

Abstract
Hereditary spastic paraplegias (HSPs), characterized by progressive and bilateral spasticity of the legs, are usually caused by developmental failure or degeneration of motor axons in the corticospinal tract. There are considerable interfamilial and intrafamilial variations in age at onset and severity of spasticity. Genetic studies also showed that there are dozens of genetic loci, on multiple chromosomes, that are responsible for HSPs. Through linkage study of a pedigree of HSP with autosomal-dominant inheritance, we mapped the causative gene to 3q24-q26. Screening of candidate genes revealed that the HSP is caused by a missense mutation in the gene for acetyl-CoA transporter (SLC33A1). It is predicted that the missense mutation, causing the change of the highly conserved serine to arginine at the codon 113 (p. S113R), disrupts the second transmembrane domain in the transporter and reverses the orientation of all of the descending domains. Knockdown of Slc33a1 in zebrafish caused a curve-shaped tail and defective axon outgrowth from the spinal cord. Although the wild-type human SLC33A1 was able to rescue the phenotype caused by Slc33a1 knockdown in zebrafish, the mutant SLC33A1 (p.S113R) was not, suggesting that S113R mutation renders SLC33A1 nonfunctional and one that wild-type allele is not sufficient for sustaining the outgrowth and maintenance of long motor axons in human heterozygotes. Thus, our study illustrated a critical role of acetyl-CoA transporter in motor-neuron development and function.
Main Text
Characterized by progressive and bilateral spasticity of the legs, the hereditary spastic paraplegias (HSPs) are a highly heterogeneous group of neurodegenerative disorders that are usually caused by developmental failure or degeneration of motor axons in the corticospinal tract.1,2 Clinically, HSPs can be classified into uncomplicated or complicated form, depending on whether spasticity occurs in isolation (uncomplicated HSP) or is associated with additional symptoms, such as mental retardation, deafness, cerebellar ataxia, epilepsy, dysarthria, and ichthyosis (complicated HSP).2 The uncomplicated HSP, which is more common than the complicated HSP, usually presents four core features of HSP: a slowly progressive spastic gait, an increased muscle tone in the lower limbs, hyperreflexia, and extensor plantar responses. Although patients with uncomplicated HSP generally have the core clinical features, considerable variation in age at onset and severity of spasticity has been observed both within and between families.1–3 Genetic studies have showed that HSPs are also extremely heterogeneous with autosomal dominant (AD), autosomal recessive (AR), and X-linked inheritance.2–4 To date, 38 HSP loci and 19 spastic-paraplegia genes have been identified, according to the HUGO and OMIM databases.2,3 Among them, SPASTIN (SPG4) (MIM 604277),5 ATLASTIN (SPG3A) (MIM 606439),6 NIPA1 (SPG6) (MIM 600363),7 KIAA0196 (SPG8) (MIM 603563),8 KIF5A (SPG10) (MIM 604187),9 HSP60 (SPG13) (MIM 605280),10 REEP1 (SPG31) (MIM 610250),11 and ZFYVE27 (SPG33) (MIM 610244)12 have been associated with AD HSP. Mutations in the genes SPASTIN (SPG4), ATLASTIN (SPG3), and REEP1 (SPG31) account for the majority of AD forms of HSP.2,13,14 Mutations in KIF5A, HSP60, KIAA0196, and NIPA1 each occur in < 1% of HSP cases.
