 500K screen. (B) shows the sample after combining all follow-up samples (NIA, NCRAD, and CAG).
We next assessed whether any of the non-APOE signals also show association with AD in three additional and independently collected family samples of self-reported European ancestry (“NIA,” “NCRAD,” and “CAG”) by genotyping the same SNPs for which association was observed in the genome-wide analyses. The vast majority of these families are made up of sibships (either concordant or discordant for AD), with a total of 2689 individuals (1816 affecteds and 845 unaffecteds). Upon combining results across all three replication samples (with Fisher's combined probability test) we observed significant association with the multivariate phenotype for two of the four SNPs tested ([one-tailed] p values 0.00002 [rs11159647] and 0.007 [rs3826656]; Table 1; Figure 2B). A third SNP showed a trend toward association in the replication samples but only in the analyses using affection status as phenotype ([one-tailed] p = 0.06 [rs179943]; Table 1). The fourth SNP (rs2049161), which was only marginally associated with AD in the primary 500K screen, did not show any consistent pattern of association in the replication samples. We next investigated whether any of the four SNPs showed association in the two recently published AD GWA analyses, for which genotype data were made publicly available. Because these data did not include subject-level age-of-onset information, only test statistics using affection status could be calculated (Table 2). rs11159647 on chromosome 14q, the SNP demonstrating the strongest association with AD in our family-based analyses, revealed nominally significant association with the same allele in the TGEN data set ([one-tailed] p = 0.04; see Reiman et al.8). Meanwhile, rs2049161 on chromosome 18p showed nominally significant association in the GSK data set ([one-tailed] p = 0.045; see Li et al.10). Interestingly, this latter SNP was the only marker not showing any consistent evidence of association in our family-based replication samples (see above). rs179943 did not show evidence of association in either of the two previously published GWA screens; no analyses could be performed for rs3826656 because it was missing from both case-control GWA data sets.
Table 1.
Results of Whole-Genome Association Screening and Follow-Up Analyses using Family-based Samples
| SNP | Model |
NIMH (500K) |
NIA |
NCRAD |
CAG |
Replication |
NIMH + Replication |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p (two-tailed) | Fams | p (one-tailed) | Fams | p (one-tailed) | Fams | p (one-tailed) | Fams | p (one-tailed) | Fams | p (two-tailed) | Fams | ||
| rs11159647 | |||||||||||||
| FBAT-GEE | add | 0.001 | 200 | 0.000005 | 176 | 0.045 | 163 | 0.35 | 89 | 0.00002 | 428 | 0.000002 | 628 |
| Affection status | add | 0.05 | 128 | 0.4 | 104 | 0.02 | 100 | 0.2 | 89 | 0.05 | 293 | 0.07 | 421 |
| Age of onset | add | 0.006 | 200 | 0.002 | 176 | 0.0035 | 163 | 0.2 | 89 | 0.0001 | 428 | 0.00005 | 628 |
| rs179943 | |||||||||||||
| FBAT-GEE | add | 0.002 | 76 | 0.065 | 48 | 0.8∗ | 53 | 0.2 | 29 | 0.15 | 130 | 0.008 | 206 |
| Affection status | add | 0.007 | 55 | 0.04 | 27 | 0.8∗ | 31 | 0.07 | 29 | 0.06 | 87 | 0.008 | 142 |
| Age of onset | add | 1 | 76 | 0.2 | 48 | 0.9∗ | 53 | 0.1 | 29 | 0.25 | 130 | 0.4 | 206 |
| rs3826656 | |||||||||||||
| FBAT-GEE | dom | 0.000004 | 123 | 0.15 | 127 | 0.25 | 110 | 0.004 | 75 | 0.007 | 312 | 0.000006 | 435 |
| Affection status | dom | 0.02 | 90 | 0.03 | 79 | 0.4 | 69 | 0.035 | 74 | 0.015 | 222 | 0.01 | 312 |
| Age of onset | dom | 0.6 | 123 | 0.07 | 127 | 0.3 | 110 | 0.07 | 75 | 0.05 | 312 | 0.3 | 435 |
| rs2049161 | |||||||||||||
| FBAT-GEE | add | 0.002 | 122 | 0.8∗ | 129 | 0.8∗ | 109 | 0.04 | 57 | 0.3 | 295 | 0.006 | 417 |
