

Abstract
Diamond-Blackfan anemia (DBA), a congenital bone-marrow-failure syndrome, is characterized by red blood cell aplasia, macrocytic anemia, clinical heterogeneity, and increased risk of malignancy. Although anemia is the most prominent feature of DBA, the disease is also characterized by growth retardation and congenital anomalies that are present in ∼30%–50% of patients. The disease has been associated with mutations in four ribosomal protein (RP) genes, RPS19, RPS24, RPS17, and RPL35A, in about 30% of patients. However, the genetic basis of the remaining 70% of cases is still unknown. Here, we report the second known mutation in RPS17 and probable pathogenic mutations in three more RP genes, RPL5, RPL11, and RPS7. In addition, we identified rare variants of unknown significance in three other genes, RPL36, RPS15, and RPS27A. Remarkably, careful review of the clinical data showed that mutations in RPL5 are associated with multiple physical abnormalities, including craniofacial, thumb, and heart anomalies, whereas isolated thumb malformations are predominantly present in patients carrying mutations in RPL11. We also demonstrate that mutations of RPL5, RPL11, or RPS7 in DBA cells is associated with diverse defects in the maturation of ribosomal RNAs in the large or the small ribosomal subunit production pathway, expanding the repertoire of ribosomal RNA processing defects associated with DBA.
Main Text
Diamond-Blackfan anemia (DBA) (MIM 105650) is an inherited congenital bone-marrow-failure syndrome, characterized by normochromic macrocytic anemia and absence or insufficiency of erythroid precursors in otherwise normocellular bone marrow.1 Although anemia is the most prominent feature of DBA, the disease is also characterized by growth retardation and congenital malformations, in particular craniofacial, upper limb, heart, and urinary-system defects, that are present in ∼30%–50% of patients, reflecting the fact that DBA is a broad disorder of development.2–4 Laboratory findings such as increased mean corpuscular volume (MCV), elevated erythrocyte adenosine deaminase activity (eADA), and hemoglobin F are observed in a majority of, but not in all, DBA patients.5,6 The disease is clinically heterogenous, and within affected families, some individuals can exhibit mild or absent anemia, with only subtle indications of erythroid abnormalities, such as macrocytosis or elevated eADA and/or HbF. Additionally, increased risk of malignancy—in particular, AML and solid tumors, including osteogenic sarcoma7,8—has been associated with DBA. Approximately 90% of affected individuals present during the first year of life or in early childhood, although patients with a “nonclassical” mild phenotype are usually diagnosed later in life.3,4 The incidence of DBA is 5–7 per million live births,2,3,9 affecting both genders equally.4 Although most cases are sporadic, about 10%–25%1,4 and up to, according to recent data, even 45%10 are familial, with disease inherited in an autosomal-dominant pattern.
Heterozygous mutations in ribosomal protein genes RPS19 (MIM 603474), RPS24 (MIM 602412), and RPS17 (MIM 180472), encoding RPs of the small ribosomal subunit, and in RPL35A (MIM 180468), encoding an RP of the large subunit, have been reported in about 30% of DBA patients,6,10–15 suggesting that DBA is a disorder of ribosomes. Haploinsufficiency of RPS19 and RPS24 has been shown to be the basis for DBA in patients with mutations of those genes. In a subset of patients, large deletions and frameshift mutations lead to degradation of the mutated transcripts.9,13,16,17 Recent studies have also demonstrated that DBA missense mutations affect RPS19 conformation and stability, triggering proteasome-mediated degradation or blocking RPS19's incorporation into preribosomes.18–20 Despite mutations identified in four genes, no significant phenotype-genotype correlations have been reported to date, because RPS24, RPS17, and RPL35A mutations are rare and have been reported in only eight, one, and six patients, respectively.
RPS19 protein has been shown to play an important role in 18S rRNA maturation in yeast and in human cells.21–24 Similarly, alterations of pre-RNA processing and small or large ribosomal-subunit synthesis were demonstrated in human cells with RPS24 and RPL35A deficiency, respectively, further indicating that DBA is a disorder of ribosomes.15,25 Deficiency of RPS19 and RPL35A was shown to cause increased apoptosis in hematopoietic cell lines and in bone-marrow cells,15,26 and it has been suggested that imbalance of the p53 family proteins is a mechanism of abnormal embryogenesis and anemia in zebrafish upon perturbation of RPS19 expression.27
Here, we report the results of a large-scale screen of 24 additional RP genes and of RPS17 (in which one mutation among 24 tested patients was previously reported14) in a cohort of DBA probands. Remarkably, we identified probable pathogenic mutations in four of these genes, RPL5 (MIM 603634), RPL11 (MIM 604175), RPS7 (MIM 603658), and RPS17, representing ∼16% of our patient cohort. In addition, we report possible single mutations in three other RP genes, RPL36, RPS15 (MIM 180535), and RPS27A (MIM 191343). In contrast to previously reported data on patients with RPS19 mutations, mutations in RPL5 are associated with multiple physical abnormalities, including craniofacial, thumb, and heart anomalies, whereas isolated thumb abnormalities are predominantly present in patients carrying mutations in RPL11. We also demonstrated defective rRNA maturation in RPL5-, RPL11-, and RPS7-mutated DBA cells.
