

Abstract
Diabetic nephropathy (DN) is the primary cause of morbidity and mortality in patients with type 1 diabetes mellitus (T1DM) and affects about 30% of these patients. We have previously localized a DN locus on chromosome 3q with suggestive linkage in Finnish individuals. Linkage to this region has also been reported earlier by several other groups. To fine map this locus, we conducted a multistage case-control association study in T1DM patients, comprising 1822 cases with nephropathy and 1874 T1DM patients free of nephropathy, from Finland, Iceland, and the British Isles. At the screening stage, we genotyped 3072 tag SNPs, spanning a 28 Mb region, in 234 patients and 215 controls from Finland. SNPs that met the significance threshold of p < 0.01 at this stage were followed up by a series of sample sets. A genetic variant, rs1866813, in the noncoding region at 3q22 was associated with increased risk of DN (overall p = 7.07 × 10−6, combined odds ratio [OR] of the allele = 1.33). The estimated genotypic ORs of this variant in all Finnish samples suggested a codominant effect, resulting in significant association, with a p value of 4.7 × 10−5 (OR = 1.38; 95% confidence interval = 1.18–1.62). Additionally, an 11 kb segment flanked by rs62408925 and rs1866813, two strongly correlated variants (r2 = 0.95), contains three elements highly conserved across multiple species. Independent replication will clarify the role of the associated variants at 3q22 in influencing the risk of DN.
Introduction
Diabetic nephropathy (DN) is the single most common cause of end-stage renal disease (ESRD) in the Western world, accounting for about 40% of new cases of ESRD in the USA.1,2 ESRD is an important cause of death in type 1 diabetes mellitus (T1DM) nephropathy.1 Clinically, DN is a syndrome characterized by persistent proteinuria. The hallmarks of DN pathology include renal extracellular-matrix accumulation and thickening of the glomerular basement membrane (GBM) and the tubular basement membrane. However, the molecular pathomechanisms of DN are currently obscure.
Accumulating evidence supports the notion that development of the devastating kidney complications in diabetes has genetic components. It has clearly been documented that only a subset (∼30%) of patients with type 1 diabetes are susceptible to DN.3,4 The incidence of DN peaks during the second decade in patients with T1DM, and it declines thereafter.3,4 Familial clustering of DN has been reported by several investigators.5–7 These family-based studies have suggested that segregation of DN does not follow simple Mendelian rules and that, instead, the disease alleles significantly increase the risk of DN among siblings. Extensive efforts have been made to identify loci for DN with the use of either genome-wide scans or candidate-gene approaches, but so far no genes have been associated with DN in replica studies.8 Our previous genome-wide scan using Finnish discordant sib pairs suggested linkage to chromosome 3q.9 The 3q locus linked to DN was repeatedly reported by linkage studies using either genome scans or a candidate-region approach.10–12 In particular, linkage signals on the 3q region were also detected by two previous large genome scans, which used concordant affected sib pairs10,12 because discordant-sib-pair analysis without parent genotypes is theoretically not robust to potential genotyping errors. Those results, together with data from others, suggest that the 3q region is likely to harbor susceptibility gene(s) for DN. In the present study, we fine mapped this locus by genotyping highly dense single-nucleotide polymorphisms (SNPs) in 1822 cases and 1874 controls from three populations (Finland, Iceland, and the British Isles). Here, we report the association of genetic variants at 3q22 with an increased risk of DN.
Subjects and Methods
Study Subjects
The cross-sectional study includes a total of 1822 patients with T1DM and nephropathy and 1874 patients with T1DM but without nephropathy (controls) from Finland, Iceland, and the British Isles. The main clinical characteristics of these individuals are summarized in Table 1. All participants provided written, informed consent to participate in the study.
Table 1.
Main Clinical Characteristics of 3696 Patients with Type 1 Diabetes Included in the Study
| Panel 1 |
Panel 2 |
Panel 3 |
Panel 4 |
Panel 5 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | |
| Number | 235 | 215 | 330 | 175 | 459 | 628 | 80 | 107 | 718 | 749 |
| Male/female (%) | 53/47 | 45/55 | 61/39 | 38/62 | 62/38 | 46/54 | 69/31 | 54/46 | 58/42 | 43/57 |
| Age (yrs) | 51 ± 9.3 | 49.6 ± 10.9 | 39.3 ± 9.5 | 42.6 ± 7.6 | 53.6 ± 10.1 | 43.8 ± 12.8 | 52.5 ± 16.4 | 54.4 ± 16.4 | 49.6 ± 10.4 | 44.9 ± 11.2 |
| Diabetes duration (yrs) | 32.9 ± 8.6 | 30.7 ± 9.1 | 27.5 ± 7.9 | 30 ± 2.8 | 31.2±8.4 | 26.8 ± 9.6 | 25.1 ± 9.3 | 25.8 ± 8.7 | 34.5 ± 9.6 | 29.6 ± 9.1 |
| Blood pressure | ||||||||||
| Systolic (mm Hg) | 152 ± 16.3 | 133.9 ± 18.3 | 144 ± 20 | 132 ± 17 | 148.4 ± 21 | 129.2 ± 15.9 | 135 ± 12.9 | 130.9 ± 14.6 | 145.0 ± 21.1 | 125.0 ± 14.6 |
| Diastolic (mm Hg) | 82.9 ± 8.6 | 78.9 ± 7.3 | 84 ± 11 | 78 ± 9 | 83.8 ± 10.9 | 78.3 ± 9.2 | 80.1 ± 7.6 | 77.9 ± 6.1 | 81.8 ± 11.4 | 75.3 ± 7.7 |
| Antihypertensive therapy (%) | 90.6 | 21.9 | 93 | 16.6 | 92.1 | 13.2 | 81.3 | 57.9 | 97.1 | 0 |
| Retinal laser treatment (%) | 89.4 | 25.5 | 83.1 | 16.9 | 80.2 | 14.5 | 50 | 17.8 | 49.3 | 2.4 |
| Renal status | ||||||||||
| Normoalbuminuriaa | 0 | 215 (100%) | 0 | 175 (100%) | 0 | 628 (100%) | 0 | 107 (100%) | 0 | 749 (100%) |
| Macroalbuminuirab | 162 (69%) | 0 | 255 (77%) | 0 | 274 (60%) | 0 | 70 (88%) | 0 | 489 (68%) | 0 |
| ESRDc | 73 (31%) | 0 | 75 (23%) | 0 | 185 (40%) | 0 | 10 (12%) | 0 | 229 (32%) | 0 |
Normoalbuminuria is defined as the albumin excretion rate (AER) < 30 mg/24 hr or the albumin/creatinine ratio (ACR) < 3 mg/mmol in at least three consecutive urine samples.