| Affection status | add | 0.04 | 78 | 0.9∗ | 83 | 0.75∗ | 72 | 0.25 | 53 | 0.7 | 208 | 0.1 | 286 |
| Age of onset | add | 1 | 122 | 0.9∗ | 129 | 0.6∗ | 109 | 0.2 | 57 | 0.65 | 295 | 0.7 | 417 |
FBAT-GEE refer to analyses using affection status and age at onset as a multivariate phenotype. The p values are nominal and two-tailed for results including NIMH families, and one-tailed for results solely based on the replication samples (NIA, NCRAD, and CAG). The p values for combined samples are one-tailed for the replication samples only (“Replication”) and two-tailed for NIMH and replication samples combined (“NIMH + Replication”) and calculated by methods described previously (see Fisher22). Fams, informative families. Asterisks represent association with opposite allele as compared to 500K analyses (associated alleles in 500K analyses were “A” [rs11159647], “T” [rs179943], “G” [rs3826656], and “C” [rs2049161]). Age of onset coding based on Wilcoxon statistic. Thresholds to achieve genome-wide significance on the basis of the method by Ionita-Laza et al.19 are p ≤ 5 × 10−3 for markers rs11159647, rs179943, and rs2049161, and p ≤ 4.88 × 10−6 for rs3826656.
Table 2.
Comparison of Family-Based versus Published Case-Control GWA Findings for the Signals Identified in the NIMH 500K Screen
| SNP | Model | NIMH (500K) |
TGEN8 |
GSK10 |
|||
|---|---|---|---|---|---|---|---|
| p (two-tailed) | Fams | p (one-tailed) | n (AD + CTRL) | p (one-tailed) | n (AD + CTRL) | ||
| rs11159647, with affection status | add | 0.05 | 128 | 0.04 | 1384 | 0.9∗ | 1315 |
| rs179943, with affection status | add | 0.007 | 55 | 0.8∗ | 1376 | 0.5∗ | 1368 |
| rs3826656, with affection status | dom | 0.02 | 90 | N.A. | N.A. | N.A. | N.A. |
| rs2049161, with affection status | add | 0.04 | 78 | 0.2 | 1407 | 0.045 | 1386 |
Family-based (NIMH) p values are two sided and identical to those of Table 1. Case-control (TGEN7 and GSK10) p values are one sided, on the basis of an allelic chi-square test (1 d.f.) with the genotype frequencies of the original publications (note that the FBAT-GEE and age of onset statistics could not be computed here because of the lack of onset-age data in the original reports). The results presented in this table are based on the complete data sets made available by the authors of the respective studies. Fams, informative families. The asterisks represent association with opposite allele as compared to 500K analyses (see identity of associated alleles in legend to Table 1). “N.A.” represents no data provided for this marker. Significant p values are represented in bold.
In two separately performed projects,24,25 we assessed whether any of the currently most promising putative AD susceptibility loci (based on a recent freeze of the AlzGene database), including all five recently pinpointed by the two high-density case-control GWA studies,8,10 showed association in the four family data sets tested here. After combining results across all four samples with the same analytical methodology applied here, we observed nominally significant association with variants in ACE (MIM 106180), CHRNB2 (MIM 118507), GAB2 (MIM 606203), TF (MIM 190000), and an as-yet-unidentified locus on chromosome 7p15.2. Of these, GAB28 and the 7p15.2 locus9 were originally implicated by GWA association analyses. Note, however, that the level of statistical support for each of these loci was several orders of magnitude smaller (i.e., combined p values ranging between 0.03 and 0.002) than that observed for the chromosome 14 locus identified here. Variants in other recently reported potential AD genes, such as SORL1 (MIM 602005) and GOLM1 (a.k.a. GOLPH2 [MIM 606804]; see AlzGene database for details), did not show any significant evidence in these analyses.
