

Abstract
Renal hypouricemia is an inherited disorder characterized by impaired renal urate (uric acid) reabsorption and subsequent low serum urate levels, with severe complications such as exercise-induced acute renal failure and nephrolithiasis. We previously identified SLC22A12, also known as URAT1, as a causative gene of renal hypouricemia. However, hypouricemic patients without URAT1 mutations, as well as genome-wide association studies between urate and SLC2A9 (also called GLUT9), imply that GLUT9 could be another causative gene of renal hypouricemia. With a large human database, we identified two loss-of-function heterozygous mutations in GLUT9, which occur in the highly conserved “sugar transport proteins signatures 1/2.” Both mutations result in loss of positive charges, one of which is reported to be an important membrane topology determinant. The oocyte expression study revealed that both GLUT9 isoforms showed high urate transport activities, whereas the mutated GLUT9 isoforms markedly reduced them. Our findings, together with previous reports on GLUT9 localization, suggest that these GLUT9 mutations cause renal hypouricemia by their decreased urate reabsorption on both sides of the renal proximal tubules. These findings also enable us to propose a physiological model of the renal urate reabsorption in which GLUT9 regulates serum urate levels in humans and can be a promising therapeutic target for gout and related cardiovascular diseases.
Main Text
Renal hypouricemia (MIM 220150) is a common inherited disorder characterized by impaired renal urate reabsorption and subsequent low serum urate levels. Typically, it is associated with severe complications such as exercise-induced acute renal failure and nephrolithiasis.1,2 We previously reported that the causative gene for renal hypouricemia is URAT1, also known as SLC22A12 (MIM 607096).3 However, the fact of renal hypouricemic patients who have no URAT1 mutations4,5 implies the existence of another important urate transporter in the human kidney. Recent genome-wide association studies have revealed that the most significant single-nucleotide polymorphisms (SNPs) associated with urate concentrations map within GLUT9 (also known as SLC2A9 [MIM 606142]), which encodes glucose transporter 9 (GLUT9) protein.6–9 Yet, neither the physiological role of GLUT9 in vivo nor human cases with functional GLUT9 deficiency has been reported previously. Generally, serum urate levels in humans are higher than those in most other mammals (such as mice), because humans lack the urate-degrading enzyme hepatic uricase.10 Therefore, to investigate the physiological importance in human urate regulation by GLUT9, it is of no use to simply employ GLUT9 gene-targeted mice, because they express active uricase. Accordingly, we decided to use an actual human health examination database to genetically identify and investigate human patients with GLUT9 deficiency.
First, the health examination database of the personnel of Japan Maritime Self-Defense Force (JMSDF)—which consists of about 850,000 sets of examination data accumulated over the past ten years—was surveyed, and 21,260 persons were selected that had serum urate data available for screening of renal hypouricemia (the flow chart is shown in Figure 1). All procedures were carried out in accordance with the standards of the institutional ethical committees involved in this project and the experiments were performed according to the Declaration of Helsinki. After written consent had been given by each participant, blood samples were obtained. Among those, there were 200 persons (0.94%) who showed serum urate levels ≤3.0 mg/dl (178 μM) (Table 1). To date, we have written consent from 50 JMSDF persons and 20 outpatients with hypouricemia, and clinicogenetic analysis was performed on these 70 hypouricemic cases. We excluded cases with the most-frequent mutation (W258X) in the URAT1 gene, which left 23 hypouricemic cases without the URAT1 W258X mutation, on which we performed genetic analysis of the GLUT9 gene. The human GLUT9 gene contains 14 exons and produces two main transcripts: GLUT9 isoform 1 (long isoform, GLUT9L, also known as GLUT911) and isoform 2 (short isoform, GLUT9S, also known as GLUT9ΔN11) (Figure 2). We performed mutational analysis of all coding regions and intron-exon boundaries of the GLUT9 gene. For determination of the GLUT9 sequence, we used primers described by Li et al.8 with slight modifications (Table S1 available online). Some primers were newly