Clinical Features
We were contacted by a family member of a large kindred with HSP for genetic counseling. The disease clearly followed a pattern of AD inheritance, though with incomplete penetrance (Figure 1). We set out to conduct a genetic study of this family. The study was approved by the Ethics Committee of the Shandong University School of Medicine, and informed consent was obtained from all participants. Clinical examinations of 57 subjects in the family were performed by an experienced neurologist (C.Y). On the basis of the clinical examination, the subjects were scored as definitely affected (all four core features of HSP—spasticity, increased muscle tone, hyperreflexia, and extensor plantar reflexes—were present), unaffected, or mildly affected (with only one or two core feature[s] of HSP). Summarized in Table 1 are the clinical features of all patients. The age at onset was listed according to the self-report of affected subjects. Of the 57 family members examined, 20 individuals were diagnosed as definitely affected and three (III-8, IV-12, and V-1) as mildly affected, displaying lower-limb hyperreflexia or mild spastic gait alone. As shown in Table 1, the age at onset varied greatly among the affected. This variation is not unexpected for AD HSP, partly because mild symptoms, which can progress slowly, might go unnoticed for years. A probable case of incomplete penetrance was also noticed for III-22. Whereas his daughter, IV-14, is definitely affected, with spastic paraplegia that started when she was 8 years old, III-22 is asymptomatic and shows no abnormal features upon neurological examination at age 43. No additional neurological symptoms were detected in this family. Two affected family members, IV-4 and IV-14, were further examined by electromyography (EMG), magnet resonance imaging (MRI), and muscle biopsy, and no abnormalities were detected. Therefore, the patients in this pedigree might represent those with an uncomplicated form of HSP.
Figure 1.
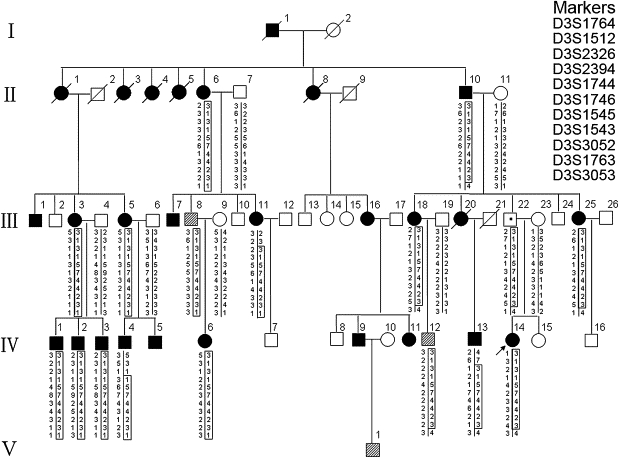
The Pedigree of a Large Family with Hereditary Spastic Paraplegia and the Haplotypes of Markers Spanning the Linked Region on Chromosome 3q24-q26
Filled squares and circles indicate definitely affected, hatched squares indicate mildly affected. The haplotype segregating with HSP is boxed. SPG42 is flanked by D3S2326 and D3S3053.
Table 1.
A Summary of Clinical Features Observed in Patients of a Chinese Family with HSP
| Patient | Sex | Age at Examination (Yrs) | Age at Onset (Yrs) | Disability Gradesa | Spastic Gait | Increased Tone in LL | Hyperreflexia in LL | Weakness in LL | Wasting in LL | Extensor Plantar Reflex | Pes Cavus |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II-6 | F | 75 | 4 | 3 | +++ | + | + | + | + | + | − |
| II-10 | M | 72 | 30 | 3 | ++ | + | + | + | + | + | − |
| III-1 | M | 56 | 12 | 3 | ++ | + | + | + | + | + | − |
| III-3 | F | 69 | 10 | 4 | +++ | + | + | + | + | + | + |
| III-5 | F | 63 | 42 | 2 | ++ | + | + | + | + | + | + |
| III-7 | M | 50 | 11 | 3 | ++ | + | + | + | + | + | + |
| III-8 | M | 43 | 26 | 1 | + | + | − | − | − | − | − |
| III-11 | F | 36 | 13 | 1 | + | + | + | + | − | + | − |
| III-16 | F | 56 | 9 | 3 | ++ | + | + | + | + | + | + |
| III-18 | F | 51 | 4 | 1 | + | + | + | − | − | + | − |
| III-25 | F | 36 | 20 | 1 | + | + | + | − | − | + | − |
| IV-1 | M | 43 | 11 | 3 | +++ | + | + | + | + | + | − |
| IV-2 | M | 40 | 4 | 3 | +++ | + | + | + | + | + | + |
| IV-3 | M | 36 | 7 | 4 | +++ | + | + | + | + | + | + |
| IV-4 | M | 41 | 40 | 1 | + | + | + | + | − | + | + |
| IV-5 | M | 38 | 10 | 1 | + | + | + | + | + | + | − |
| IV-6 | F | 21 | 8 | 1 | + | + | + | + | + | + | + |
| IV-11 | F | 31 | 11 | 2 | ++ | + | + | + | − | + | − |
| IV-12 | M | 27 | 20 | 1 | − | − | − | + | − | − | − |
| IV-13 | M | 26 | 15 | 1 | + | + | + | + | + | + | − |
| IV-14 | F | 20 | 8 | 2 | ++ | + | + | + | − | + | + |
| V-1 | M | 9 | 9 | 1 | + | − | + | − | − | − | − |
“−” indicates absent, “+” indicates mild, “++” indicates moderate, and “+++” indicates severe. “LL” denotes lower limbs.
