

Abstract
Although high-density SNP genotyping platforms generate a momentum for detailed genome-wide association (GWA) studies, an offshoot is a new insight into population genetics. Here, we present an example in one of the best-known founder populations by scrutinizing ten distinct Finnish early- and late-settlement subpopulations. By determining genetic distances, homozygosity, and patterns of linkage disequilibrium, we demonstrate that population substructure, and even individual ancestry, is detectable at a very high resolution and supports the concept of multiple historical bottlenecks resulting from consecutive founder effects. Given that genetic studies are currently aiming at identifying smaller and smaller genetic effects, recognizing and controlling for population substructure even at this fine level becomes imperative to avoid confounding and spurious associations. This study provides an example of the power of GWA data sets to demonstrate stratification caused by population history even within a seemingly homogeneous population, like the Finns. Further, the results provide interesting lessons concerning the impact of population history on the genome landscape of humans, as well as approaches to identify rare variants enriched in these subpopulations.
Main Text
The abundance of high-resolution, genome-wide SNP data has recently provided a new level of insight into population structure across populations.1–4 However, only limited knowledge exists concerning the population structure within population isolates, which have traditionally been considered to show reduced genetic diversity. The Finnish population provides an interesting example of population history in which the structure has been molded by both old and relatively recent events. Compared to mainland Europe, the genome of Finns exhibits a decrease of genetic diversity5 and an increase in linkage disequilibrium (LD)6,7 that are hallmarks of populations with a recent founding bottleneck. Numerous Mendelian disease genes have been identified taking advantage of this fact and it has been proposed that gene mapping for more complex traits should also be especially advantageous in this population.6,8
The population history of Finland is well known. The region has been inhabited for 10,000 years, but two major migration waves have mostly molded the gene pool of current Finns. The first wave approximately 4000 years ago came from the east, whereas the second came from the south and west some 2000 years ago. For centuries, only the coastal regions were inhabited, often referred to as an early-settlement region (Figure S1 available online). A third, major migratory movement was internal and originated from a limited region in the early settlement in the sixteenth century resulting in the late settlement (Figure S1)—geographically wide inland areas in the northern and eastern parts of the country became slowly inhabited, each village established by a small number of settlers resulting in genetically distinct subpopulations isolated by distance.9
Thus, the populations of the subisolates all originate from the initial early-settlement population representing the outcomes of classical bottleneck effects. In this study, we use samples carefully ascertained from these subisolates in different regions of Finland to demonstrate the impact of fine-scale population history on genetic substructure of an isolated population by utilizing genome-wide SNP data.
We sampled individuals on the basis of the birthplaces of the parents (91% known) from ten geographical regions that represent distinct eras in the population history of Finland to study the effect of bottlenecks and isolation. The parents were born before the 1950s excluding most of the genetic admixture by the migratory movement of industrialization after the Second World War. Studies on rare Finnish genetic diseases have demonstrated that the birthplaces of grandparents reveal the geographical origin of the founder mutation, even though patients themselves show no distinct geographical clustering.10–12
We ascertained samples from four regions of the early settlement: the south coastal region (early-settlement south [ESS]), South Oulu (early-settlement west [ESW1]), North Oulu (ESW2), and the Tornio-River valley in West Lapland (early-settlement north [ESN]) (Figure 1B). Samples from South Ostrobothnia (late-settlement west [LSW]), Central Finland (late-settlement central [LSC]) and Central Lapland (late-settlement north [LSN]) were ascertained to represent older regions of the late settlement established 500–1000 years ago. To represent the youngest subisolates in northeastern Finland established 300–400 years ago as a result of the internal migration in the sixteenth century (from south to north), we ascertained individuals from South Kainuu (isolate south [ISS]), North Kainuu (isolate central [ISC]), and East Lapland (isolate north [ISN]) (Figure 1B). We constructed all the subisolates according to time scale of inhabitation, taking into account the Finnish dialect borders and their subdivision, which is known to correspond precisely to their population history. In addition, 162 anonymous individuals from the capital region of Helsinki (HEL), which has experienced substantial migration from other regions, especially after World War II, were selected to represent the general admixture of the current Finnish population. To compare the data with the neighboring country that has shared hundreds of years of history with Finland, we included 302 Swedish individuals (SWE). Thus, the total study sample is 1395 individuals.
Figure 1.
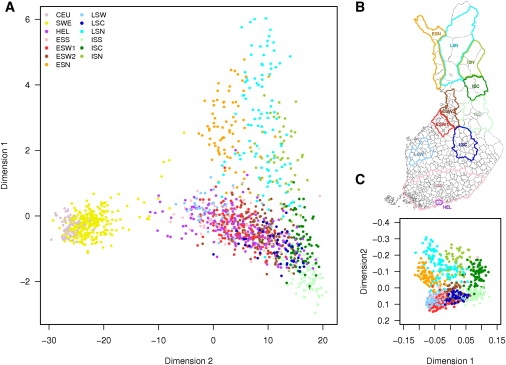
Population Substructure
(A) Results of multidimensional scaling in the subisolates as well as the Helsinki, Swedish, and CEU populations.