selected according to the genomic structure of human GLUT9 (see Figure 2). For determination of the URAT1 sequence, primers as described elsewhere3,5 were used. High-molecular-weight genomic DNA was extracted from whole peripheral blood cells12,13 as a template and was amplified by PCR. The PCR products were sequenced in both directions13 with a 3130xl Genetic Analyzer (Applied Biosystems). Through this analysis, we identified two distinct heterozygous missense mutations (R380W and R198C in GLUT9L, corresponding to R351W and R169C in GLUT9S) in three patients with hypouricemia (Figures 3A–3C). By digesting the PCR products with restriction enzyme BtsCI (Table S1), the cosegregation of the R380W substitution genotype with the low-urate phenotype was confirmed, being seen in both an affected mother (serum urate: 1.5 mg/dl, 70 years old, I-2) and her affected son (serum urate: 2.7 mg/dl, 43 years old, II-1), but not in unaffected members of the same family (Figure 3D). The fractional excretion of urate was 15.7% in I-2 and 14.6% in II-1, respectively. A pyrazinamide loading test1,5 of the patient II-1 revealed a normal response, indicating that there is no functional alternation in URAT1. The R198C substitution (Figure 3E) was observed in a hypouricemic 32-year-old female patient whose serum urate level was 2.1 mg/dl. Whole-sequence analysis of the URAT1 gene was performed in patients having GLUT9 mutations (R380W or R198C), and it was confirmed that they have no mutations in the URAT1 gene. Together with other clinical data, they were diagnosed as renal hypouricemia without URAT1 mutation (Figure 1). For the genotyping of Japanese controls and family members, the mutations of GLUT9 in exon 10 (R380W) and in exon 6 (R198C) were detected by restriction digestion of the PCR product with BtsCI and AlwI, respectively (Table S1). Neither R380W nor R198C mutations were detected in any samples from 130 randomly selected Japanese controls (260 total chromosomes). For prediction of human GLUT9 membrane topology model, the transmembrane regions of GLUT9L and GLUT9S were predicted based on the TMpred algorithm. N-glycosylation sites and phosphorylation sites were predicted by PROSITE (Figure 3A).
Figure 1.
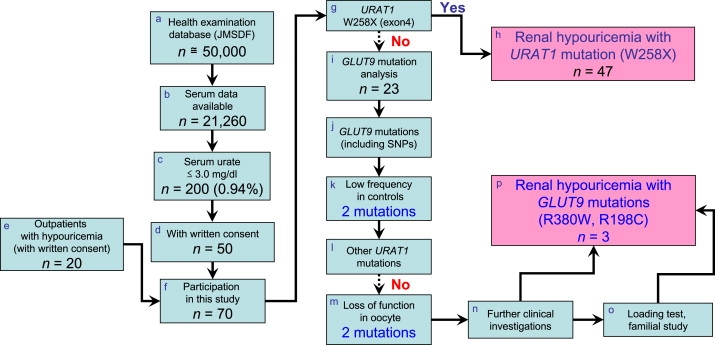
The Flowchart for Clinicogenetic Analysis of Hypouricemia with GLUT9 Mutations
(a) We surveyed the health examination database of about 50,000 personnel of Japan Maritime Self-Defense Force (JMSDF). The database contains data for about 850,000 sets of examinations over 10 years (Apr. 1, 1997 to Mar. 31, 2007). To screen for renal hypouricemia, we selected 21,260 personnel data sets from 2006 in which serum urate data were available (b). (c) The number of persons who showed serum urate levels ≤ 3.0 mg/dl (178 μM) was 200 (0.94%) out of 21,260 persons. (d) 50 JMSDF persons who gave written consent and (e) an additional 20 outpatients with hypouricemia ([f] 70 hypouricemic cases in sum) participated in this clinicogenetic study. (g) First, we performed mutational analysis of URAT1 exon 4, to detect the most frequent mutation (W258X) in URAT1. (h) After 47 cases having the URAT1 W258X mutation were excluded, (i) remaining 23 hypouricemic cases were analyzed to find mutations in GLUT9. (j) After exclusion of known frequent SNPs or high-frequency mutations in Japanese controls, (k) 2 missense mutations (R380W and R198C) in GLUT9 were selected. (l) Subsequently, the patients with these GLUT9 mutations were confirmed to have no URAT1 mutations by whole-sequence analysis of URAT1. (m) Urate uptake activity was measured using oocytes, and these two mutations were proved to be loss-of-function mutations. (n) Further clinical investigations including a hypoxanthine assay and fractional excretion of urate (FEUA) were performed to confirm that the cases are true renal hypouricemia. (o) A pyrazinamide loading test and further familial studies were performed in possible cases. (p) Finally, two loss-of-function mutations in GLUT9 were identified as a cause of renal hypouricemia.
Table 1.
Frequency of hypouricemia of the Japan Maritime Self-Defense Force
| Urate (mg/dl) | Frequency | Cumulative Frequency | Relative Frequency (%) | Cumulative Relative Frequency (%) |
|---|---|---|---|---|
| 0.0–0.5 | 3 | 3 | 0.01 | 0.01 |
| 0.6–1.0 | 27 | 30 | 0.13 | 0.14 |
| 1.1–1.5 | 2 | 32 | 0.01 | 0.15 |
| 1.6–2.0 | 7 | 39 | 0.03 | 0.18 |
| 2.1–2.5 | 29 | 68 | 0.14 | 0.32 |
| 2.6–3.0 | 132 | 200 | 0.62 | 0.94 |
| 3.1– | 21060 | 21260 | 99.06 | 100.00 |
Figure 2.