Disability grades: 1, no mobility problems or slight stiffness of the legs; 2, moderate gait stiffness; 3, problems running but able to walk alone; 4, problems walking; 5, wheelchair-bound.
Linkage Studies
Blood samples were obtained from 21 family members (17 definitely affected, three mildly affected, and one unaffected) and seven spouses (all unaffected), and leukocyte genomic DNA was extracted via standard techniques. We first tested, by linkage analysis, whether the HSP in this family was caused by a mutation in any of the known HSP loci. In view of the high variability in phenotype and the age-dependent penetrance of AD HSP, pairwise logarithm of odds (LOD) scores were calculated with a conservative approach, in which only definitely affected subjects and unaffected spouses were included in the analysis. Linkage to any of the known AD HSP loci was excluded. Negative LOD scores, as well as obligate recombinants in patients, were observed for all tested loci (Table S1, available online). Subsequently, we performed a genome-wide scan using 200 microsatellite markers spaced, on average, 15–20 cM apart on autosomes. Among the 205 markers genotyped, six markers produced positive LOD scores, of which two mapped to chromosome 3q and the other four were on chromosome 4 (D4S2431), 6 (D6S1959), 7 (D7S2201), and 20 (D20S162) (Table S2). The regions surrounding these loci were then further analyzed with additional microsatellite markers at a higher density. For the regions on chromosomes 4, 6, 7, and 20, all additional markers analyzed generated LOD scores less than −2. In contrast, seven additional markers genotyped on chromosome 3q gave significant pairwise LOD scores > 3, with a peak of 5.085 at θ = 0 for D3S1746, showing strong evidence of linkage between the chromosomal region and the disease (Table 2). Multipoint linkage analysis with the use of 11 markers on chromosome 3q reached a peak LOD score between D3S2326 and D3S3053 (Figure 2). Haplotype analysis indicated that all of the markers flanked by D3S2326 and D3S3053 cosegregated with the disease (Figure 1). This 22 cM interval flanked by D3S2326 and D3S3053 corresponds to a physical distance of 27.54 Mb. These results clearly establish the existence of a locus for AD HSP within chromosome 3q24-q26. This locus was named SPG42, in accordance with HUGO nomenclature.
Table 2.
Two-Point LOD Score for Linkage between the HSP Locus and Markers on Chromosome 3q24-q26
| Marker | Position |
Za at θ = 0 | |
|---|---|---|---|
| cM | Mb | ||
| D3S1764 | 152.62 | 140.67 | −7.710 |
| D3S1512 | 158.38 | 145.82 | −7.547 |
| D3S2326 | 159.8 | 145.69 | −1.532 |
| D3S2394 | 160.4 | 147.03 | 3.035 |
| D3S1744 | 161.04 | 148.57 | 3.569 |
| D3S1746 | 169.6 | 153.21 | 5.085 |
| D3S1545 | 173.72 | 163.15 | 5.069 |
| D3S1543 | 173.84 | 162.15 | 5.068 |
| D3S3052 | 174.94 | 165.68 | 5.045 |
| D3S1763 | 176.54 | 168.72 | 4.975 |
| D3S3053 | 181.87 | 173.23 | 0.797 |
A gene frequency of 0.0001 and a penetrance of 90% were assumed for the disease locus. Marker order and genetic distances were based on the chromosome 3 genetic map of the Center for Medical Genetics at the Marshfield Medical Research Foundation.
Figure 2.