Our findings are noteworthy for a number of reasons. First, to our knowledge, this is the first GWA analysis using family-based methods to be reported in the field of AD. For two of the four non-APOE SNPs, the initial evidence for genetic association was replicated in three independent collections of AD families of self-reported European descent, whereas a third SNP showed at least a trend toward association in the analyses limited to affection status. Moreover, for the SNP exhibiting the strongest and most consist family-based association with AD in our analyses, rs11159647, we also observed statistically significant association of the same risk allele with AD in an independent collection of cases and controls that had been probed with the same 500K SNP array.8 Collectively, these data strongly argue for the presence of a genuine AD susceptibility locus in the vicinity of marker rs11159647 on chromosome 14q31.2. In addition, our analyses highlight two further putative AD loci located on chromosomes 6p22 and 19q13. Second, we used a quantitative analysis approach combining the two most widely available phenotypes in AD samples, i.e., age of onset and affection status. This has the advantage of increasing power while ensuring consistency of the findings across both phenotypes.18,21 Power calculations reveal that minimally ∼700 combined cases and controls are required for detection of the additively transmitted rs11159647 risk effect (i.e., an allelic OR of ∼1.4) at α = 0.05 in order to achieve 80% power and minimally ∼2,300 (∼8,600) samples for the more modest risk effects of SNPs rs3826656 and rs179943. Third, the association signal for rs11159647 maps to the distal end of a genetic linkage region identified earlier by our group in a whole-genome linkage screen of the NIMH sample,12 as well as in an independent collection of Caribbean Hispanic families that used age of onset as a phenotype.26 In our own previous report, most of the linkage evidence originated from families with an “early/mixed” onset age, i.e., those families in which at least one affected family member, showed an onset age prior to 65 years.23 This is in good agreement with the decrease in onset age observed here in individuals carrying the A allele at rs11159647. A similar observation was made with the whole-genome linkage signal encompassing the APOE region on chromosome 19q13, which was also most pronounced in families with an “early/mixed” onset age.23 Interestingly, the other two putative signals implied by our GWA and follow-up analyses map to chromosomes 6p22 and 19q13, which were also highlighted by genome-wide linkage analyses of our and other groups.7,23
Despite the compelling statistical and genetic epidemiological evidence strongly implicating the presence of a putative AD gene on chromosome 14q, and possibly additional loci on chromosomes 6p22 and 19q13, the potential functional and pathophysiological consequences of our findings remain elusive. According to the UCSC genome browser (hg18, NCBI Build 36.1), the genomic region in the vicinity of the AD-associated SNP, rs11159647, on chromosome 14q31 does not contain any known RefSeq genes. This SNP resides at position 83,844,962 bp on chromosome 14 