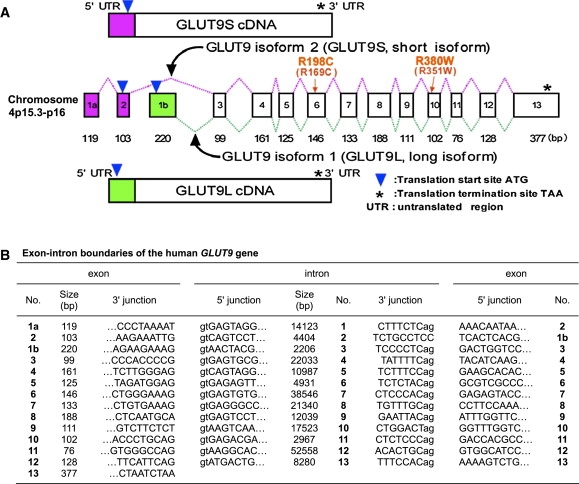
Genomic Structure of the Human GLUT9 Gene
(A) The structure of the GLUT9 gene and cDNAs. The human GLUT9 gene contains 14 exons (1 noncoding and 13 coding) and is located on chromosome 4p15.3-p16. The alternative splicing of the GLUT9 gene results in two main transcripts: GLUT9 isoform 1 (long isoform, GLUT9L) and isoform 2 (short isoform, GLUT9S).
(B) Exon-intron boundaries of the GLUT9 gene.
Figure 3.
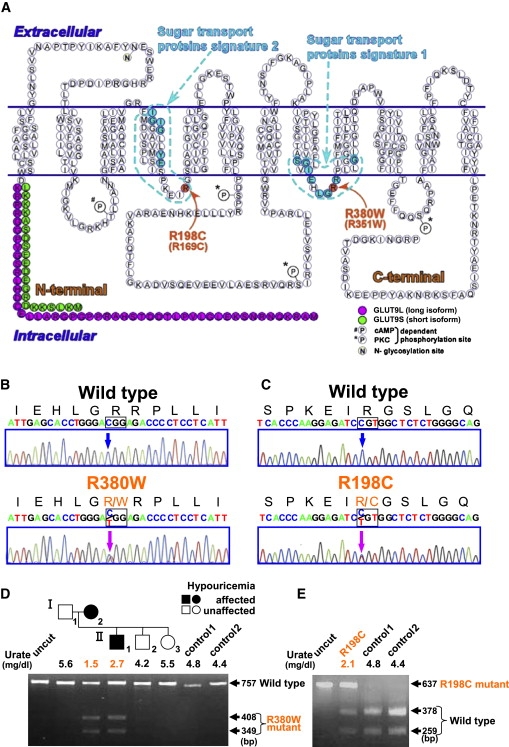
GLUT9 Mutations in Patients with Renal Hypouricemia
(A) Mutation positions in a predicted human GLUT9 membrane topology model. Both mutations are present at equivalent positions within the cytoplasmic loops, which result in loss of positive charges.
(B and C) Heterozygous mutations (c.1138C → T [p.R380W] and c.592C → T [p.R198C]; indicated by magenta arrows) were identified in the patients with renal hypouricemia.
(D) The R380W mutation results in the gain of a BtsCI restriction site. A family tree of the hypouricemic patients having the R380W mutations is shown with serum urate levels.
(E) The R198C mutation results in the loss of an AlwI site.