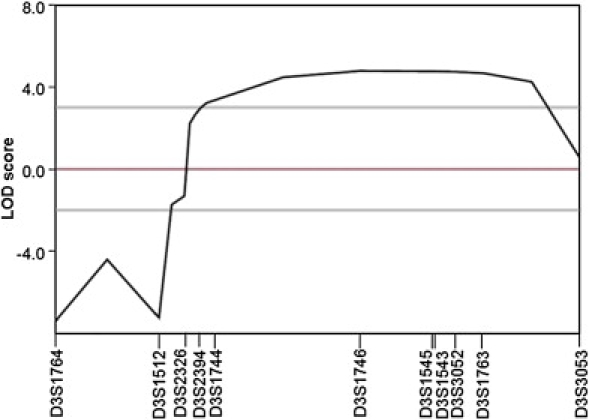
Multipoint Linkage Analysis for Markers on Chromosome 3q24-q26
Mutation Analysis
A total of 130 transcripts have been mapped to the 22 cM interval containing the SPG42 locus (UCSC Genome Browser), none of which stood out as a clear candidate for the disease. We selected a group of genes, on the basis of their expression pattern in neuron and their possible relevance to the disease, for mutation analysis. The coding region, exon-intron boundaries, and 5′- and 3′-UTRs of the candidate genes (Figure 3A) were amplified by PCR from genomic DNA of one patient and sequenced. The obtained DNA sequences were compared with GenBank sequences by BLAST. No noticeable mutation was found in the first five genes sequenced (PFN2 [MIM 176590, GenBank accession number NM_053024], SCHIP1 [MIM 611622, GenBank accession number NM_014575], SLITRK3 [MIM 609679, GenBank accession number NM_014926], MYNN [MIM 606042, GenBank accession number NM_018657], CLDN11 [MIM 601326, GenBank accession number NM_005602]). However, sequence analysis of SLC33A1 (MIM 603690, GenBank accession number NM_004733) revealed a heterozygous c.339 T→G transversion in exon 1. This substitution was not present in the unaffected spouses (Figure 3B). Because the c.339T→G single-nucleotide change did not result in a gain or loss of a restriction site, we used an allele-specific tetra-primer PCR assay to determine whether the mutant G allele cosegregated with the disease. Sequencing analysis of 35 DNA samples from affected subjects and controls confirmed the validity of the tetra-primer PCR assay (data not shown). As shown in Figure 3C, the mutant G allele was present in all of the definitely affected individuals tested but not in any of the unaffected spouses. This mutant G allele was also present in three mildly affected subjects and in III-22, a likely obligate carrier given that he has an affected daughter. Furthermore, the G allele was excluded as a SNP, because it was not detected in a panel of 200 normal, unrelated, ethnically matched controls (i.e., 400 chromosomes). Together, our genetic data strongly implicated the c.339T→G transversion in exon 1 of SLC33A1 as the causative mutation.
Figure 3.
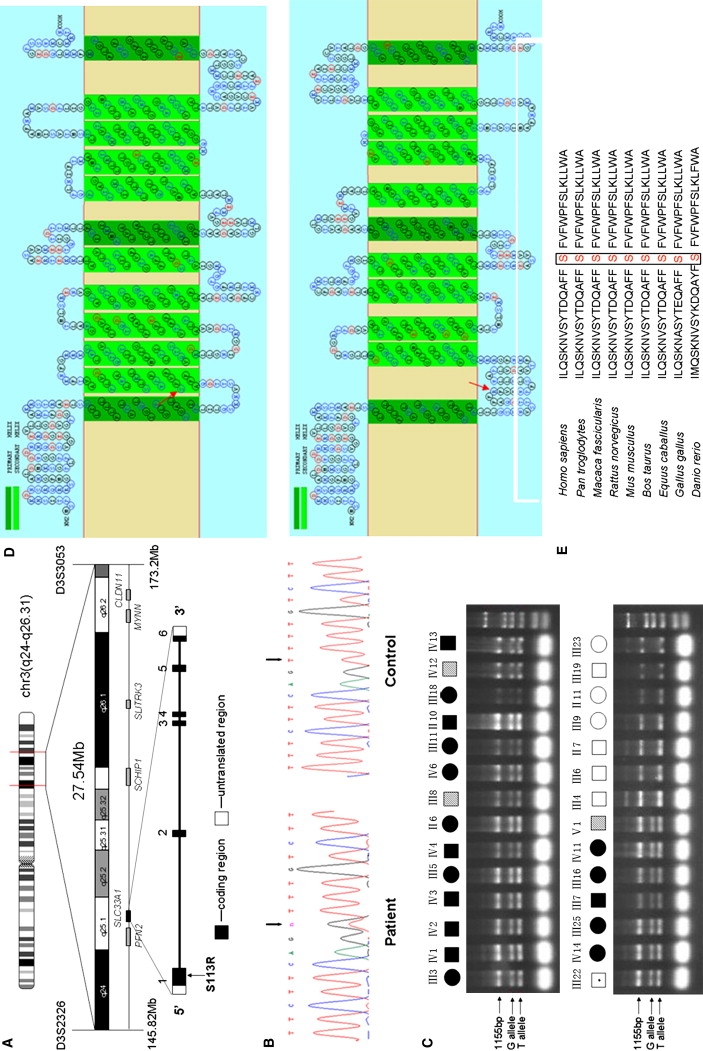
Mutation in SLC33A1 Causes HSP
(A) The candidate region of 27.54 Mb between D3S2326 and D3S3053. The candidate genes screened are shown at the bottom. The location of SLC33A1 is highlighted.
(B) Partial sequence chromatograms of exon 1 of SLC33A1 in a patient and a healthy control. The arrows mark the position of the SLC33A1 mutation. Sequence analysis was done on an ABI 3100 automated sequencer (Applied Biosystems).