(B) Geographical locations of the subisolates.
(C) Results of multidimensional scaling in the subisolates shown on the map in 1b. The following abbreviations are used: CEU, HapMap CEPH; SWE, Sweden; HEL, Helsinki; ESS, early-settlement south; ESW1, early-settlement west 1; ESW2, early-settlement west 2; ESN, early-settlement north; LSW, late-settlement west; LSC, late-settlement central; LSN, late-settlement north; ISS, isolate south; ISC, isolate central; and ISN, isolate north.
Samples were collected across Finland and Sweden as part of larger studies and the summary data for each population group are presented in Table S1 available online. The Swedish samples (SWE, n = 302) are a subset of the GenomeEUtwin study and consist of female monozygotic twins13 with no data for the birthplace of the parents. The Health2000 cohort is a national sample originally collected to provide a comprehensive picture of health in the population aged over 18 in Finland in the years 2000–2001.14 Genome-wide (GW) SNP data as well as information concerning parents' birthplaces was available for a subset of 341 individuals. The Northern Finland Birth Cohort 1966 (NFBC66) is a longitudinal birth cohort of individuals born in the two most northern provinces in Finland in 1966.15 GW SNP data as well as information about parents' birthplaces was available for 1869 individuals. GW SNP data, but only limited information about parents' birthplaces, were available for samples from South Ostrobothnia (LSW) and Central Finland (LSC). Thus, we had samples that were collected within a particular geographically limited region in Finland, LSW and LSC, and these groups were augmented by samples from the population-based cohorts. Sample groups for the remaining regions (ESS, ESW1, ESW2, ESN, LSN, ISS, ISC, and ISN) were created with samples from NFBC66 and Health2000 and only individuals with both parents born within the geographically restricted regions were included. The population controls from Helsinki are part of a Finnish-US collaborative project studying the genetic basis of brain aneurysms. All study samples were kept anonymous with no possibility of identification of individual subjects. This study has been approved by the ethical committees of the Joint Authority for the Hospital District of Helsinki and Uusimaa, Finland.
Genomic DNA was genotyped with the Illumina 300K platforms (Table S1). The genotyping for three groups was performed at the Broad Institute, Cambridge, MA, USA. Samples from LSW and LSC were genotyped with the Illumina HumanHap300 chip and NFBC66 with the HumanCNV370-duo chip (Illumina, San Diego, CA USA). Health2000 samples were genotyped at DeCode Genetics in Reykjavik, Iceland, with the Illumina HumanHap300-duo chip. The genotyping for the Swedish samples was performed with the HumanHap300-duo chip in Uppsala, Sweden, (Table S1). The genotyping for Helsinki samples was performed with the HumanCNV370-duo chip at Yale University, School of Medicine, New Haven, CT, USA. The genotyping was performed according to the manufacturer's instruction.
Given that the samples were genotyped with three different SNP chips, we first compared the SNP content between Illumina HumanHap300 (317K SNPs), HumanHap300-duo (318K SNPs), and HumanCNV370-duo chips. All the SNPs on the HumanHap300-duo were present on the HumanCNV370 chip, whereas ∼314K SNPs from the HumanHap300 were also present on the HumanHap300-duo and the HumanCNV370-duo chips. All SNPs were called on TOP-strand orientation with same GeneCall criteria and Illumina's default genotype cluster positions in each project. Common SNPs between all chip types were extracted from each data set after quality check: SNPs and samples with success rate <95% in each study set were excluded. Genotype data were combined with PLINK and a strict threshold of success (≥99%) was required for SNPs to be accepted. The final data set consisted of 231,116 SNPs and the average genotyping success rate was >99.6% in each subpopulation (Table S1). All genomic positions are given according to NCBI build 35.
Analyses of the genome-wide SNP data were performed with PLINK16 and Eigensoft 2.0.17 Estimation of the proportion of the genome shared identical by descent (IBD) was performed with PLINK to identify closely related individuals. We excluded one individual from all pairs sharing > 10% of their genome IBD (n = 50) to remove first, second and third degree relatives. Hardy-Weinberg equilibria were calculated in PLINK using the exact option and including all unrelated samples in each subpopulation.