Macroalbuminuria is defined as AER ≥ 300 mg/24 hr or ACR ≥ 30 mg/mmol in two of three consecutive measurements on sterile urine.
End-stage renal disease.
All study subjects had had T1DM for at least 10 years, with the age at onset ≤ 30 years. The renal status was based on the albumin excretion rate (AER) in a 24 hr urine collection or the albumin/creatinine ratio (ACR) in a random, spot urine collection. The presence of ESRD was defined according to whether patients were undergoing dialysis or had had a kidney transplant. In addition to persistent macroalbuminuria, the concomitance of retinopathy is one of the criteria required for a clinical diagnosis of nephropathy in T1DM.1,13 Retinopathy status was assessed on the basis of fundoscopy or retinal photography and information about laser treatment. Established DN was defined by (1) persistent macroalbuminuria (AER ≥ 300 mg/24 hr or ACR ≥ 30 mg/mmol) in two of three consecutive measurements or the presence of ESRD; (2) the presence of retinopathy; and (3) the absence of clinical or laboratory evidence of nondiabetic renal or urinary-tract disease. Control status was defined by the normoalbuminuria (AER < 30 mg/24 hr or ACR < 3 mg/mmol) despite duration of diabetes for at least 15 years. The diagnostic criteria for the presence of nephropathy were established prior to SNP genotyping.
The Finnish samples came from two sources: (1) the screening panel (panel 1) and (2) follow-up cohorts from the Finnish Diabetic Nephropathy study, FinnDiane (panels 2 and 3). Sample collections from these two sources were carried out separately and independently of each other. Panel 1 samples, used for the screening stage, were ascertained and recruited by the Department of Public Health, University of Helsinki, Finland, as described elsewhere,9 and close relatives of samples, including siblings and parent-offspring, were excluded from this panel. The FinnDiane study is a comprehensive, multicenter, nationwide project with the aim to characterize 25% of all adult patients with T1DM in Finland, and this project has been described elsewhere.14 A small-sized Icelandic cohort (panel 4), recruited at Landspítali University Hospital in Reykjavík, Iceland, and a British Isles cohort (panel 5), recruited in the UK and Ireland as part of multicenter DN-research projects, were included as additional follow-up samples. As a multistage association study, individuals of panel 2 were genotyped for the top SNPs that met the significance threshold of 0.01 at the screening stage. The additional follow-up study was performed on panels 3, 4, and 5 if SNPs that met the significance threshold of 0.05 in panel 2 involved the same risk allele in panels 1 and 2. Because panel 1 and the two FinnDiane cohorts were recruited independently throughout Finland, we excluded any overlap of the sample collections prior to genotyping the top SNPs in panel 2. This was done with the implementation of a unique personal identification (ID) code, containing information on date of birth and sex, as well as control digits.
The Ethical Committees of the Finnish National Public Institute and the Karolinska Institutet approved the protocol. Regarding the FinnDiane study, the local ethical committee at each FinnDiane center approved the protocol. The Icelandic Data Protection Commission and the National Bioethics Committee in Iceland have approved the study. Ethical approval for recruitment of the British Isles cohort was obtained from the appropriate Research Ethics Committees in the UK and Ireland, and written, informed consent was obtained from individuals prior to the study's inception.
SNP Selection and Genotyping
At the screening stage, we took the region at 3q ranging from 124 to 152 Mb (NCBI build 35), corresponding to the region between D3S1267 and D3S1308. This region covered our linkage peak and also extended the coverage to include the linkage peak reported by another study.11 We used the linkage disequilibrium (LD)-based approach to select SNPs.15 In brief, SNP genotype data typed in the Centre d'Etude du Polymorphisme Humain (CEPH) collection (CEU; 30 trios of northern and western European ancestry living in Utah) in the region between 124 and 152 Mb was downloaded from the HapMap database (see Web Resources). The Tagger program, implemented in Haploview (see Web Resources), was applied for selection of tag SNPs with three-marker haplotype tests (LOD ≥ 2.0), so that all SNPs with a minor-allele frequency (MAF) > 2% were captured with r2 ≥ 0.7. We finally determined a set of 3072 SNPs that passed Illumina quality design scores for genotyping. The SNP density in the region was, on average, 1.1 SNPs per 10 kb, although the density is highly dependent upon LD extent in this region.