(C) Confirmation of the c.339T→G mutation by allele-specific tetra-primer PCR assay. All definitely affected and mildly affected individuals exhibited the G allele. Primers sequences are as follows: forward outer primer 5′-GTGCCCTTATCGCTCTGA-3′, reverse outer primer 5′-GTGTTATTTGATGGGTTGC-3′, forward inner primer (G allele) 5′-TTAGCTATACAGACCAAGCTTTCTTCGGG-3′, and reverse inner primer (T allele) 5′-TTGAGACTGAAGGGCCAAAAGACAGAA-3′.
(D) Topological prediction of the mutant SLC33A1 protein, with use of the SOSUI program. The S113R mutation resulted in the loss of the original second TM motif and reversed the orientation of all domains, starting from 113. Upper panel, normal SLC33A1; bottom panel, mutant SLC33A1 (S113R). The mutated amino acid is indicated by the red arrow.
(E) Amino acid sequence alignment of human SLC33A1 and orthologs from other species, showing phylogenetic conservation of S113. The sequences were retrieved from the Entrez protein database and aligned to each other with the use of Clustal W.
SLC33A1 encodes the acetyl-CoA transporter, which consists of 549 amino acids and contains multiple transmembrance domains, with a leucine zipper motif in transmembrane domain III.15,16 The c.339T→G transversion in exon 1 resulted in a missense mutation that changed serine to arginine at the codon 113 (p. S113R). The effect of this variation on gene function was predicted to be nonneutral by the SNAP program and to be probably damaging by the PolyPhen program. In silico topology prediction, with SOSUI software, indicated that SLC33A1 contains 12 transmembrane domains. The S113 residue of SLC33A1 is located at the beginning of the second transmembrane domain. The SOSUI software predicted that the S113R mutation would dislodge the second transmembrane domain (Figure 3D), leading to its expulsion from the membrane bilayer. Furthermore, it would also cause all domains starting from 113R to be placed in an opposite orientation across the membrane when compared to the normal transporter (Figure 3D). Cross-species alignment of the amino acid sequences for SLC33A1 also shows that the serine at position 113 is highly conserved among vertebrates (Figure 3E). Thus, the S113R mutation probably renders SLC33A1 nonfunctional and leads to a functional haploinsufficiency in the heterozogtes. Alternatively, S113R could act as a dominant-negative mutation in causing the phenotypes in the heterozygotes. However, as shown below in the functional study of S113R in zebrafish, the latter was less likely.