Population stratification analyses were performed with all autosomal SNPs passing QC measures. First the proportion of alleles shared IBS between all pairs of individuals was determined and standard classical (metric) multidimensional scaling was used to extract the first four dimensions from the data in the IBS-sharing matrix for visualization. To estimate the difference of IBS-sharing between each of the subisolates and the general Finnish population, we permuted the group membership labels between the subisolates and the general Finnish population 10,000 times. Empirical p values for the difference between groups were calculated from the average IBS sharing within and between groups. Principal components and their statistical significance were determined with Eigensoft 2.0 with autosomal SNPs.17 Statistical significance between populations was evaluated by summing the Anova significance statistics for the ten most significant eigenvectors. The result is approximate to a chi-square test with 10 degrees of freedom. Fst analysis was performed for all pairs of populations with allele frequencies for all autosomal SNPs as described in Li et al.2
We performed multidimensional scaling (MDS) of pairwise identity by state (IBS) sharing data to delineate and visualize the population structure both within Finland as well as with respect to the Swedish and the HapMap European (CEU) population (Figure 1A and Figure S2). As expected on the basis of the origins of Scandinavian populations, both Finns and Swedes resemble CEU, with some of the most northern Finns clustering slightly closer to the Asians, potentially reflecting the “Eastern” migration wave in the inhabitation of Finland. However, PCA analysis of several European populations reported that Finns do not cluster with other European populations.18 Average genome-wide IBS sharing was higher within the CEU, Finns, and Swedes than between groups, with the highest similarities within the Finns (Figure S3). Interestingly, the two primary dimensions of the MDS analysis of Finns correspond remarkably well to the east-west and north-south directions, respectively, in concordance with the direction of the internal migration (Figure 1C). Principal component analysis with Eigensoft show that the first two principal components explain 29% of the variance observed in the data (Table S2 and Figure S4). The first three principal components reflect small differences over the whole genome and are not driven by a few highly significant loci (Figures S5 and S6). The additional principal components added little information for the resolution of subgroups but show high statistical significance (Figure S7 and Table S2). The youngest subisolates in northeastern Finland (ISS, ISC, and ISN) showed highest IBS similarity (Figure S3) and in these subisolates separation into neighboring municipalities is possible on an exceptionally fine scale even on the north-south gradient (Figure S8). This reflects the time scale of migration and founder effect followed by strong isolation. Accordingly, recent genome-wide SNP data collected from the study sample ascertained from the western coast of Finland reported that the first two principal components correspond to geographical origin of samples.19 Similar results suggesting east-west differences were recently reported in a small-scale study of genome-wide association (GWA) data.20
An east-west boundary line dividing Finland from northwest to southeast and reflecting a relative migration block was formed in 1323 by the border defined in the peace treaty between Russia and Sweden and lasted for several centuries ending with the internal migration in the sixteenth century. This border defined “Eastern” and Western parts of the country for centuries and has created differences in anthropological features, dialects, and prevalence of various traits such as cardiovascular diseases still observed today.21 Our high-density genome-wide SNP data would relate the western Finland early settlement close to other European populations, as previously suggested by Y chromosome data.22 This would be in line with the recent GWA-based studies of the substructure of European populations that revealed principal components corresponding to north-south distinction and, if only the northern European populations were included, distinction on an east-west gradient.3,4
In general, the average IBS similarity within subgroups was greater than between groups, suggesting true genetic subpopulations agreeing with the well-documented multiple bottlenecks in Finnish population history. We determined an empirical p value for the differences in IBS sharing between each of the subgroups and the general Finnish population (HEL) by permuting the group membership 10,000 times and calculated average IBS sharing within and between groups. Significant differences were observed except for ESS and LSC (Table 1 and Figure S3), and these results were confirmed with the population differentiation test of Eigensoft (Table S3). To further illustrate the effects this substructure would have on a case-control GWA study in which cases were genealogically ascertained from a strictly defined subisolate and controls represent a more admixed population, we calculated the genomic inflation factor (λ)23 in each subisolate versus the HEL population (Table 1 and Figure S9). All subisolates except for ESS show λ > 1.05, an indication of inflation of the median test statistic. Thus, a priori information of the geographical origin of cases and controls is beneficial when association studies are planned and performed, even within population isolates. We also calculated Fst for all pairs of populations to obtain a measure of subpopulation difference that is less sensitive to sample size. The results agree with the MDS- and IBS-sharing results, with largest differences between the most eastern and most western subpopulations, and when compared to the HEL population, highest Fst was observed in the youngest subisolates (Table 2). The Fst values separating the most-western Finnish populations from the most-eastern ones are of the same magnitude as Fst values obtained with GWA data from individuals with Northwest and Southeast European ancestry, indicating substantial differences in subpopulations within Finland.4
Table 1.
Tests of Differences between the Subisolates and the General Finnish Population
| Between-Group IBS Difference Test | Genomic Inflation Factor | |
|---|---|---|
| CEU | <1.0E-05 | 2.063 |
| SWE | <1.0E-05 | 2.941 |
| ESSa | 0.484 | 1.023 |
| ESW1a | <1.0E-05 | 1.331 |
| ESW2a | <1.0E-05 | 1.220 |
| ESNa | <1.0E-05 | 1.503 |
| LSWb | 0.001 | 1.248 |
| LSCb | 0.998 | 1.100 |
| LSNb | <1.0E-05 | 1.563 |
| ISSc | <1.0E-05 | 1.570 |
| ISCc | <1.0E-05 | 1.782 |
| ISNc | 1.5E-04 | 1.562 |
Values for the IBS test are p values, and median chi-square values for the genomic inflation factor. Population abbreviations are the same as those used in Figure 1.