To test the hypothesis that mutations in other RP genes can also cause DBA, we carried out direct sequencing of RP genes, starting with what we considered to be the most probable candidates. One hundred ninety-six DBA families participated in the study. Thirty-six of them were multiplex families, and 160 included only one clinically affected individual. All probands were negative for mutations in RPS19 and RPS24, and 132 tested probands were negative for mutations in RPL35A. Informed consent was obtained from all patients and their family members participating in the study under a protocol at Children's Hospital Boston. The diagnosis of DBA in all probands was based on normochromic, often macrocytic, anemia; reticulocytopenia; a low number or lack of erythroid precursors in bone marrow; and, in some patients, congenital malformations and elevated eADA.
Genomic DNA samples from 95 unrelated DBA probands enrolled in the study were amplified by polymerase chain reaction (PCR) and sequenced for mutations in 25 RP genes. Initially, we sequenced three genes, RPS3A, RPS13, and RPS16, which encode RPs and, like RPS19 and RPS24, are involved in binding of initiation factor eIF-2 to the 40S subunit.28 Subsequently, we sequenced 11 RP genes located on chromosome 19 (RPS5, RPS9, RPS11, RPL13A, RPL18, RPL18A, RPL28, RPL40, RPS28, RPL36, RPS15), because previous studies had reported linkage of DBA to this chromosome in families that did not segregate RPS19 mutations.11 Next, we screened five genes on chromosome 1 (RPL5, RPL11, RPL22, RPS27, RPS8), because Heyn et al. previously reported a pericentric inversion of chromosome 1 in a DBA patient with cleft lip and palate and a ventricular septal defect.29 RPS17 (chromosome 15) was screened because Cmejla et al. reported one mutation among 24 tested DBA patients,14 and RPL23 (chromosome 17) was screened because the RPS23 protein inhibits MDM2-mediated p53 ubiquitination and degradation in response to ribosomal stress in the same manner as RPL5 and RPL11.30 Subsequently, we have started sequencing RP genes according to their chromosomal location, starting with four genes on chromosome 2 (RPL31, RPL37A, RPS7, RPS27A). Primers (sequences available on request) were designed with Primer3 software for amplification of the coding exons and intron-exon boundaries of the above genes. PCR products, between 200 and 600 bp, were robotically prepared with a Tecan Genesis Workstation (Tecan, Duram, NC), purified with Exo-SAP enzyme (USB, Cleveland, OH) and sequenced on both strands with an Applied Biosystems 3730 DNA Analyzer (Applied Biosystems, Foster City, CA). The chromatograms were analyzed with Sequencher software, version 4.7 (Gene Codes, Ann Arbor, MI). When a sequence change was identified in a given gene, we sequenced an additional 101 samples from unrelated probands for mutations in this gene. Duplicate, independent PCR products were sequenced to confirm the observed nucleotide changes in the probands. We sequenced DNA samples from at least 150 control individuals, i.e., 300 chromosomes, to determine whether the observed sequence variations were nonpathogenic variants. Subsequently, we sequenced DNA samples from available family members to determine whether the mutation cosegregated with the DBA phenotype within the pedigree.
Interestingly, Heyn and colleagues reported a DBA patient with cleft lip and palate, VSD, and pericentric inversion of chromosome 1, inv (1p-; 1q+), resulting in the shortened p arm and lengthened q arm.29 These findings suggest that the gene causing DBA might be located on chromosome 1. In accord with these findings, direct sequencing revealed multiple sequence changes in DBA patients in coding regions and intron-exon boundaries in two genes, RPL5 and RPL11, located on chromosome 1p (1p22.1 and 1p35-p36.1, respectively). In addition, screening of 23 other RP genes revealed single sequence changes in five of them, RPS7, RPS17, RPL36, RPS15, and RPS27A, in DBA probands. In total, we found sequence changes in seven out of 25 screened RP genes (Table 1). DNA samples from available family members were also screened, demonstrating that the identified sequence changes cosegregated with the DBA phenotype within the pedigrees (Tables 1–4).
Table 1.
Summary of Sequence Changes in Seven RP Genes Identified in 196 DBA Families
| Gene Symbol | No. of Tested DNA Samples from Unrelated Probands | No. of Probands with Mutations (%) | No. of Subjects with Mutations | Mutation Types |
|---|---|---|---|---|
| RPL5 | 196 | 18 (9%) | 24 | nonsense, missense, splice site, small indel |
| RPL11 | 196 | 13 (6.5%) | 18 | nonsense, splice site, small indel |
| RPS7 | 159 | 1 < 1% | 1 | splice site |
| RPS17 | 193 | 1 < 1% | 1 | small del |
| RPL36 | 132 | 1 < 1% | 1 | small del |
| RPS15 | 151 | 1 < 1% | 1 | missense |
| RPS27A | 159 | 1 < 1% | 2 | missense |
“Indel” denotes insertion-deletion; “del” denotes deletion.