Effect of SLC33A1 Mutation on Zebrafish Development
Zebrafish have recently been used in the study of pathological mechanisms underlying HSP.8,17 Decreased levels of SPG4 in the developing zebrafish embryos were shown to cause motor-axon-specific outgrowth defects that can only be rescued by wild-type SPG4, not by mutated SPG4.17 Similar phenotype was observed when SPG8 was knocked down.8 Considering that SLC33A1 is ubiquitously expressed and that human and zebrafish SLC33A1 proteins are 69% identical, we used zebrafish as a model to test the function of slc33a1 and the consequence of the missense mutation S113R. First, we determined whether reduced levels of SLC33A1 would affect the development of zebrafish. An antisense morpholino (MO) was designed to specifically inhibit translation of zebrafish slc33a1 mRNA (5′-TGTGAGAGATTCAGACAGTTC(CAT)C-3′), and a 5 bp mismatch control (TGTcAGAcATTCAcACAcTTC(gAT)C) was used to titer a MO-specific nontoxic injection dose. The specificity and validity of slc33a1 MO and control MO were verified by TNT Quick Coupled Translation System (Promega) (data not shown). Slc33a1 MO or control MO was injected into embryos at one- to four-cell stage, and embryos were allowed to develop until the desired stage. Approximately 78% (n = 216) of embryos injected with slc33a1 MO survived, compared to 91% (n = 256) survival when injected with the same concentration of control MO. Among the slc33a1 MO-injected embryos that survived, many showed curve-shaped tails similar to those observed in SPG4 and SPG8 MO knockdown experiments in the zebrafish.8,17 As shown in Figure 4A, at 36 hr after fertilization (hpf), dechorionated wild-type zebrafish had a straight tail (Figure 4Aa) and no significant change was observed in the fish injected with mismatch control MO (Figure 4Ab). In contrast, slc33a1 MO-injected fish exhibited severely or slightly curly tail phenotypes (Figures 4Ac and 4Ad). When the injected embryos were divided into four categories—normal, slightly curly tail, severely curly tail, and lethality—at 36 hpf, 50.5% (109/216) embryos were observed as having a slightly curly tail and 22.2% (48/216) as having a severely curly tail in the slc33a1 MO-injected group, in comparison to 12.5% (32/256) and 10.9% (28/256), respectively, in the control MO-injected group. Next, we determined whether human SLC33A1 could correct the defects observed in slc33a1 MO-injected zebrafish. One- to four-cell embryos were injected with a mixture of slc33a1 MO and full-length human SLC33A1 mRNA, which is not recognized by the slc33a1 MO. In contrast to the high percentage, 72.7%, of embryos with abnormal tails occurring when slc33a1 MO alone was injected, coinjection of human SLC33A1 mRNA and slc33a1 MO reduced the percentage of embryos with abnormal tails to 24.4%, which was comparable to that in the control MO-injected group, 23.4% (Figures 4Ae, 4Af, and 4B). These results suggest that the increased occurrence of the curved-tail phenotype was caused by a reduction of slc33a1 protein and that human SLC33A1 mRNA can compensate for the loss of the endogenous zebrafish mRNA.
Figure 4.