To investigate the functional aspect of these mutations, either wild-type or mutated GLUT9 cRNA was injected into Xenopus oocytes and the urate transport activity was measured. Complementary DNAs (cDNAs) of human GLUT9L (long isoform) and GLUT9S (short isoform) were obtained from Open Biosystems (also see Accession Numbers section). The GLUT9L cDNA insert was subcloned into pcDNA3.1(+) (Invitrogen) at the KpnI and NotI restriction enzyme cleavage sites. The GLUT9S cDNA insert was subcloned into pcDNA3.1(+) at the EcoRI and XhoI restriction enzyme sites. Site-directed mutagenesis of GLUT9L and GLUT9S cDNAs in the pcDNA3.1(+) plasmid was carried out with the QuickChange kit (Stratagene).14 Oligonucleotide primers for the generation of R380W mutant in GLUT9L or R351W mutant in GLUT9S were 5′-GGTCATTGAGCACCTGGGA(T)GGAGACCCCTCCTCATTGG-3′ and 5′-CCAATGAGGAGGGGTCTCC(A)TCCCAGGTGCTCAATGACC-3′. The mutagenic primers for the generation of R198C (GLUT9L) or R169C (GLUT9S) mutants were 5′-CACCCAAGGAGATC(T)GTGGCTCTCTGGGGC-3′ and 5′-GCCCCAGAGAGCCAC(A)GATCTCCTTGGGTG-3′. The accuracy of the mutated cDNAs was confirmed by complete sequencing of the constructs with the dye terminator cycle sequence method (Applied Biosystems). For the evaluation of urate uptake activity, Xenopus laevis oocytes were injected with 25 ng of cRNA that was synthesized in vitro from linearized GLUT9L or GLUT9S cDNAs. In vitro transcription of the cRNA was performed with the T7 mMESSAGE mMACHINE Kit (Ambion). The urate transport activity was measured 2 days after cRNA injection.7,15 The uptake of 14C-labeled urate (1.25 μCi ml−1) by the oocytes was measured in the regular uptake solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES [pH 7.4]) containing 0.9 mM of urate for 1 hr. The uptake activity was measured in triplicate and expressed as pmol per oocyte per min. The oocytes expressing wild-type GLUT9L showed a 44-fold uptake of 14C-labeled urate relative to water-injected control oocytes. In the oocytes injected with mutant GLUT9L (R380W or R198C) cRNA, urate transport was dramatically suppressed (Figure 4A). The oocytes expressing wild-type GLUT9S also showed high urate uptake relative to water-injected control oocytes, whereas the oocytes injected with mutant GLUT9S (R351W or R169C) cRNA showed markedly suppressed urate transport (Figure 4B). These results were consistent with those from mutant URAT14,5 that cause renal hypouricemia. These results indicate the malfunction of both mutated GLUT9 isoforms (GLUT9L and GLUT9S) in the pathophysiological condition. The expression of wild-type or mutated GLUT9 proteins on the plasma membrane was confirmed by immunofluorescence staining with anti-GLUT9 (Figure S1). Immunofluorescent detection of GLUT9 protein expressed in Xenopus laevis oocytes was performed as described previously,16,17 with minor modifications. In brief, 2 days after injection of cRNAs, Xenopus oocytes were fixed with 4% paraformaldehyde in phosphate buffer at 4°C overnight. The sections (4 μm) were incubated with rabbit anti-human GLUT9 (Alpha Diagnostic Intl. Inc; diluted 1:500) for 2 hr at room temperature. Thereafter, they were treated with FITC-conjugated goat anti-rabbit IgG (Immunotech; diluted 1:100). The images were acquired with a Nikon C1 laser scanning confocal microscope (Nikon, Tokyo, Japan). An Argon laser beam at 488 nm was used for the excitation of FITC and visualization.
Figure 4.
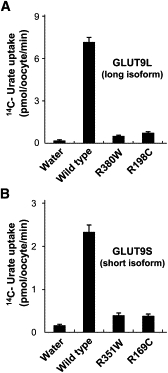
Markedly Reduced Urate Transport Activities in Oocytes that Express Mutant GLUT9 Isoforms
High urate transport activities were observed in oocytes that express each wild-type GLUT9 isoform. In contrast, urate transport activity in oocytes was markedly reduced both in GLUT9L mutants (R380W and R198C) (A) and in GLUT9S mutants (R351W and R169C, which correspond to R380W and R198C in GLUT9L) (B). Results are expressed as mean ± SEM.
Here, by using the large human database, we identified loss-of-function mutations of GLUT9 in renal hypouricemic patients who have no URAT1 mutations. The findings first show the genetic heterogeneity of renal hypouricemia. To collect enough cases of hypouricemia for the analysis of pathogenic GLUT9 mutations, the serum urate data from the health examination database of the JMSDF were investigated. Although there have been few reports on the frequency of hypouricemia among large healthy populations, we could demonstrate the frequency of individuals with lower urate levels as shown in Table 1. The use of an actual human health examination database enabled us to analyze larger populations than those of the recent genome-wide association studies,6–8 which have usually analyzed populations of less than 5000 having known SNPs. The use of this large JMSDF human database in this study was essential in identifying pathogenic mutations in the GLUT9 gene. We believe that the large-database approach used in this study is a reliable and powerful way to identify novel causative genes in such studies.