in an intron of the Genscan-predicted gene, NT_026437.1360 (Figure 3), which spans 723,153 bp. The coding region of this predicted gene in the region of rs11159647 reveals no significant homologies to other genes or coding regions in GenBank. Interestingly, the 3′ end of this predicted gene contains exons with homology to the C2H2-type kruppel-like zinc-finger protein 268 (ZNF268 [MIM 604753]; see Gou et al.27). However, the AD-associated SNP, rs11159647, is >350 Kb from the ZNF268 homologous region, and SNPs in this region reveal no linkage disequilibrium with rs11159647. There are three expressed sequence tags (ESTs) residing within 60 Kb on either side of rs11159647. These include ESTs, M85511, CA390254, and AI003603. All three ESTs are expressed in the brain and are encoded within the same region as the predicted gene, NT_026437.1360. However, the predicted exon structure of these ESTs does not align with the predicted exons of NT_026437.1360. Thus, these ESTs may represent exons of separate gene(s) in this region, which are expressed in the brain. It is also worth noting that SNPs in these three ESTs display varying degrees of LD with rs11159647. BLAST analyses of these ESTs reveal no significant homologies with any known genes. Figure 3 illustrates the LD patterns in the region surrounding rs11159647, whereas Figure 4 shows that there are several other SNPs within ∼200 kb yielding evidence for association with AD on the 500K array, thereby delineating the chromosomal region that should be targeted by subsequent fine-mapping efforts. SNP rs179943, on 6p22.3 at position 16,506,297 bp, resides within an intron of the ataxin 1 (ATXN1) gene, in which an elongated polyglutamine tract causes the progressive neurodegenerative disease spinocerebellar ataxia (SCA1 [MIM 164400]), characterized by progressive degeneration of the cerebellum, brain stem, and spinal cord.28 SNP, rs3826656, on 19q13.33 at position 56,418,175 bp, resides in a region that contains no known RefSeq genes. However, this SNP resides in a predicted Genscan gene, NT_011109.848, spanning 126,319 bp. The 3' portion of this locus overlaps with the gene encoding human protein CD33 (MIM 159590). CD33, also known as SIGLEC3, encodes a cell-surface receptor on cells of monocytic or myeloid lineage. It is also a member of the SIGLEC family of lectins that bind sialic acid and regulate the innate immune system via the activation of caspase-dependent and caspase-independent cell-death pathways.29 Finally, rs2049161, on 18p11.31 at position 4,117,583 bp, resides within an intron of BC040718, a gene of currently unknown function.
Figure 3.
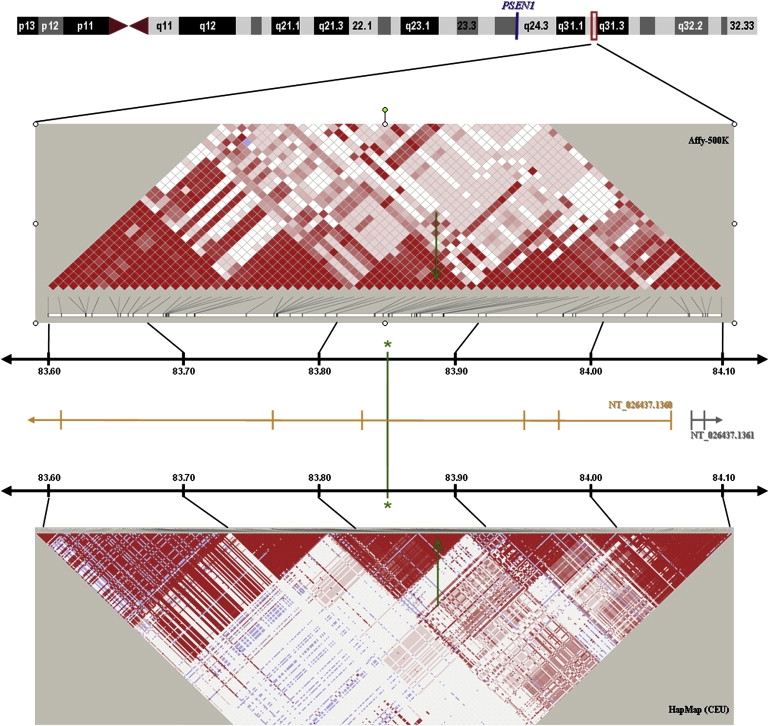
Genomic Context of the Chromosome 14q31 Association Signal
Linkage disequilibrium structure and location of Genscan Gene predictions (NTSs) in a 500 kb interval encompassing rs11159647 on chromosome 14q31.2.
Figure 4.
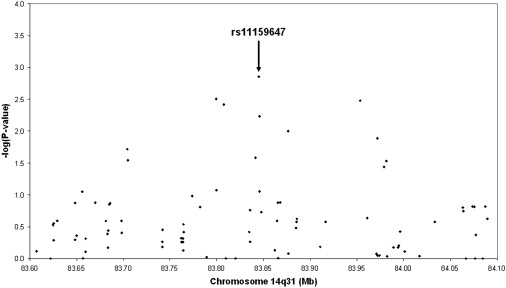
Association Results of Markers within 500 kb of the Chromosome 14q31 Association Signal
Distribution of association results of SNPs on the 500K array within ±250 kb of SNP rs11159647 on chromosome 14q31 showing genome-wide significance in the NIMH-CAU sample (FBAT-GEE statistic, additive model).