At the screening stage, the selected tag SNPs were genotyped in panel 1 with the Illumina BeadArrays, 96-array matrix, at the Wallenberg Consortium North (WCN) SNP platform (Uppsala, Sweden). At least 0.5 μg of genomic DNA, quantified with the PicoGreen assay, was used for genotyping of each sample. The genotype quality was evaluated with the PLINK program16 (see Web Resources). We excluded SNPs if they showed (1) a genotyping success rate < 90% in panel 1 cases and controls (2) an MAF < 2% in panel 1 cases and controls, and (3) significant deviation from Hardy-Weinberg equilibrium (HWE) in the controls (p < 0.001). Any samples that yielded < 85% of genotype rates were excluded. For the follow-up study, we used the standard TaqMan allelic-discrimination method (Applied Biosystems) to genotype all of the putative loci that reached the significance thresholds. The TaqMan SNP-genotyping assays were obtained from Applied Biosystems. Some SNPs were genotyped via the sequencing method with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) if the TaqMan genotyping assays were not available. We also checked whether genotypes of the follow-up SNPs showed significant deviation from HWE in controls.
Statistical Analysis
Statistical analyses were performed with the PLINK program (see Web Resources). We assessed allelic association of the single markers between cases and controls using standard χ2 tests. ORs for each individual allele and the corresponding 95% confidence intervals [CI] were calculated. All p values presented were two-tailed. We used Fisher's exact test to assess deviation of the genotype frequency from that expected under HWE in the controls. To avoid any bias leading to significant association, we checked whether missing rates between cases and controls for each SNP differed significantly. The raw data combined from different populations was analyzed jointly with a Cochran-Mantel-Haenszel model under the assumption of common relative risks. The Pearson χ2 statistic was used to test for evidence of heterogeneity of the ORs across studies. For detection of potential population stratification that might lead to spurious association, two methods, implemented in the PLINK program, were performed, with the use of all valid tag SNPs in panel 1: (1) the genomic-control method,17 which estimates the genomic inflation factor, λ, on the basis of the median distribution of the χ2 statistics; and 2) the structured-association method,18 which clusters individuals into homogenous subsets with the distance-based clustering approach. We performed clustering of individuals, by which each cluster consists of at least one case and one control, with a threshold of 0.01 for the pairwise population concordance (PPC) test. λ = 1 indicates a null distribution with no inflation of test statistics.
Logistic-regression analysis was performed to estimate genotypic OR and corresponding 95% CI for individual SNP genotypes, with the Statistical Analysis System (SAS) program, version 9.1.3. We estimated the regression coefficients (β), logarithms of the ORs for the genotypic effect, with the use of dummy variables (χ1, χ2) to code the genotypes.
Pairwise LD was calculated by the Haploview program (see Web Resources), with the use of the standard measures D′ and r2. We used the expectation-maximization algorithm, implemented in the Haploview program, to estimate haplotype frequencies. We performed the χ2 test for association of haplotypes, as well as 10,000 permutations, to obtain empirical p values in order to correct for multiple-testing bias.
Power calculations were performed with the use of a 1% significance level for detecting an association, assuming an additive model with relative risk of 1.5 and disease-allele frequency of 20%. The calculations were performed with the online Genetic Calculator software (see Web Resources).
We utilized the UCSC multiple-species genome alignment of 28 vertebrate species (20 mammals, including human, and 8 nonmammalian vertebrates) of the UCSC genome browser (see Web Resources) to localize evolutionarily conserved elements.
Results
Before the high-density SNP genotyping, we estimated the power in panel 1 assuming an additive model with relative risk of 1.5, yielding 75% power to find association with a p value of 0.01 (unadjusted), which was determined as the significance threshold for the follow-up study. At the screening stage, we genotyped 3072 tag SNPs, spanning the 28 Mb region from 124 to 152 Mb (NCBI build 35), in panel 1. After performing the genotype quality control (see Subjects and Methods), we included 2805 SNPs (91.3%) and 444 samples (98.7%) in subsequent analyses, resulting in initial associations (p < 0.01) of 27 SNPs with DN in the allele-based test (Table 2). Two boundary SNPs (rs13094003 and rs8052) of the targeted region were still included among valid SNPs, suggesting that the coverage of the successfully genotyped SNPs had no significant loss. We assessed population stratification of panel 1 (444 samples) on the basis of 2805 SNPs using two approaches. With the use of the genomic-control method, the inflation factor, λ, was 1.06. With the structured-association approach, λ was 1.06, based on the median distribution of test statistics after individual clustering in which each cluster had at least one case and one control. These results from two different methods indicated little evidence for inflation in association-test statistics as a result of population stratification in panel 1.
Table 2.