The sites of mutation identified in this study (R380W and R198C) are both perfectly conserved in GLUT9 vertebrate orthologs (Figure S2). They are also highly conserved in the GLUT family transporters (Figure 5). Interestingly, GLUT1 deficiency syndrome (MIM 606777) is caused by heterozygous mutations in human GLUT1 (R333W and R153C),18 which correspond to the mutation sites in human GLUT9 (R380W and R198C, respectively). Moreover, both the GLUT9 and GLUT1 mutations occur in “sugar transport proteins signatures 1 and 2,” which are highly conserved in most sugar transport proteins among yeast, bacteria, plants, and mammals (Figure S3). One of the sugar transport proteins signatures contains a well-known conserved amino acid motif (D/E-x(2)-G-R-R)19 (Figure 5), and pathogenic mutations in GLUT9 (R380W) and GLUT1 (R333W) are identified in this motif. Sato and Mueckler showed that positive charge (arginine residues) in this conserved motif plays a critical role in forming cytoplasmic anchor points that are involved in determining the membrane topology of GLUT1.20 The loss of these positive charges in GLUT1 completely abolished transport activities as a result of a local perturbation in the membrane topology, resulting in aberrant “flipping” of the corresponding cytoplasmic loop into the exogenous compartment.20 They suggested that this finding provides a simple explanation for the presence of this conserved motif in hundreds of membrane transporters that share a common predicted membrane topology.20 The marked reduction of the urate transport activity in mutated GLUT9 may be ascribed to the loss of a cytoplasmic anchor point and the perturbation of the membrane topology. Arginine residues in GLUT4, which correspond to R380 and R198 in GLUT9, have also been shown to be essential for its appropriate comformation.21
Figure 5.
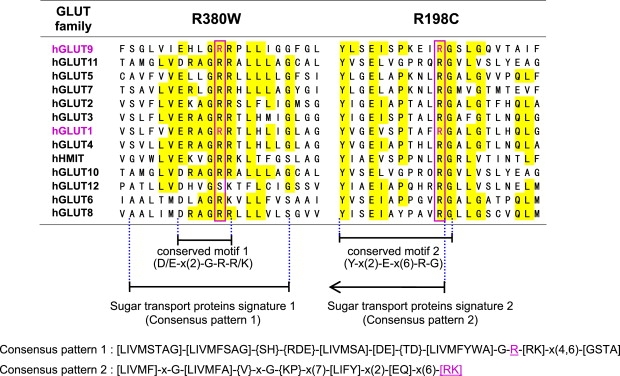
Amino Acid Conservation in the GLUT Family Transporters
Arginine residues homologous to human GLUT9 amino acid positions 380 and 198, the sites of missense mutations identified in hypouricemia patients, are boxed in magenta. These mutations are observed at the well-known conserved motif (D/E-x(2)-G-R-R/K) and another conserved motif (Y-x(2)-E-x(6)-R-G) that is 100% conserved in all GLUT family transporters. These motifs are a part of the consensus patterns 1/2 that are demonstrated in the PROSITE database as “sugar transport proteins signatures 1/2” (also see Figure S3). The mutation sites in GLUT9 are found to be key residues in these consensus patterns. Interestingly, mutations of human GLUT1 at amino acid positions 333 and 153 (magenta), which correspond to the human GLUT9 mutation sites, are reported to cause GLUT1 deficiency syndrome. hHMIT represents human H+-coupled myo-inositol transporter, also known as SLC2A13.
To test whether heterozygous GLUT9 mutations cause hypouricemia because of dominant-negative effects, we performed additional functional studies with oocytes coinjected with wild-type and mutant GLUT9 cRNA. Coexpression of wild-type and mutant GLUT9 did not reduce the urate transport (Figure S4), which suggests that GLUT9 mutants may not induce dominant-negative effects. GLUT1 deficiency syndrome is caused by GLUT1 heterozygous mutations because of haploinsufficiency,22,23 which was confirmed by the uptake studies with patient-derived cells such as erythrocytes. In the case of GLUT9, however, there are no available patient-derived cells that are known to show sufficient GLUT9 functions. GLUT9 mutations may also cause hypouricemia because of haploinsufficiency rather than dominant-negative effects, which should be confirmed by further experiments.
Because humans lack uricase, there are considerable differences in urate metabolism between humans and mice.10 Furthermore, the expression of GLUT9 in human kidney is quite different from that in mouse kidney. Human GLUT9 is expressed in the proximal tubules,11 whereas mouse GLUT9 is expressed in the distal convoluted tubules or connecting tubules of the renal cortex.24 Moreover, the isoforms of mouse GLUT9 (GLUT9a and GLUT9b) show only basolateral localization in polarized MDCK cells,24 whereas the isoforms of human GLUT9 show a different expression pattern; i.e., human GLUT9L (long isoform) shows basolateral localization and human GLUT9S (short isoform) shows apical localization in MDCK cells.11 Because of these considerable differences, the investigation of human patients with loss-of-function GLUT9 mutations has a great advantage for the analysis of the urate transporting activity by GLUT9 relative to using GLUT9 gene-targeted mice.