In conclusion, to our knowledge this is the first study to employ a family-based GWA approach to AD. In addition to a likely APOE ɛ4-related effect, we obtained compelling evidence for genome-wide significant association between AD and at least two additional SNPs. The replication of these associations in three independent AD family samples—and in the case of rs11159647 also in one independent case-control GWA data set—strongly implies the existence of AD susceptibility loci that warrant follow up in additional independent samples as well as in functional genomic analyses.
Acknowledgments
The authors wish to thank all families for participating in this study. This work was sponsored by grants from the Cure Alzheimer Fund (as part of the “Alzheimer Genome Project”), the NIMH, and the Extendicare Foundation. Work completed at TorreyPines Therapeutics and was funded by Eisai. L.B. and R.E.T. are consultants to, and equity holders in, TorreyPines Therapeutics and Prana Biotechnology. R.E.T. is also a consultant to Prana Biotechnology. R.E.T. is a cofounder of both TorreyPines Therapeutics and Prana Biotechnology. K.L.O., K.E., X.W., D.H.-L., M.R., K.D.B., and S.W. are employees of, and hold stock options in, TorreyPines Therapeutics. K.E. and S.W. also hold stock in Merck.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Affymetrix, http://www.affymetrix.com
AlzGene, http://www.alzgene.org
Broad Institute, http://www.broad.mit.edu
International HapMap Consortium, http://www.hapmap.org
National Cell Repository for Alzheimer's Disease (NCRAD), http://www.ncrad.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC genome browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 2.Levy-Lahad E., Wasco W., Poorkaj P., Romano D.M., Oshima J., Pettingell W.H., Yu C.E., Jondro P.D., Schmidt S.D., Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 3.Rogaev E.I., Sherrington R., Rogaeva E.A., Levesque G., Ikeda M., Liang Y., Chi H., Lin C., Holman K., Tsuda T. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 4.Sherrington R., Rogaev E.I., Liang Y., Rogaeva E.A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 5.Saunders A.M., Strittmatter W.J., Schmechel D., George-Hyslop P.H., Pericak-Vance M.A., Joo S.H., Rosi B.L., Gusella J.F., Crapper-MacLachlan D.R., Alberts M.J. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 6.Gatz M., Reynolds C.A., Fratiglioni L., Johansson B., Mortimer J.A., Berg S., Fiske A., Pedersen N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 7.Bertram L., McQueen M.B., Mullin K., Blacker D., Tanzi R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 8.Reiman E.M., Webster J.A., Myers A.J., Hardy J., Dunckley T., Zismann V.L., Joshipura K.D., Pearson J.V., Hu-Lince D., Huentelman M.J. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grupe A., Abraham R., Li Y., Rowland C., Hollingworth P., Morgan A., Jehu L., Segurado R., Stone D., Schadt E. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007;16:865–873. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- 10.Li H., Wetten S., Li L., St Jean P.L., Upmanyu R., Surh L., Hosford D., Barnes M.R., Briley J.D., Borrie M. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 11.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 12.Blacker D., Haines J.L., Rodes L., Terwedow H., Go R.C., Harrell L.E., Perry R.T., Bassett S.S., Chase G., Meyers D. ApoE-4 and age at onset of Alzheimer's disease: The NIMH genetics initiative. Neurology. 1997;48:139–147. doi: 10.1212/wnl.48.1.139. [DOI] [PubMed] [Google Scholar]
- 13.Bertram L., Hiltunen M., Parkinson M., Ingelsson M., Lange C., Ramasamy K., Mullin K., Menon R., Sampson A.J., Hsiao M.Y. Family-based association between Alzheimer's disease and variants in UBQLN1. N. Engl. J. Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- 14.Rabbee N., Speed T.P. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 15.Chen X., Levine L., Kwok P.Y. Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res. 1999;9:492–498. [PMC free article] [PubMed] [Google Scholar]