Summary of the Screening Stage in Panel 1 and the Follow-Up Study in Panel 2
| Screening Stage in Panel 1 |
Follow-Up Study in Panel 2 |
|||||||
|---|---|---|---|---|---|---|---|---|
| SNP ID | Position (bp) | Minor Allele | MAF Case | MAF Control | p Value | MAF Case | MAF Control | p Value |
| rs1049296 | 134977044 | T | 0.069 | 0.146 | 1.8×10−4 | 0.111 | 0.107 | 0.68 |
| rs4678015 | 124583153 | G | 0.524 | 0.403 | 3.3×10−4 | 0.473 | 0.459 | 0.36 |
| rs12492285 | 144436293 | A | 0.168 | 0.094 | 0.0012 | 0.092 | 0.084 | 0.69 |
| rs4395444 | 126210437 | A | 0.353 | 0.46 | 0.0012 | 0.414 | 0.418 | 0.91 |
| rs12492170 | 138359517 | A | 0.299 | 0.21 | 0.0025 | 0.271 | 0.233 | 0.23 |
| rs6801610 | 144331065 | G | 0.442 | 0.54 | 0.0034 | 0.524 | 0.53 | 0.86 |
| rs1871349 | 144018107 | G | 0.154 | 0.231 | 0.0041 | 0.194 | 0.22 | 0.34 |
| rs2712421 | 129770441 | G | 0.27 | 0.358 | 0.0042 | 0.332 | 0.302 | 0.37 |
| rs6439127 | 129651111 | A | 0.052 | 0.102 | 0.0048 | 0.074 | 0.066 | 0.65 |
| rs2587025 | 147731609 | C | 0.308 | 0.399 | 0.0048 | 0.359 | 0.345 | 0.68 |
| rs1866813 | 138284628 | C | 0.218 | 0.144 | 0.0049 | 0.212 | 0.144 | 0.0088 |
| rs3796180 | 138314818 | T | 0.296 | 0.215 | 0.0053 | 0.273 | 0.232 | 0.2 |
| rs4679257 | 126239279 | T | 0.106 | 0.17 | 0.0053 | 0.107 | 0.146 | 0.082 |
| rs6789065 | 144623118 | A | 0.274 | 0.36 | 0.0056 | 0.339 | 0.345 | 0.85 |
| rs6805170 | 150328788 | G | 0.114 | 0.061 | 0.0057 | 0.146 | 0.112 | 0.15 |
| rs4974501 | 135592551 | A | 0.42 | 0.33 | 0.0059 | 0.358 | 0.373 | 0.64 |
| rs7648426 | 151504183 | A | 0.131 | 0.075 | 0.0065 | 0.092 | 0.094 | 0.94 |
| rs1382270 | 138461351 | C | 0.418 | 0.33 | 0.0069 | 0.409 | 0.395 | 0.66 |
| rs7628692 | 144564710 | G | 0.522 | 0.431 | 0.0071 | 0.455 | 0.436 | 0.58 |
| rs7611217 | 144327151 | G | 0.394 | 0.308 | 0.0076 | 0.339 | 0.317 | 0.51 |
| rs6803636 | 147599739 | C | 0.209 | 0.141 | 0.0084 | 0.17 | 0.201 | 0.27 |
| rs33264 | 124892478 | G | 0.366 | 0.283 | 0.0086 | 0.361 | 0.364 | 0.93 |
| rs4974491 | 135538571 | C | 0.312 | 0.396 | 0.0091 | 0.36 | 0.366 | 0.85 |
| rs6440067 | 143407729 | T | 0.345 | 0.264 | 0.0092 | ND | ||
| rs349558 | 141744377 | T | 0.086 | 0.141 | 0.0092 | 0.097 | 0.095 | 0.94 |
| rs2659690 | 129664467 | A | 0.22 | 0.296 | 0.0093 | 0.278 | 0.251 | 0.37 |
| rs7632370 | 146980190 | T | 0.487 | 0.401 | 0.0099 | 0.392 | 0.468 | 0.024 |
The 27 SNPs that met the significance threshold (p < 0.01) at the screening stage were genotyped in the follow-up study, and they are listed in order of significance. Initial significances are highlighted in boldface if they reached the significance threshold (p < 0.05) in panel 2. Abbreviations are as follows: MAF, minor allele frequency; ND, not done. Follow up was not done for rs6440067, because the SNP assay is not available from Applied Biosystems.
To eliminate false-positive associations occurring by chance, we then evaluated the 27 SNPs that met the significance threshold of 0.01 using panel 2. A single SNP, rs1866813 (p = 0.0049 at the screening stage), among the 26 genotyped SNPs in panel 2 (one TaqMan SNP assay for rs6440067 was unavailable) met the threshold for moving on to panels 3–5 for further genotyping. The minor allele C of rs1866813 in panel 2 was associated with increased risk of DN (p = 0.0088; OR = 1.60; 95% CI = 1.12–2.28) (Table 2). At the second follow-up stage, we were also able to replicate the association for rs1866813 (Table 3) in panel 3 (p = 0.03) and panel 4 (p = 0.021). The combination of all Finnish samples (panels 1–3) resulted in significant association (p = 2.41 × 10−5; OR = 1.42). Association of this SNP in panel 5 (the British Isles cohort) was not significant (p = 0.235). To interpret the results as a whole, we analyzed data from all cohorts (panels 1–5), using the Cochran-Mantel-Haenszel model, which yielded an overall two-tailed p value of 7.07 × 10−6 (OR = 1.33; 95% CI = 1.17–1.51) (Table 3). There was no evidence for heterogeneity of ORs across studies (p > 0.05 for all) with the Pearson χ2 statistic used. The allelic association of rs1866813 with DN at 3q22 remained significant, even after a Bonferroni adjustment for 2805 SNPs tested (padjusted = 0.0198). The consistent finding of association in four out of five cohorts provided evidence against the possibility that our finding was due to population stratification, because it is unlikely that the same kind of stratification exists in each of these disparate population samples. However, the association of rs1866813 did not reach the genome-wide significance level, and further replication is necessary for confirmation of our findings.
Table 3.
Association Results for rs1866813 in All DN Cohorts
| Population | Cases |
Controls |
p Value | Odds Ratio (95% CI) | |
|---|---|---|---|---|---|
| MAFa AA/AC/CCb | MAFa AA/AC/CCb | ||||
| Panel 1 | Finland | 0.218 143/77/12 | 0.144 155/51/5 | 0.0049 | 1.65 (1.16–2.34) |
| Panel 2 | Finland | 0.212 208/98/20 | 0.144 128/42/4 | 0.0088 | 1.60 (1.12–2.28) |
| Panel 3 | Finland | 0.202 296/131/26 | 0.165 427/161/20 | 0.03 | 1.28 (1.02–1.60) |
| Finnish combinedc | 0.209 647/306/58 | 0.157 710/254/29 | 2.41 × 10−5 | 1.42 (1.20–1.66) | |
| Panel 4 | Iceland | 0.194 52/25/3 | 0.109 85/19/2 | 0.021 | 1.98 (1.10–3.54) |
| Panel 5 | British Isles | 0.143 516/180/11 | 0.127 561/155/16 | 0.235 | 1.14 (0.92–1.41) |
| All combinedd | 0.182 1215/511/72 | 0.142 1356/428/47 | 7.07 × 10−6 | 1.33 (1.17–1.51) |
Minor-allele frequency and genotype counts in the cases and controls; the allelic ORs with 95% confidence intervals (95% CI) and the two-tailed p values on an allele-based test are presented.