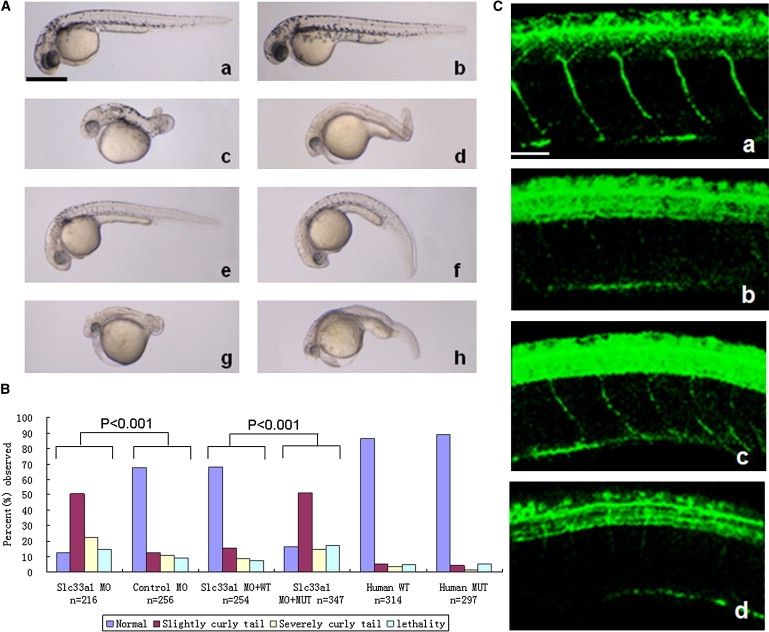
Rescue of slc33a1 Knockdown-Induced Abnormal-Tail Phenotype in Zebrafish by Wild-Type, But Not Mutant, Human SLC33A1 mRNA
(A) Morphological features of zebrafish at 36 hpf from knockdown and rescue experiments. a, Untreated wild-type zebrafish; b, Zebrafish injected with mismatch control MO; c and d, Slc33a1 MO-injected zebrafish with severely curved tail (c) and slightly curved tail (d); e and f, zebrafish coinjected with slc33a1 MO and wild-type human SLC33A1 mRNA—the curly-tail phenotype was rescued completely (e) and partially (f); g and h, zebrafish coinjected with slc33a1 MO and mutated human SLC33A1 mRNA, with severely curved tail (g) or slightly curved tail (h). Scale bar: represents 1 mm.
(B) Phenotype profile from zebrafish knockdown and rescue experiments. The embryos were classified as normal, slightly curly tail, severely curly tail, and lethality, and the percentage for each group is shown.
(C) Defective motor-neuron outgrowth in the reduction of slc33a1. Spinal motor axons were stained with an anti-acetylated tubulin antibody at 36 hr after fertilization. a, motor neurons in spinal cord of zebrafish injected with mismatch control MO; b, injection of slc33a1 MO dramatically impaired outgrowth of motor axons from the spinal cord; c, coinjection of human wild-type SLC33A1 mRNA with slc33a1 MO rescued motor-axon defects; d, Coinjection of mutated human SLC33A1 mRNA did not rescue motor axon defects. Scale bar represents 50 μm.
If the defects observed in slc33a1 MO-injected fish reflect spastic paraplegia in humans, it should be predicted that the mutant SLC33A1 that causes HSP would not be able to rescue the developmental defect caused by slc33a1 knockdown. Indeed, when slc33a1 MO and human mRNA with S113R mutation were coinjected, the frequency of tail abnormality was not significantly different from that in slc33a1 MO injection alone, indicating that the human SLC33A1 with S113R mutation can not compensate for the loss of the endogenous zebrafish mRNA. Furthermore, injection of S113R mutant mRNA alone did not cause an increase in the occurrence of developmental defects (Figure 4B), suggesting that S113R mutation might not act in a dominant-negative manner.
HSPs are caused by developmental failure or degeneration of motor axons in the corticospinal tract.1,2 To determine whether the tail abnormality and impaired mobility observed upon slc33a1 knockdown were due to motor-axon defects, we examined the motor axon by staining embryos with an antibody against acetylated tubulin, which marks the growing spinal motor neurons. As shown in Figure 4Cb, by 36 hpf, spinal motor neurons in slc33a1 MO embryos were scarce and poorly organized, in contrast to the orderly downward outgrowth from the spinal cord observed in embryos injected with a control MO (Figure 4Ca), indicating that inhibition of slc33a1 significantly impaired outgrowth of motor axons. Furthermore, the motor-axon defects could be rescued by wild-type human SCL33A1 mRNA (Figure 4Cc) but not by mutant human SLC33A1 mRNA (Figure 4Cd).
Therefore, the results obtained with zebrafish substantiated the notion that S113R in SCL33A1 is a loss-of-function mutation. The fact that spastic paraplegia manifests in S113R heterozygous individuals suggests that SLC33A1 is haploinsufficient. Acetyl-CoA transporter, the protein product of SLC33A1, is positioned to carry acetyl-CoA into the lumen of Golgi apparatus, where acetyl-CoA is transferred to the sialyl residues of gangliosides and glycoproteins.15,16 The modification of gangliosides and glycoproteins by acetylation probably plays a critical role in the outgrowth and maintenance of the axons of the motor neurons. Inadequate supply of acetyl-CoA, as caused by a reduced flow of acetyl-CoA into the Golgi apparatus, can result in misprocessing of gangliosides and glycoproteins. Additional work is obviously needed to elucidate the molecular mechanism of the protein and to determine how it relates to other proteins implicated in HSP during the disease development. As more genes causing HSP are identified, we will have a better understanding of the molecular pathology of HSP.