Early settlement.
Late Settlement.
Isolate.
Table 2.
Fst Values for Subpopulations
| CEU | SWE | HEL | ESS | ESW1 | ESW2 | ESN | LSW | LSN | LSC | ISS | ISC | ISN | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CEU | 0.001 | 0.007 | 0.006 | 0.007 | 0.008 | 0.008 | 0.007 | 0.009 | 0.009 | 0.012 | 0.012 | 0.011 | |
| SWE | 0.001 | 0.005 | 0.004 | 0.005 | 0.006 | 0.006 | 0.005 | 0.007 | 0.007 | 0.010 | 0.010 | 0.009 | |
| HEL | 0.007 | 0.005 | 0.000 | 0.001 | 0.001 | 0.002 | 0.001 | 0.002 | 0.001 | 0.002 | 0.004 | 0.003 | |
| ESS | 0.006 | 0.004 | 0.000 | 0.001 | 0.001 | 0.003 | 0.002 | 0.002 | 0.001 | 0.003 | 0.004 | 0.004 | |
| ESW1 | 0.007 | 0.005 | 0.001 | 0.001 | 0.001 | 0.003 | 0.002 | 0.003 | 0.001 | 0.003 | 0.004 | 0.004 | |
| ESW2 | 0.008 | 0.006 | 0.001 | 0.001 | 0.001 | 0.003 | 0.002 | 0.002 | 0.001 | 0.002 | 0.003 | 0.003 | |
| ESN | 0.008 | 0.006 | 0.002 | 0.003 | 0.003 | 0.003 | 0.003 | 0.002 | 0.004 | 0.005 | 0.006 | 0.005 | |
| LSW | 0.007 | 0.005 | 0.001 | 0.002 | 0.002 | 0.002 | 0.003 | 0.004 | 0.003 | 0.005 | 0.006 | 0.005 | |
| LSN | 0.009 | 0.007 | 0.002 | 0.002 | 0.003 | 0.002 | 0.002 | 0.004 | 0.003 | 0.004 | 0.004 | 0.002 | |
| LSC | 0.009 | 0.007 | 0.001 | 0.001 | 0.001 | 0.001 | 0.004 | 0.003 | 0.003 | 0.002 | 0.003 | 0.004 | |
| ISS | 0.012 | 0.010 | 0.002 | 0.003 | 0.003 | 0.002 | 0.005 | 0.005 | 0.004 | 0.002 | 0.003 | 0.004 | |
| ISC | 0.012 | 0.010 | 0.004 | 0.004 | 0.004 | 0.003 | 0.006 | 0.006 | 0.004 | 0.003 | 0.003 | 0.004 | |
| ISN | 0.011 | 0.009 | 0.003 | 0.004 | 0.004 | 0.003 | 0.005 | 0.005 | 0.002 | 0.004 | 0.004 | 0.004 |
Population abbreviations are the same as those used in Figure 1.
Next, we addressed the LD to further explore the genomic structure of the subpopulations. For analysis of LD, the square correlation coefficient (r2)24 was calculated in 70 SNP windows for all SNPs passing QC on chromosomes 22 and 3q (n = 3960 and n = 7824, respectively) as chromosome 22 has been used in a previous study addressing LD in global population isolates.6 Chromosome 3q was randomly chosen to confirm that the LD structure was not a chromosome-specific artifact for chromosome 22. Linkage disequilibrium unit (LDU) maps were constructed for the same chromosomes with the LDMAP program.6,25 The LDU scale is constructed from the product of physical (kb) distance and a parameter describing the exponential decline in association with distance computed for each interval between adjacent SNPs. The resultant map has additive distances and is an LD analog of the linkage map. Whereas the contours of the map correspond strongly to the linkage map, the overall map lengths reflect time since an effective population bottleneck. Map intervals with LDU ≥ 2.5 are termed “holes,” and these align closely with regions of intense recombination6 but may also reflect local variations in marker coverage.
The proportion of SNP pairs in different r2 bins are presented in Figure 2A. Similar results were obtained for chromosome 3q (Figure S10). The late-settlement regions display more SNP pairs in high LD bins compared to both the general Finnish and Swedish populations. The extent of LD, as well as the number of SNP pairs in high LD (r2 > 0.7) located >20 kb apart, was highest in the youngest subisolates and significantly higher in all subisolates compared to the general Finnish, Swedish, and CEU populations (Figures 2B and 2C and Figure S11).
Figure 2.