Minor-allele frequency.
Genotype counts.
Samples include only Finnish samples (panels 1–3).
Association for the C allele of rs1866813 was calculated in all samples (panels 1–5) with the Cochran-Mantel-Haenszel model.
We used logistic-regression analysis to estimate the genotypic effects of rs1866813 on DN on the basis of all Finnish samples (panels 1–3). The heterozygous carriers have significantly higher risk than do noncarriers (OR = 1.32; 95% CI = 1.08–1.61; p = 0.0056), whereas individuals who are homozygous with respect to the C allele have increased risk in comparison to heterozygous carriers (OR = 1.66; 95% CI = 1.03–2.67; p = 0.037). This implicates a codominant allele-dose effect. We then tested for association using a codominant model (additive genotype coding), which resulted in a p value of 4.7 × 10−5 (OR = 1.38; 95% CI = 1.18–1.62). The effect size of 1.38 was not strong enough to account for the LOD score of 2.67 observed in our linkage study.9
In the haplotype analysis, we focused on the segment in which rs1866813 is located. Tag SNPs flanking 100 kb of rs1866813 were selected. In total, 24 SNPs genotyped in panel 1, spanning about 250 kb, were included for analysis using the expectation-maximization algorithm (the Haploview program). This analysis estimated five haplotype blocks. A 44 kb block containing rs1866813 (138250708–138294816 bp, NCBI build 36), flanked by recombination hotspots with multiallelic LD measures (D′) of 0.73 was observed. This block consisted of four tag SNPs (rs17374749, rs6766709, rs1866813, and rs16844489). Only five haplotypes with a frequency > 5% in cases and controls in this block were estimated, and they made up 99% of the total chromosomes observed. The rs1866813 C-allele-carrying haplotype (GTCT) was more frequently observed in the cases than in the controls (21% in cases versus 14% in controls, p = 0.01) (Table 4). However, no haplotypes observed showed stronger association than the single SNP rs1866813 (p = 0.0049) genotyped in panel 1.
Table 4.
Estimated Haplotypes of Four SNPs, Consisting of rs17374749, rs6766709, rs1866813, and rs16844489; and Three SNPs, Consisting of rs62408925, rs9826507, and rs1866813, in the rs1866813-Carrying Block in Panel 1
| Haplotype | Cases | Controls | χ2 | p Value | pperm Valueb |
|---|---|---|---|---|---|
| Four SNP Haplotypes | (n = 468a) | (n = 426) | |||
| GTAT | 185 (0.395) | 203 (0.477) | 6.062 | 0.0138 | 0.1795 |
| GAAT | 92 (0.196) | 71 (0.166) | 1.338 | 0.2474 | 0.9997 |
| GTCT | 98 (0.21) | 61 (0.144) | 6.377 | 0.0116 | 0.1526 |
| ATAT | 58 (0.125) | 65 (0.152) | 1.343 | 0.2465 | 0.9997 |
| GAAC | 28 (0.06) | 22 (0.052) | 0.283 | 0.5947 | 1.0 |
| Three SNP Haplotypes | (n = 450) | (n = 420) | |||
| CGA | 220 (0.49) | 216 (0.513) | 0.495 | 0.4819 | 0.8214 |
| CAA | 127 (0.281) | 139 (0.332) | 2.59 | 0.1076 | 0.2716 |
| TGC | 100 (0.222) | 61 (0.145) | 8.537 | 0.0035 | 0.0062 |
Estimated haplotype counts in cases and controls are presented, and corresponding frequencies are shown in parentheses.
Total chromosomal number in the group.
The empirical p values were obtained on the basis of 10,000 permutations.
The variant identified is located in a noncoding region (∼750 kb) between IL20RB [MIM 605621] and SOX14 [MIM 604747] and resides about 70 kb downstream of a cluster of three genes; IL-20RB, NCK1 [MIM 600508], and TMEM22 (Figure 1A). IL-20RB encodes the interleukin-20 receptor B that is generally expressed in endothelial cells,19 but its expression in human glomeruli was undetectable by immunofluorescence staining with two polyclonal IL-20RB antibodies (K-13 and K-17; Santa Cruz Biotechnology) (data not shown). Nck1 is an intracellular adaptor protein that is involved in actin polymerization. In podocytes, Nck1 links the slit-diaphragm protein nephrin to the actin cytoskeleton, and this interaction has been shown to be essential for nephrin-dependent reorganization of actin in podocyte injury.20,21 Nck1 is also expressed in endothelial cells, but its expression or role in DN has not been reported. TMEM22 encodes a transmembrane protein of unknown functions, but by RT-PCR, we were able to detect a low level of Nck1 expression in mammalian glomeruli (data not shown).
Figure 1.