As mentioned above, the human GLUT9 protein is supposed to be expressed on both sides of renal proximal tubules, which should be confirmed by the further immunohistochemical analysis with specific antibodies against human GLUT9 isoforms. In contrast, the human URAT1 protein expresses only on the apical side. Together with this renal localization, our data of reduced function in both mutant GLUT9 isoforms indicate that impaired urate reabsorption occurs on both sides of the proximal tubules in patients with these mutations (Figure 6). Based on our findings and previous reports on renal localization of GLUT9, we propose a physiological model of renal urate transport, in which two isoforms of GLUT9 play a key role in urate reabsorption on both urine and blood sides of the renal proximal tubules (Figure 6). Because there had been no clear in vivo evidence of the molecules that mediate urate reabsorption on the basolateral side, our findings provide the first evidence for the these molecules on the basolateral side (Figure 6). The presence of SNPs associated with gout found in recent studies6–9 implies that these SNPs relate to increased function of GLUT9 (renal urate reabsorption), but no functional evidence is shown. Increased renal urate reabsorption would result from increased protein expression of GLUT9, which should be proved by further experiments.
Figure 6.
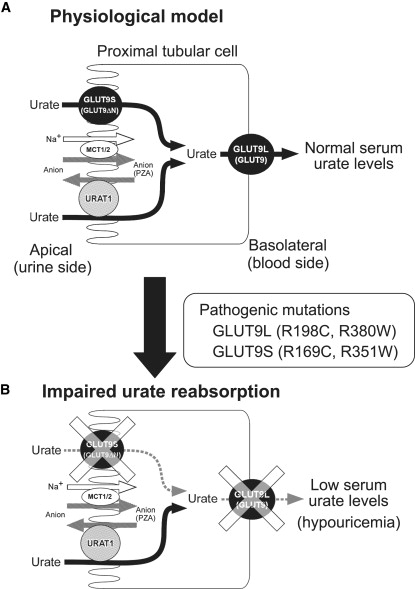
Proposed Model of Renal Urate Reabsorption in Humans
(A) Based on the findings from the hypouricemic patients with pathogenic GLUT9 mutations, we propose a physiological model of renal urate transport via human GLUT9 molecules. The localization of GLUT9L and GLUT9S is based on the previous observation from polarized MDCK cells. Here, GLUT9 mediates renal urate reabsorption on both sides of proximal tubular cells. URAT1 is expressed only on the apical side and is indirectly coupled with Na+-anion cotransporters, such as monocarboxylic acid transporter1/2 (MCT1/2).
(B) An impaired urate reabsorption model in the renal proximal tubular cells. Pathogenic mutations in GLUT9L and GLUT9S on both sides of proximal tubules markedly reduce the urate reabsorption and cause hypouricemia. GLUT9L or “GLUT9” represents GLUT9 isoform 1 (long isoform) and the GLUT9S or GLUT9ΔN represents GLUT9 isoform 2 (short isoform). PZA represents pyrazinecarboxylic acid, a metabolite of pyrazinamide that is used for loading test of hypouricemic patients.
Our results confirmed the ability of both GLUT9L and GLUT9S to transport urate and showed the existence of loss-of-function mutations of GLUT9 in hypouricemic patients via the oocyte expression system. The ability of GLUT9 to transport glucose was also reported via a high-sensitivity intramolecular FRET glucose sensor,25 suggesting multifunctional aspects of this transporter. Although urate did not inhibit D-glucose or D-fructose uptake via an oocyte expression system, Caufield et al. suggested that GLUT9 can exchange extracellular glucose for intracellular urate.26 Further investigation of the functional properties of GLUT9 will be helpful in elucidating the other physiological and pathophysiological roles of this transporter.
Very recently, functional properties of GLUT9 have been reported,27 which support the biological significance of our findings on GLUT9 pathogenic mutations. Also reported was a case of hypouricemia with a GLUT9 amino acid alternation (P412R)27 that had less effect on the transport function. Our data suggest that P412R is unlikely to be a pathogenic mutation for renal hypouricemia, because it does not reduce the function (Figure S5). Taken together, we have identified GLUT9 as a causative gene for renal hypouricemia and demonstrated that human GLUT9 physiologically regulates serum urate levels in vivo. Our results also indicate that GLUT9 can be a promising therapeutic target for hyperuricemia, gout, and related cardiovascular diseases.