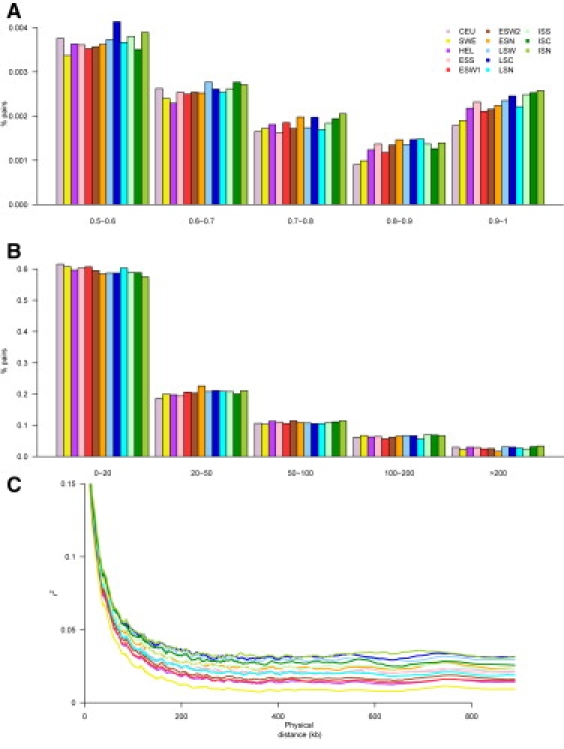
Distribution of Correlation, Represented by r2, between Pairs of SNPs on Chromosome 22
(A) Proportion of SNP pairs within different r2 bins the subpopulations.
(B) Proportion of SNP pairs with r2 > 0.7 within different SNP distance bins in the subpopulations.
(C) Correlation between physical distance and r2. The average r2 was estimated in successive windows of 5000 SNP pairs (4000 SNP pair overlap). Population abbreviations are the same as those used in Figure 1.
To complement the pairwise LD analysis, and quantitatively describe the effects of recombination on LD over the whole chromosome, we constructed comprehensive LD maps for chromosome 22.25 The length of the LD map is inversely related to the extent of LD over a given chromosomal segment, and therefore shorter LD maps are observed in recently founded population isolates compared with older and more heterogeneous populations. As have previous investigations, we observed different map lengths between Finnish subpopulations (Figure 3A). Indeed, the map lengths observed in this study agree with those reported earlier, with ISC having a LD map length of only 393 LDU corresponding to 368 LDU observed previously in the Eastern subisolate of Kuusamo and HEL here showing similar map length (606.8) compared to 606.5 LDU in a previous sample representing the general Finnish population (Table 3).6 The relatively large map lengths observed in LSW and LSC do not agree with results obtained in the pairwise r2 comparisons (Figure 2) and may reflect a greater degree of genetic heterogeneity within these groups (for which genealogical information was limited). Maps were also constructed for chromosome 3q, and map lengths between chromosomes show high correlation (Figure S12). We also investigated the number and chromosomal distribution of LD “holes,” which corresponds to a gap of >2.5 LDU between adjacent SNPs (Figures 3B and 3C and Table 3), and populations with longer maps displayed in general more LD holes.
Figure 3.
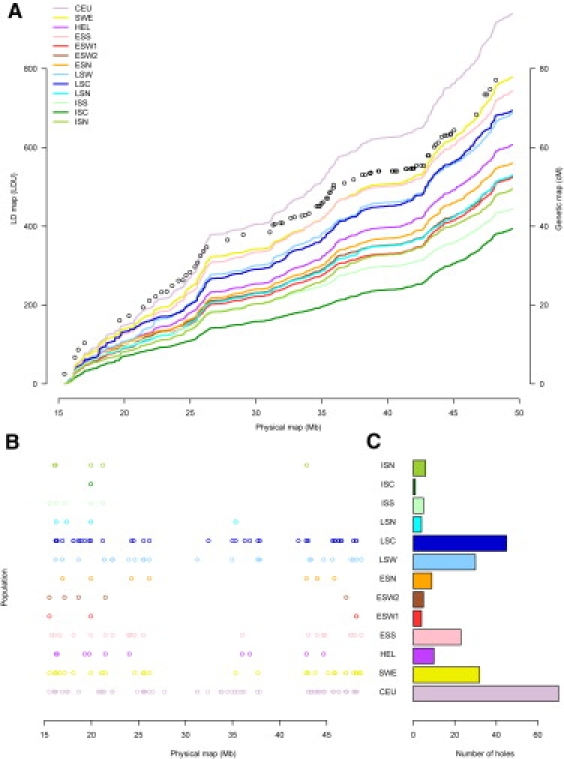
LD Maps for Chromosome 22
(A) Comparison of physical distance to LDUs using LD maps. Open circles represent the genetic map and corresponds to the x axis on the right side of the figure.
(B) Physical distribution of LD holes, defined by gaps of >2.5 LDU in the LD map.
(C) Total number of LD holes. Population abbreviations are the same as those used in Figure 1.
Table 3.