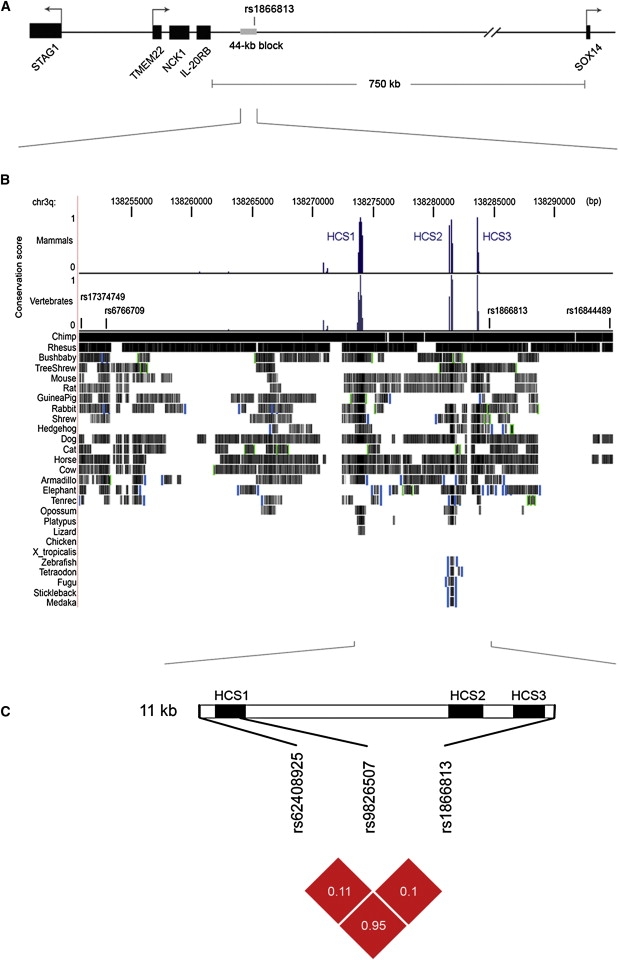
Schematic View of the rs1866813-Carrying Region and Comparative Genomic Analysis
(A) Genomic structure in a 1.2 Mb interval (137.8–139 Mb, NCBI build 36) on chromosome 3q22. Black bars indicate five genes, and a gray bar indicates the 44 kb haplotype block carrying rs1866813. Orientation of the genes is indicated with arrows.
(B) Multiple alignments of 28 vertebrate species, created with the 44 kb LD block in the UCSC Genome Browser (chr3: 138,250,700–138,294,820 bp). Conservation scores for the placental mammal subset (17 species plus human) and a subset of ten other vertebrates (eight nonmammalian species, platypus, and opossum) are displayed as a histogram in the upper panel, in which the height reflects the size of the score. Short vertical lines indicate positions of four SNPs. Pairwise alignments of each species with the human genome are displayed below the histogram, where the species aligned are listed on the left side. A grayscale density plot indicates alignment quality. Vertical blue bar and green square brackets suggest discontinuities in the genomic context of the aligned DNA in the aligning species.
(C) Schematic view of three conserved elements (HCS1, HCS2, and HCS3) in an 11 kb region between rs62408925 and rs1866813 and pairwise LD measures of rs62408925, rs9826507, and rs1866813 were illustrated in the lower panel. Black bars indicate conserved elements. Red blocks indicate D′ = 1.0, and the white number in the blocks indicates the value of r2.
It is unclear how the associated variant could influence the development of DN, given that it resides about 70 kb from the nearest genes, but it is possible that long-range regulatory sequences are a part of the mechanism. To explore this, we analyzed the 44 kb block for the presence of evolutionarily conserved genomic elements. Three elements, designated HCS1, HCS2, and HCS3, which are highly conserved (conservation scores > 75%) among both mammals and nonmammalian vertebrates, were identified in close vicinity of rs1866813 (Figure 1B).
We then sequenced the three conserved elements in 46 samples. Interestingly, a SNP, rs62408925, 500 bp upstream of HCS1, showed a very high correlation with the associated SNP, rs1866813 (r2 = 0.95 and D′ = 1.0). After we genotyped this SNP (rs62408925) in panel 1 by sequencing, the T allele of rs62408925 was also significantly associated with increased risk of DN (p = 0.0051). Thus, the second associated variant in strong LD with rs1866813 was identified. Because of its high correlation with rs1866813, SNP rs62408925 can be estimated as giving almost the same associations as rs1866813 in our remaining panels. The three conserved sequences (HCS1, HCS2, and HCS3) reside in the high-LD region (11 kb in size) between rs62408925 and rs1866813. In addition, a SNP, rs9826507, was detected within HCS1 and showed no allelic association (p = 0.10). However, individuals homozygous for the A allele of rs9826507 had significantly reduced risk of DN, assuming the recessive model (p = 0.006; OR = 0.38; 95% CI = 0.19–0.77). Pairwise LD measures (D′ and r2) of these three SNPs were illustrated (Figure 1C). We also performed haplotype analysis of three SNPs (rs62408925, rs9826507, and rs1866813) on panel 1, resulting in three common haplotypes (accounting for 99% of total haplotypes observed). The at-risk TGC haplotype was significantly associated with DN (p = 0.0035, pperm = 0.0062), showing a frequency of 22% in cases and of 14.5% in controls. The frequency distribution of the at-risk TGC haplotype between cases and controls was very similar to that of the at-risk GTCT haplotype (Table 4).