Acknowledgments
We would like to thank all the patients involved in this study. The authors thank Y. Kusanagi, S. Tadokoro, K. Nakanishi, M. Nudejima, M. Kobayashi, T. Iwamoto, A. Kudo, M. Miyazawa, J. Inoue, M. Watanabe, and M. Wakai for genetic analysis; H. Horikoshi, S. Kubo, M. Yoshida, S. Watanabe, O. Tajima, M. Fujita, and Y. Tadano for patient analysis; T. Kasahara, M. Kasahara, Y. Kobayashi, and J. Fukuda for helpful discussion; and D.E. Nadziejka for technical editing of this manuscript. This work was supported in part by grants from the Ministry of Defense of Japan (to H.M. and K.K.), the Kawano Masanori Memorial Foundation for Promotion of Pediatrics, and the Uehara Memorial Foundation (to H.M.). All authors declare absence of conflict of interests. The opinions and assertions contained herein are the private ones of the authors and not to be constructed as official or reflecting the views of the Japan Maritime Self-Defense Force.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PROSITE, http://au.expasy.org/prosite/
Accession Numbers
The amino acid sequences of GLUT9L and GLUT9S shown in Figure 3A were obtained from GenBank (accession codes BC110414 and BC018897, respectively). Their sequences are identical to those from the GLUT9 clones used in this study.
PROSITE accession codes for consensus patterns shown in Figure 5 are PS00216 (Sugar transport proteins signature 1) and PS00217 (Sugar transport proteins signature 2).
Note Added in Proof
In the version of Table 1 published online ahead of the print issue, the final entry in the first column, “Frequency,” was incorrectly edited. The correct entry is 21060. The corrected version of the table appears in this final version of the manuscript.
References
- 1.Kikuchi Y., Koga H., Yasutomo Y., Kawabata Y., Shimizu E., Naruse M., Kiyama S., Nonoguchi H., Tomita K., Sasatomi Y. Patients with renal hypouricemia with exercise-induced acute renal failure and chronic renal dysfunction. Clin. Nephrol. 2000;53:467–472. [PubMed] [Google Scholar]
- 2.Diamond H.S., Paolino J.S. Evidence for a postsecretory reabsorptive site for uric acid in man. J. Clin. Invest. 1973;52:1491–1499. doi: 10.1172/JCI107323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enomoto A., Kimura H., Chairoungdua A., Shigeta Y., Jutabha P., Cha S.H., Hosoyamada M., Takeda M., Sekine T., Igarashi T. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 4.Wakida N., Tuyen D.G., Adachi M., Miyoshi T., Nonoguchi H., Oka T., Ueda O., Tazawa M., Kurihara S., Yoneta Y. Mutations in human urate transporter 1 gene in presecretory reabsorption defect type of familial renal hypouricemia. J. Clin. Endocrinol. Metab. 2005;90:2169–2174. doi: 10.1210/jc.2004-1111. [DOI] [PubMed] [Google Scholar]
- 5.Ichida K., Hosoyamada M., Hisatome I., Enomoto A., Hikita M., Endou H., Hosoya T. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J. Am. Soc. Nephrol. 2004;15:164–173. doi: 10.1097/01.asn.0000105320.04395.d0. [DOI] [PubMed] [Google Scholar]
- 6.Doring A., Gieger C., Mehta D., Gohlke H., Prokisch H., Coassin S., Fischer G., Henke K., Klopp N., Kronenberg F. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat. Genet. 2008;40:430–436. doi: 10.1038/ng.107. [DOI] [PubMed] [Google Scholar]
- 7.Vitart V., Rudan I., Hayward C., Gray N.K., Floyd J., Palmer C.N., Knott S.A., Kolcic I., Polasek O., Graessler J. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008;40:437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 8.Li S., Sanna S., Maschio A., Busonero F., Usala G., Mulas A., Lai S., Dei M., Orru M., Albai G. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007;3:e194. doi: 10.1371/journal.pgen.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mcardle P.F., Parsa A., Chang Y.P., Weir M.R., O'Connell J.R., Mitchell B.D., Shuldiner A.R. Association of a common nonsynonymous variant in GLUT9 with serum uric acid levels in old order amish. Arthritis Rheum. 2008;58:2874–2881. doi: 10.1002/art.23752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X.W., Lee C.C., Muzny D.M., Caskey C.T. Urate oxidase: Primary structure and evolutionary implications. Proc. Natl. Acad. Sci. USA. 1989;86:9412–9416. doi: 10.1073/pnas.86.23.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Augustin R., Carayannopoulos M.O., Dowd L.O., Phay J.E., Moley J.F., Moley K.H. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J. Biol. Chem. 2004;279:16229–16236. doi: 10.1074/jbc.M312226200. [DOI] [PubMed] [Google Scholar]