Properties of LD Maps
| Population | Length of LD Map (LDU) | LD Holes (number) | LD Holes (kb span) |
|---|---|---|---|
| CEU | 939.7 | 70 | 1239 |
| SWE | 778.1 | 32 | 1536 |
| HEL | 606.8 | 10 | 580 |
| ESSa | 743.3 | 23 | 1178 |
| ESW1a | 524.2 | 4 | 535 |
| ESW2a | 527.4 | 5 | 684 |
| ESNa | 560.0 | 9 | 390 |
| LSWb | 686.9 | 30 | 732 |
| LSCb | 693.2 | 45 | 1141 |
| LSNb | 529.6 | 4 | 369 |
| ISSc | 443.9 | 5 | 817 |
| ISCc | 393.1 | 1 | 338 |
| ISNc | 494.7 | 6 | 384 |
LD holes are defined as a gap of >2.5 LDU between adjacent markers. Population abbreviations are the same as those used in Figure 1.
Early settlement.
Late Settlement.
Isolate.
Our results agree with previous results in which Finns, particularly those from late-settlement regions of Northern Finland, exhibited high LD and fewer LD holes compared with other well-defined isolates worldwide.6 Furthermore, the fact that LD maps obtained here closely resemble earlier observations from Finnish subpopulations supports the hypothesis that LD map lengths can be considered characteristic for a given population.
The results of this study imply genetic heterogeneity of the Finnish subpopulations, and the role of multiple bottlenecks and isolation on the patterns of genetic variance observed today. In particular, the increased LD in these subisolates could be beneficial for shared segment-based gene identification studies of rare alleles behind common diseases, especially since the prevalence of some complex disorders and especially their familial forms is distinctly higher in some subisolates compared to the general Finnish population.26,27 Conversely, the genetic variability observed here between subpopulations suggests that in the design of case-control association studies attention be paid to the degree to which cases and controls are matched with respect to subpopulation of origin.
Next, we determined the number and the length of extended regions of homozygosity (ROHs). A ROH was defined as a segment exceeding 1 Mb and having 100 consecutive homozygous SNPs with a SNP density of at least 1 SNP per 50 kb. In each homozygous segment, two heterozygous and/or missing genotypes were allowed. The B allele frequency and log R ratio was visualized with Illumina BeadStudio 3.1.0 Genome viewer and inspected for each sample for regions with homozygous segments over 10 Mb to exclude structural variation.28 Inbreeding coefficients were estimated for each individual, and means for each subpopulation were calculated to compare populations.
First, we identified ROHs of over 1 Mb and 100 SNPs in the autosomes in concordance with methods used elsewhere.29 The number of ROHs and their median length was highest in the youngest subpopulations gradually diminishing in older and more outbred populations (Figure 4). First-cousin marriages were illegal for centuries and have thus been rare in Finland.30 We estimated inbreeding coefficients (F) for each subpopulation and only 1.65% of individuals revealed F corresponding to first-cousin marriage (F = 0.025–0.07), whereas 8.7% had F corresponding to second-cousin marriage (F = 0.01–0.025). F estimates did not vary between subpopulations (data not shown). Common overlapping ROHs (≥60 Finns) over 1 Mb include 6p21 and 2q21-q22, which are known to be under selection and correspond with previous findings29 (Table S4).
Figure 4.
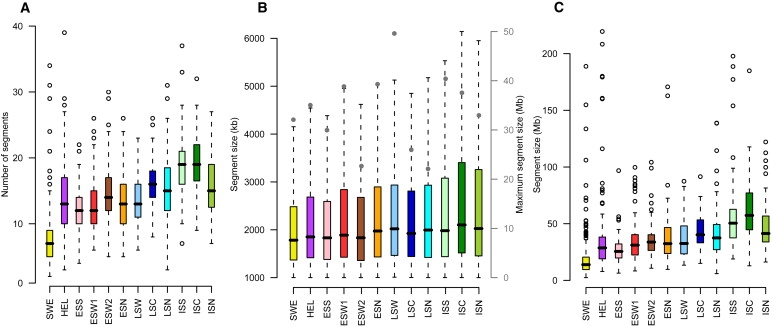
Properties of Extended Runs of Homozygosity in the Different Populations
(A) Distribution of number of homozygous segments over 1 Mb per individual.
(B) Distribution of the length of homozygous segments over 1 Mb. For clarity, outliers are omitted and is shown in Figure S15. Maximum homozygous segment size is indicated with circles, corresponding to the x axis on the right-hand side of the figure.
(C) The total length of chromosomal regions (in Mbs) covered by homozygous segments per individual. The median is indicated by the horizontal line, bars extend to the first and third quartile and error bars extend to 1.5× the interquartile range from the first or third quartile. Open circles indicate observations located more than 1.5× the interquartile range from the first or third quartile. Population abbreviations are the same as those used in Figure 1.