Discussion
In the present study, we have identified genetic variants, in the noncoding region at 3q22, that are associated with type 1 diabetic nephropathy in three Finnish cohorts. The association was confirmed in an Icelandic set. Furthermore, we found that a region very high in LD between two correlated variants, associated with DN, contains three elements conserved across multiple species. It suggests that the variants identified, together with susceptibility loci at other chromosomal regions, might influence the risk of DN through potential regulatory mechanisms. Our results identifying associated variants in a noncoding region illustrate the advantage of unbiased fine-mapping approaches using high-density SNP genotyping, because a candidate-gene approach at 3q might overlook this locus.22
Special emphasis was made to ensure authenticity of the phenotypes of cases and controls. Thus, only cases with macroalbuminuria or ESRD were accepted, whereas microalbuminria cases were excluded from the study. Also, although a minimum of 15 years of normoalbuminuria after the onset of diabetes is commonly used as criterion for controls, some DN patients with a long duration of diabetes might be incorrectly defined as controls,23 especially as a result of treatment with an ACE inhibitor or an angiotensin-receptor blocker.24 Therefore, efforts were made to ensure that none of control individuals had had proteinuria at any stage despite their long duration of diabetes. The MAF of rs1866813 in cases and controls matched perfectly between panel 1 and panel 2. In panel 3, the MAF (16.5%) of rs1866813 in controls was increased, compared to that (14.4%) in panels 1 and 2, but the MAF in cases was comparable between the three Finnish panels. Here, control individuals in panels 1 and 2 were thoroughly and repeatedly followed up and characterized for at least 20 years. Therefore, the phenotypes of these panels have higher confidence. However, panel 3 fulfilled only a single prospective review, with relatively short follow-up after diagnosis of diabetes (15 years). Mild misclassification in panel 3 might lead to inconsistent MAF in controls, compared to that of panels 1 and 2. This could explain why the associated SNP (rs1866813) shows a very similar result between panels 1 and 2, whereas only weak significant association was observed in panel 3. Another possibility for this result is random fluctuation in allele frequencies of the SNP in panel 3, given that there was no evidence for heterogeneity of ORs across the three Finnish panels. In addition, the “jackpot” effect25 could be a possible explanation for the failure to find association in the British Isles cohort; the result of sampling variations. The effect size might somehow be overestimated in early samples, such as panels 1 and 2, so that the true effect might not be easily detected in subsequent studies. Thus, further replication across populations might clarify our findings.
The DN-associated variants are intriguing, although it is positioned quite distant from genes within an apparent noncoding region. A similar result has been reported for a 9q locus in a genome-wide association study in patients with type 2 diabetes.26 Here, the variants identified are located about 70 kb downstream from a three-gene cluster (IL-20RB, NCK1, and TMEM22). Two highly correlated variants (r2 = 0.95) associated with DN make up a strong LD region that covers three conserved elements. It remains to be shown whether or not genotypes of the associated variants are correlated with gene-expression levels of the nearby genes or whether these conserved elements are really cis-acting sequences that regulate expression of the nearby genes.
The current understanding is that risk alleles for DN exist in the general population but carriers do not develop DN until long-term exposure to hyperglycemia has occurred. Given that the first manifestation of DN is microalbuminuria, glomerular endothelial cells and podocytes might be affected first. Therefore, expression of target gene(s) influenced by the risk variants should be detectable in these cells. The IL-20RB gene may be excluded, because its expression in glomeruli was not observed. Both NCK1 and TMEM22 genes were expressed in glomeruli. Nck1 has been shown to be a crucial link between phosphorylated nephrin and the actin cytoskeleton during the development of podocyte foot processes, as well as in their regeneration during repair of effaced foot processes after glomerular injury.20,21
At the present, we cannot explain how the variants identified can contribute to abnormal renal extracellular-matrix accumulation and consequent GBM thickening in hyperglycemia. However, the present results indicate that the associated variants might influence DN risk through potential remote gene-control27 mechanisms.
Supplemental Data
Supplemental Data include a list of Finnish Diabetic Nephropathy Study Group members and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
HapMap, http://www.hapmap.org/
Haploview, http://www.broad.mit.edu/mpg/haploview/
Online Genetic Power Calculator, http://pngu.mgh.harvard.edu/∼purcell/gpc/
UCSC Genome Browser, http://genome.ucsc.edu/
Acknowledgments
We thank all study subjects and physicians for their cooperation. The authors thank Bei Yang from Stockholm University for her statistical and logistic support and her helpful discussions. This work was supported in part by grants from the Foundations of Novo Nordisk, Knut and Alice Wallenberg, Söderberg and Hedlund, the Swedish Medical Research Council, the Swedish Foundation for Strategic Research, the Swedish Diabetes Association, the Sigrid Juselius Foundation, the Academy of Finland (grants 38387 and 46558), the Finnish Medical Society, and the Northern Ireland Kidney Research Fund. We acknowledge all the physicians and nurses at each center participating in the collection of the FinnDiane patients (see ref. 14 or online appendix). We acknowledge the technical assistance of Jill Kilner. The Warren 3 / UK GoKinD Study Group was jointly funded by Diabetes UK and the Juvenile Diabetes Research Foundation and includes the following investigators: A. P. Maxwell, A. J. McKnight, D. A. Savage (Belfast); J. Walker (Edinburgh); S. Thomas, G. C. Viberti (London); A. J. M. Boulton (Manchester); S. Marshall (Newcastle); A. G. Demaine, B. A. Millward (Psymouth); and S. C. Bain (Swansea). The All-Ireland Diabetic Nephropathy collection is supported by the Northern Ireland Research and Development Office and the Health Research Board, Republic of Ireland, and additional investigators include D. Sadlier and H. Brady (University College Dublin, Republic of Ireland).