Acknowledgments
We thank the patients and their families for their participation, and we thank Hongwei Zhang and Ming Shao (Shandong University School of Life Science), for expert technical assistance. This work was supported by the National Basic Research Program of China (grant no. 2007CB512001) and the National Science Foundation Research Grant (grant no. 30771201).
Supplemental Data
Web resources
The URLs for data presented herein are as follows:
ClustalW, http://www.ebi.ac.uk/clustalw
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Merlin, http://www.sph.umich.edu/csg/abecasis/Merlin/index.html
NCBI BLAST, http://www.ncbi.nlm.nih.gov/blast/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SOSUI, http://www.expasy.ch
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: An update. Curr. Opin. Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 3.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 4.Fink J.K. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003;184(Suppl 1):S106–S110. doi: 10.1016/j.expneurol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Hazan J., Fonknechten N., Mavel D., Paternotte C., Samson D., Artiguenave F., Davoine C.S., Cruaud C., Dürr A., Wincker P. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C.H., Tukel T., Apak M., Heiman-Patterson T., Ming L. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 7.Rainier S., Chai J.H., Tokarz D., Nicholls R.D., Fink J.K. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6) Am. J. Hum. Genet. 2003;73:967–971. doi: 10.1086/378817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valdmanis P.N., Meijer I.A., Reynolds A., Lei A., MacLeod P., Schlesinger D., Zatz M., Reid E., Dion P.A., Drapeau P., Rouleau G.A. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am. J. Hum. Genet. 2007;80:152–161. doi: 10.1086/510782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid E., Kloos M., Ashley-Koch A., Hughes L., Bevan S., Svenson I.K., Graham F.L., Gaskell P.C., Dearlove A., Pericak-Vance M.A. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am. J. Hum. Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen J.J., Dürr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M.N., Davoine C.S., Brice A., Fontaine B., Gregersen N. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Züchner S., Wang G., Tran-Viet K.N., Nance M.A., Gaskell P.C., Vance J.M., Ashley-Koch A.E., Pericak-Vance M.A. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am. J. Hum. Genet. 2006;79:365–369. doi: 10.1086/505361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mannan A.U., Krawen P., Sauter S.M., Boehm J., Chronowska A., Paulus W., Neesen J., Engel W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006;79:351–357. doi: 10.1086/504927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauter S.M., Engel W., Neumann L.M., Kunze J., Neesen J. Novel mutations in the Atlastin gene (SPG3A) in families with autosomal dominant hereditary spastic paraplegia and evidence for late onset forms of HSP linked to the SPG3A locus. Hum. Mutat. 2004;23:98. doi: 10.1002/humu.9205. [DOI] [PubMed] [Google Scholar]
- 14.Beetz C., Schüle R., Deconinck T., Tran-Viet K.N., Zhu H., Kremer B.P., Frints S.G., van Zelst-Stams W.A., Byrne P., Otto S. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131:1078–1086. doi: 10.1093/brain/awn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanamori A., Nakayama J., Fukuda M.N., Stallcup W.B., Sasaki K., Fukuda M., Hirabayashi Y. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter. Proc. Natl. Acad. Sci. USA. 1997;94:2897–2902. doi: 10.1073/pnas.94.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirabayashi Y., Kanamori A., Nomura K.H., Nomura K. The acetyl-CoA transporter family SLC33. Pflugers Arch. 2004;447:760–762. doi: 10.1007/s00424-003-1071-6. [DOI] [PubMed] [Google Scholar]
- 17.Wood J.D., Landers J.A., Bingley M., McDermott C.J., Thomas-McArthur V., Gleadall L.J., Shaw P.J., Cunliffe V.T. The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 2006;15:2763–2771. doi: 10.1093/hmg/ddl212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.