A high number of extremely long ROHs were identified (1523 ROHs ≥ 5 Mb and 389 ROHs ≥ 10 Mb in length) in Finns, distributed seemingly evenly without obvious hotspots (Figure S13 and Table S5). Manual inspection of the raw intensity plots of these regions revealed no large-scale deletions. The proportion of individuals having at least one ROH exceeding 5 Mb was highest (up to 90%) in the youngest subpopulations (Figure S14). In comparison, only 9.5% of individuals from the US population had similar ROHs,28 and ROHs of over 3 Mb were found only in 18.9% of HapMap samples.31 Individuals of Pakistani and Arab origin whose parents were first cousins were reported to have mean genome homozygosity of 11% and each individual had on average 20 ROHs over 3 cM, when Affymetrix 10K SNP data were used.32 In comparison, when ROHs greater than 1 Mb are considered, Finns have mean genome homozygosity ranging from 0.9% in the early-settlement (ESS) population to 2.0% in ISC (Table S6). Thus, the Finns exhibit a substantial degree of homozygosity as expected on the basis of the population history, but this is not comparable to the extent observed for tribes and populations with the culture of consanguineous marriages. The increased number of extended ROHs and their even distribution across the genome in younger subisolates is most probably due to autozygosity and reflects the fewer number of founders and subtle increases in relatedness in individuals from subisolates as seen in IBS-sharing comparisons. The opportunity to use homozygous segments to identify rare alleles has already been utilized successfully in Finland to identify Mendelian mutations such as those behind Meckel syndrome (MIM 612284).33 This strategy could offer an avenue also for the tagging of recessive variants in complex disorders with common SNPs.
In conclusion, we demonstrate the power of genome-wide SNP data in revealing fine-scale population variation even within a founder population such as Finland that is overall substantially more genetically homogeneous than most populations. The patterns identified in such genetic data correlate remarkably well with population subdivisions based on historical and linguistic information and clarify the impact of consecutive historical bottlenecks and founder effects on the current population today, as reflected in measures of LD and homozygosity. In Iceland, another population representing multiple founder effects, in a study of autosomal STR data, revealed significant substructure similarly corresponding to geographical origin.34 We further conclude that dense and genome-wide information is crucial to draw reliable conclusions about fine population substructure in the design of gene-mapping strategies. In this study, several measures of genome-wide properties strongly agree with results from previous genetic studies and reveal significant heterogeneity in subpopulations corresponding with known historical migration patterns. These special populations may be helpful in identifying possible rare variants that are likely to be enriched in particular subpopulations and thus aid in unraveling the genetic architecture of complex traits.
Acknowledgments
We thank Murat Gunel and Richard Lifton for provision of control Helsinki genotypes prior to publication, and Arpo Aromaa for access to the Health2000 samples. Jun Z. Li is gratefully acknowledged for providing tools for Fst analysis. The genotyping for this study has been financially supported by National Institutes of Health grant 1R01HL087679-01, STAMPEED, GenomEUtwin (QLG2-CT-2002-01254), EU grant LSHM-CT-2006-037761 (Project SGENE), the Nordic Centre of Excellence in Disease Genetics (NCoEDG), the Center of Excellence in Complex Disease Genetics of the Academy of Finland, and the Helsinki University Finnish Genome Center. The Swedish samples were collected with support from the Swedish Foundation for Strategic Research and the Swedish Research Council (M-2005-1112). The Broad Institute Center for Genotyping and Analysis is supported by grant U54 RR020278 from the National Center for Research Resources.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
EIGENSOFT, http://genepath.med.harvard.edu/∼reich/Software.htm
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 2.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 3.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price A.L., Butler J., Patterson N., Capelli C., Pascali V.L., Scarnicci F., Ruiz-Linares A., Groop L., Saetta A.A., Korkolopoulou P. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sajantila A., Salem A.H., Savolainen P., Bauer K., Gierig C., Paabo S. Paternal and maternal DNA lineages reveal a bottleneck in the founding of the Finnish population. Proc. Natl. Acad. Sci. USA. 1996;93:12035–12039. doi: 10.1073/pnas.93.21.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Service S., Deyoung J., Karayiorgou M., Roos J.L., Pretorious H., Bedoya G., Ospina J., Ruiz-Linares A., Macedo A., Palha J.A. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat. Genet. 2006;38:556–560. doi: 10.1038/ng1770. [DOI] [PubMed] [Google Scholar]