References
- 1.Parving H.-H., Mauer M., Ritz E. Diabetic nephropathy. In: Brenner B.M., editor. The Kidney. Seventh Edition. Saunders; Philadelphia, USA: 2004. pp. 1777–1818. [Google Scholar]
- 2.American Diabetes Association Diabetic nephropathy. Diabetes Care. 2002;25 (supp 1):S85–S89. [Google Scholar]
- 3.Andersen A.R., Christiansen J.S., Andersen J.K., Kreiner S., Deckert T. Diabetic nephropathy in type 1 (insulin-dependent) diabetes: an epidemiological study. Diabetologia. 1983;25:496–501. doi: 10.1007/BF00284458. [DOI] [PubMed] [Google Scholar]
- 4.Krolewski A.S., Warram J.H., Christlieb A.R., Busick E.J., Kahn C.R. The changing natural history of nephropathy in type 1 diabetes. Am. J. Med. 1985;78:785–794. doi: 10.1016/0002-9343(85)90284-0. [DOI] [PubMed] [Google Scholar]
- 5.Seaquist E.R., Goetz F.C., Rich S., Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N. Engl. J. Med. 1989;320:1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 6.Quinn M., Angelico M.C., Warram J.H., Krolewski A.S. Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia. 1996;39:940–945. doi: 10.1007/BF00403913. [DOI] [PubMed] [Google Scholar]
- 7.Harjutsalo V., Katoh S., Sarti C., Tajima N., Tuomilehto J. Population-based assessment of familial clustering of diabetic nephropathy in type 1 diabetes. Diabetes. 2004;53:2449–2454. doi: 10.2337/diabetes.53.9.2449. [DOI] [PubMed] [Google Scholar]
- 8.Iyengar S.K., Freedman B.I., Sedor J.R. Mining the genome for susceptibility to diabetic nephropathy: the role of large-scale studies and consortia. Semin. Nephrol. 2007;27:208–222. doi: 10.1016/j.semnephrol.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Österholm A.-M., He B., Pitkäniemi J., Albinsson L., Berg T., Sarti C., Tuomilehto J., Tryggvason K. Genome-wide scan for type 1 diabetic nephropathy in the Finnish population reveals suggestive linkage to a single locus on chromosome 3q. Kidney Int. 2007;71:140–145. doi: 10.1038/sj.ki.5001933. [DOI] [PubMed] [Google Scholar]
- 10.Imperatore G., Hanson R.L., Pettitt D.J., Kobes S., Bennett P.H., Knowler W.C. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes. 1998;47:821–830. doi: 10.2337/diabetes.47.5.821. [DOI] [PubMed] [Google Scholar]
- 11.Moczulski D.K., Rogus J.J., Antonellis A., Warram J.H., Krolewski A.S. Major susceptibility locus for nephropathy in type 1 diabetes on chromosome 3q: results of novel discordant sib-pair analysis. Diabetes. 1998;47:1164–1169. doi: 10.2337/diabetes.47.7.1164. [DOI] [PubMed] [Google Scholar]
- 12.Bowden D.W., Colicigno C.J., Langefeld C.D., Sale M.M., Williams A., Anderson P.J., Rich S.S., Freedman B.I. A genome scan for diabetic nephropathy in African Americans. Kidney Int. 2004;66:1517–1526. doi: 10.1111/j.1523-1755.2004.00915.x. [DOI] [PubMed] [Google Scholar]
- 13.Parving H.-H., Hommel E., Mathiesen E., Skøtt P., Edsberg B., Bahnsen M., Lauritzen M., Hougaard P., Lauritzen E. Prevalence of microalbuminuria, arterial hypertension, retinopathy, and neuropathy in patients with insulin dependent diabetes. BMJ. 1988;296:156–160. doi: 10.1136/bmj.296.6616.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorn L.M., Forsblom C., Fagerudd J., Thomas M.C., Pettersson-Fernholm K., Saraheimo M., Wadén J., Rönnback M., Rosengård-Bärlund M., Björkesten C.G. Metabolic syndrome in type 1 diabetes: association with diabetic nephropathy and glycemic control (the FinnDiane study) Diabetes Care. 2005;28:2019–2024. doi: 10.2337/diacare.28.8.2019. [DOI] [PubMed] [Google Scholar]
- 15.de Bakker P.I., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat. Genet. 2006;38:663–667. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 16.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 18.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh M.Y., Chen W.Y., Jiang M.J., Cheng B.C., Huang T.Y., Chang M.S. Interleukin-20 promotes angiogenesis in a direct and indirect manner. Genes Immun. 2006;7:234–242. doi: 10.1038/sj.gene.6364291. [DOI] [PubMed] [Google Scholar]
- 20.Jones N., Blasutig I.M., Eremina V., Ruston J.M., Bladt F., Li H., Huang H., Larose L., Li S.S., Takano T. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 21.Verma R., Kovari I., Soofi A., Nihalani D., Patrie K., Holzman L.B. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vionnet N., Tregouet D., Kazeem G., Gut I., Groop P.H., Tarnow L., Parving H.H., Hadjadj S., Forsblom C., Farrall M. Analysis of 14 candidate genes for diabetic nephropathy on chromosome 3q in European populations. Diabetes. 2006;55:3166–3174. doi: 10.2337/db06-0271. [DOI] [PubMed] [Google Scholar]
- 23.Arun C.S., Stoddart J., Mackin P., MacLeod J.M., New J.P., Marshall S.M. Significance of mircoalbuminuria in long-duration type 1 diabetes. Diabetes Care. 2003;26:2144–2149. doi: 10.2337/diacare.26.7.2144. [DOI] [PubMed] [Google Scholar]
- 24.Ritz E., Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat. Clin. Pract. Nephrol. 2006;2:562–567. doi: 10.1038/ncpneph0298. [DOI] [PubMed] [Google Scholar]
- 25.Hirschhorn J.N., Lohmueller K., Byrne E., Hirschhorn K. A comprehensive review of genetic association studies. Genet. Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Diabetes Genetics Initiative Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 27.De Gobbi M., Viprakasit V., Hughes J.R., Fisher C., Buckle V.J., Ayyub H., Gibbons R.J., Vernimmen D., Yoshinaga Y., de Jong P. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312:1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.