- 16.Lange C., DeMeo D., Silverman E.K., Weiss S.T., Laird N.M. PBAT: tools for family-based association studies. Am. J. Hum. Genet. 2004;74:367–369. doi: 10.1086/381563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laird N.M., Horvath S., Xu X. Implementing a unified approach to family-based tests of association. Genet. Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 18.Lange C., Silverman E.K., Xu X., Weiss S.T., Laird N.M. A multivariate family-based association test using generalized estimating equations: FBAT-GEE. Biostatistics. 2003;4:195–206. doi: 10.1093/biostatistics/4.2.195. [DOI] [PubMed] [Google Scholar]
- 19.Ionita-Laza I., McQueen M.B., Laird N.M., Lange C. Genomewide weighted hypothesis testing in family-based association studies, with an application to a 100K scan. Am. J. Hum. Genet. 2007;81:607–614. doi: 10.1086/519748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Steen K., McQueen M.B., Herbert A., Raby B., Lyon H., Demeo D.L., Murphy A., Su J., Datta S., Rosenow C. Genomic screening and replication using the same data set in family-based association testing. Nat. Genet. 2005;37:683–691. doi: 10.1038/ng1582. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H., Harrington D., Raby B.A., Bertram L., Blacker D., Weiss S.T., Lange C. Family-based association test for time-to-onset data with time-dependent differences between the hazard functions. Genet. Epidemiol. 2006;30:124–132. doi: 10.1002/gepi.20132. [DOI] [PubMed] [Google Scholar]
- 22.Fisher R.A. Oliver and Boyd; Edinburgh: 1925. Statistical Methods for Research Workers. [Google Scholar]
- 23.Blacker D., Bertram L., Saunders A.J., Moscarillo T.J., Albert M.S., Wiener H., Perry R.T., Collins J.S., Harrell L.E., Go R.C. Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum. Mol. Genet. 2003;12:23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- 24.Schjeide B.-M.M., McQueen M.B., Mullin K., Divito J., Hogan M.F., Parkinson M., Lange C., Blacker D., Tanzi R., Bertram L. Assessment of Alzheimer's disease case-control associations using family-based methods. Neurogenetics. 2008 doi: 10.1007/s10048-008-0151-3. in press. Published online October 2, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schjeide B.-M.M., Hooli B., Parkinson M., Hogan M.F., Divito J., Mullin K., Blacker D., Tanzi R., Bertram L. Follow-up of genome-wide association results suggests GAB2 as an Alzheimer's disease susceptibility gene. Arch. Neurol. 2009 doi: 10.1001/archneurol.2008.552. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J.H., Barral S., Cheng R., Chacon I., Santana V., Williamson J., Lantigua R., Medrano M., Jimenez-Velazquez I.Z., Stern Y. Age-at-onset linkage analysis in Caribbean Hispanics with familial late-onset Alzheimer's disease. Neurogenetics. 2008;9:51–60. doi: 10.1007/s10048-007-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gou D.M., Sun Y., Gao L., Chow L.M., Huang J., Feng Y.D., Jiang D.H., Li W.X. Cloning and characterization of a novel Kruppel-like zinc finger gene, ZNF268, expressed in early human embryo. Biochim. Biophys. Acta. 2001;1518:306–310. doi: 10.1016/s0167-4781(01)00194-4. [DOI] [PubMed] [Google Scholar]
- 28.Orr H.T., Zoghbi H.Y. SCA1 molecular genetics: a history of a 13 year collaboration against glutamines. Hum. Mol. Genet. 2001;10:2307–2311. doi: 10.1093/hmg/10.20.2307. [DOI] [PubMed] [Google Scholar]
- 29.von Gunten S., Simon H.U. Sialic acid binding immunoglobulin-like lectins may regulate innate immune responses by modulating the life span of granulocytes. FASEB J. 2006;20:601–605. doi: 10.1096/fj.05-5401hyp. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.