- 7.Varilo T., Paunio T., Parker A., Perola M., Meyer J., Terwilliger J.D., Peltonen L. The interval of linkage disequilibrium (LD) detected with microsatellite and SNP markers in chromosomes of Finnish populations with different histories. Hum. Mol. Genet. 2003;12:51–59. doi: 10.1093/hmg/ddg005. [DOI] [PubMed] [Google Scholar]
- 8.Peltonen L., Palotie A., Lange K. Use of population isolates for mapping complex traits. Nat. Rev. Genet. 2000;1:182–190. doi: 10.1038/35042049. [DOI] [PubMed] [Google Scholar]
- 9.Varilo T. National Public Health Institute; Helsinki: 1999. The age of the mutations in the Finnish disease heritage; a genealogical and linkage equilibrium study. [Google Scholar]
- 10.Norio R. The Finnish Disease Heritage III: the individual diseases. Hum. Genet. 2003;112:470–526. doi: 10.1007/s00439-002-0877-1. [DOI] [PubMed] [Google Scholar]
- 11.Peltonen L., Jalanko A., Varilo T. Molecular genetics of the Finnish disease heritage. Hum. Mol. Genet. 1999;8:1913–1923. doi: 10.1093/hmg/8.10.1913. [DOI] [PubMed] [Google Scholar]
- 12.Pastinen T., Perola M., Ignatius J., Sabatti C., Tainola P., Levander M., Syvanen A.C., Peltonen L. Dissecting a population genome for targeted screening of disease mutations. Hum. Mol. Genet. 2001;10:2961–2972. doi: 10.1093/hmg/10.26.2961. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen N.L., Lichtenstein P., Svedberg P. The Swedish Twin Registry in the third millennium. Twin Res. 2002;5:427–432. doi: 10.1375/136905202320906219. [DOI] [PubMed] [Google Scholar]
- 14.Aromaa A., Koskinen S. Baseline results of the Health 2000 Health Examination Survey, B12. Publications of the National Public Health Institute; Helsinki: 2004. Health and functional capacity in Finland. [Google Scholar]
- 15.Rantakallio P. The longitudinal study of the northern Finland birth cohort of 1966. Paediatr. Perinat. Epidemiol. 1988;2:59–88. doi: 10.1111/j.1365-3016.1988.tb00180.x. [DOI] [PubMed] [Google Scholar]
- 16.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lao O., Lu T.T., Nothnagel M., Junge O., Freitag-Wolf S., Caliebe A., Balascakova M., Bertranpetit J., Bindoff L.A., Comas D. Correlation between genetic and geographic structure in Europe. Curr. Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 19.Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I., Chen H., Roix J.J., Kathiresan S., Hirschhorn J.N., Daly M.J. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 20.Salmela E., Lappalainen T., Fransson I., Andersen P.M., Dahlman-Wright K., Fiebig A., Sistonen P., Savontaus M.L., Schreiber S., Kere J. Genome-wide analysis of single nucleotide polymorphisms uncovers population structure in Northern Europe. PLoS ONE. 2008;3:e3519. doi: 10.1371/journal.pone.0003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norio R. Finnish Disease Heritage II: population prehistory and genetic roots of Finns. Hum. Genet. 2003;112:457–469. doi: 10.1007/s00439-002-0876-2. [DOI] [PubMed] [Google Scholar]
- 22.Lappalainen T., Koivumaki S., Salmela E., Huoponen K., Sistonen P., Savontaus M.L., Lahermo P. Regional differences among the Finns: a Y-chromosomal perspective. Gene. 2006;376:207–215. doi: 10.1016/j.gene.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 24.Hill W.G., Robertson A. Linkage Disequilibrium in Finite Populations. Theor. Appl. Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 25.Maniatis N., Collins A., Xu C.F., McCarthy L.C., Hewett D.R., Tapper W., Ennis S., Ke X., Morton N.E. The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc. Natl. Acad. Sci. USA. 2002;99:2228–2233. doi: 10.1073/pnas.042680999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hovatta I., Terwilliger J.D., Lichtermann D., Makikyro T., Suvisaari J., Peltonen L., Lonnqvist J. Schizophrenia in the genetic isolate of Finland. Am. J. Med. Genet. 1997;74:353–360. [PubMed] [Google Scholar]
- 27.Sumelahti M.L., Tienari P.J., Wikstrom J., Palo J., Hakama M. Increasing prevalence of multiple sclerosis in Finland. Acta Neurol. Scand. 2001;103:153–158. doi: 10.1034/j.1600-0404.2001.103003153.x. [DOI] [PubMed] [Google Scholar]
- 28.Simon-Sanchez J., Scholz S., Fung H.C., Matarin M., Hernandez D., Gibbs J.R., Britton A., de Vrieze F.W., Peckham E., Gwinn-Hardy K. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- 29.Lencz T., Lambert C., DeRosse P., Burdick K.E., Morgan T.V., Kane J.M., Kucherlapati R., Malhotra A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA. 2007;104:19942–19947. doi: 10.1073/pnas.0710021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorde L.B., Pitkanen K.J. Inbreeding in Finland. Am. J. Phys. Anthropol. 1991;84:127–139. doi: 10.1002/ajpa.1330840203. [DOI] [PubMed] [Google Scholar]
- 31.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woods C.G., Cox J., Springell K., Hampshire D.J., Mohamed M.D., McKibbin M., Stern R., Raymond F.L., Sandford R., Malik Sharif S. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tallila J., Jakkula E., Peltonen L., Salonen R., Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helgason A., Yngvadottir B., Hrafnkelsson B., Gulcher J., Stefansson K. An Icelandic example of the impact of population structure on association studies. Nat. Genet. 2005;37:90–95. doi: 10.1038/ng1492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.