- 12.Matsuo H., Kamakura K., Saito M., Okano M., Nagase T., Tadano Y., Kaida K., Hirata A., Miyamoto N., Masaki T. Familial paroxysmal dystonic choreoathetosis: Clinical findings in a large Japanese family and genetic linkage to 2q. Arch. Neurol. 1999;56:721–726. doi: 10.1001/archneur.56.6.721. [DOI] [PubMed] [Google Scholar]
- 13.Matsuo H., Kamakura K., Matsushita S., Ohmori T., Okano M., Tadano Y., Tsuji S., Higuchi S. Mutational analysis of the anion exchanger 3 gene in familial paroxysmal dystonic choreoathetosis linked to chromosome 2q. Am. J. Med. Genet. 1999;88:733–737. doi: 10.1002/(sici)1096-8628(19991215)88:6<733::aid-ajmg27>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Kleta R., Romeo E., Ristic Z., Ohura T., Stuart C., Arcos-Burgos M., Dave M.H., Wagner C.A., Camargo S.R., Inoue S. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet. 2004;36:999–1002. doi: 10.1038/ng1405. [DOI] [PubMed] [Google Scholar]
- 15.Kanai Y., Hediger M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 16.Chairoungdua A., Kanai Y., Matsuo H., Inatomi J., Kim D.K., Endou H. Identification and characterization of a novel member of the heterodimeric amino acid transporter family presumed to be associated with an unknown heavy chain. J. Biol. Chem. 2001;276:49390–49399. doi: 10.1074/jbc.M107517200. [DOI] [PubMed] [Google Scholar]
- 17.Matsuo H., Kanai Y., Kim J.Y., Chairoungdua A., Kim D.K., Inatomi J., Shigeta Y., Ishimine H., Chaekuntode S., Tachampa K. Identification of a novel Na+-independent acidic amino acid transporter with structural similarity to the member of a heterodimeric amino acid transporter family associated with unknown heavy chains. J. Biol. Chem. 2002;277:21017–21026. doi: 10.1074/jbc.M200019200. [DOI] [PubMed] [Google Scholar]
- 18.Pascual J.M., Wang D., Yang R., Shi L., Yang H., De Vivo D.C. Structural signatures and membrane helix 4 in GLUT1: inferences from human blood-brain glucose transport mutants. J. Biol. Chem. 2008;283:16732–16742. doi: 10.1074/jbc.M801403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joost H.G., Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members. Mol. Membr. Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 20.Sato M., Mueckler M. A conserved amino acid motif (R-X-G-R-R) in the Glut1 glucose transporter is an important determinant of membrane topology. J. Biol. Chem. 1999;274:24721–24725. doi: 10.1074/jbc.274.35.24721. [DOI] [PubMed] [Google Scholar]
- 21.Schurmann A., Doege H., Ohnimus H., Monser V., Buchs A., Joost H.G. Role of conserved arginine and glutamate residues on the cytosolic surface of glucose transporters for transporter function. Biochemistry. 1997;36:12897–12902. doi: 10.1021/bi971173c. [DOI] [PubMed] [Google Scholar]
- 22.Seidner G., Alvarez M.G., Yeh J.I., O'Driscoll K.R., Klepper J., Stump T.S., Wang D., Spinner N.B., Birnbaum M.J., De Vivo D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998;18:188–191. doi: 10.1038/ng0298-188. [DOI] [PubMed] [Google Scholar]
- 23.Ho Y.Y., Yang H., Klepper J., Fischbarg J., Wang D., De Vivo D.C. Glucose transporter type 1 deficiency syndrome (Glut1DS): Methylxanthines potentiate GLUT1 haploinsufficiency in vitro. Pediatr. Res. 2001;50:254–260. doi: 10.1203/00006450-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 24.Keembiyehetty C., Augustin R., Carayannopoulos M.O., Steer S., Manolescu A., Cheeseman C.I., Moley K.H. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol. Endocrinol. 2006;20:686–697. doi: 10.1210/me.2005-0010. [DOI] [PubMed] [Google Scholar]
- 25.Takanaga H., Chaudhuri B., Frommer W.B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim. Biophys. Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caulfield M.J., Munroe P.B., O'Neill D., Witkowska K., Charchar F.J., Doblado M., Evans S., Eyheramendy S., Onipinla A., Howard P. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5:e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anzai N., Ichida K., Jutabha P., Kimura T., Babu E., Jin C.J., Srivastava S., Kitamura K., Hisatome I., Endou H. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 2008;283:26834–26838. doi: 10.1074/jbc.C800156200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.