Table 2.
RPL5 Mutations in 18 Probands and Six Family Members from a Total of 196 Families Studied
| Mutation Type | Proband's ID (Gender) Inheritance | Family Members | DNA Mutation | Exon/ Intron | Predicted Amino Acid Change | Age at Diagosis | Malformation Status | Response at First Steroid Therapy | Present Therapy |
|---|---|---|---|---|---|---|---|---|---|
| Nonsense mutation | P1 (F) sporadic | m: normal seq | c.48C→G | Ex2 | Y16Stop | at birth | NA | responsive to high doses only | RBC trx |
| P2 (F) de novo | f, m: normal seq | c.67C→T | Ex2 | R23Stop | 2mo | cleft lip, cleft palate | responsive SD | RBC trx | |
| P3 (F) sporadic | c.228C→A | Ex4 | C76Stop | 10 mo | cleft palate, ASD, hypoplastic thumb, micrognathia, tracheomalacia | initially responsive | stem cell transplant | ||
| Missense mutation | P4 (M) familial | m, s, b: normal seq | c.418G→A | Ex5 | G140S | 3 mo | none | unresponsive | RBC trx |
| f | c.418G→A | Ex5 | G140S | no anemia | NA | no therapy | no therapy | ||
| Deletion/ insertion | P5 (M) sporadic | c.46_47 insA | Ex2 | Y16Stop | at birth | micrognathia, hypertelorism, soft cleft palate, triphalangeal right thumb, widened webbed spaced between first and second toes, hypospadias | no therapy | no therapy | |
| P6 (F) de novo | f, m: normal seq | c.156_159 delAGTT | Ex3 | Frameshift at codon 52; stop at 54 | 10 mo | small jaw, cleft palate, triphlangeal thumb, hip dysplasia, rib anomalies | responsive SD | steroid therapy | |
| P7 (M) sporadic | c.169_172 delAACA | Ex3 | Frameshift at codon 57; stop at 68 | at birth | partial anomalous pulmonary venous return | unresponsive | RBC trx | ||
| P8 (M) de novo | f, m, b: normal seq | c.173 delG | Ex3 | Frameshift at codon 58; stop at 69 | 9 mo | multiple congenital heart defectsa | responsive SD | RBC trx | |
| P9 (M) sporadic | c.173_4 delGA | Ex3 | Frameshift at codon 58; stop at 111 | NA | long proximal thumb phalanges bilateral, multiple congenital heart defectsb | NA | NA | ||
| P10 (M) sporadic | c.173_4 delGA | Ex3 | Frameshift at codon 58; stop at 111 | 10 mo | small jaw, cleft palate, bronchiopharyngeal malacia, mild hydrocephalus | responsive SD | steroid therapy | ||
| P11 (F) de novo | f, m, b, s: normal seq | c.173_4 delGA | Ex3 | Frameshift at codon 58; stop at 111 | NA | NA | NA | NA | |
| P12 (F) familial | c.235_236 insT | EX4 | Frameshift at codon 79; stop at 112 | 11 yc | cleft soft palate | unresponsive | RBC trx | ||
| m: familial | c.235_236 insT | EX4 | Frameshift at codon 79; stop at 112 | 55 y | none | refused steroid trial | RBC trx | ||
| P13 (F) familial | c.235_236 insT | EX4 | Frameshift at codon 79; stop at 112 | 12 mo | cleft palate, bifid uvula, hypoplastic thumb, | NA | RBC trx | ||
| m: familial | c.235_236 insT | EX4 | Frameshift at codon 79; stop at 112 | NA | none | NA | RBC trx | ||
| P14 (F) familial | 348_351 insTGGA | Ex5 | Frameshift at codon 117; stop at 121 | 12 mo | mandibular hypoplasia with retrognathia, cleft palate with bifid uvula, dysplastic thumbs, ASD type II | unresponsive | RBC trx | ||
| s: familial | ND | ND | 12 mo | mandibular hypoplasia with retrognathia, cleft palate, triphalangeal thumbs, persistent foramen ovale, ASD type II | no therapy | no therapy | |||
| m: familial | 348_351 insTGGA | Ex5 | Frameshift at codon 117; stop at 121 | 20 yr | none | no therapy | no therapy | ||
| P15 (M) familial | f, m, s: normal seq | c.498_502 delTGTGG and 39bp ins | Ex5 | NA | triphalangeal thumbs, VSD | responsive | no therapy | ||
| b: familial | c.498_502 delTGTGG and 39bp ins | Ex5 | NA | cleft lip, triphalangeal thumbs | unresponsive | RBC trx | |||
| d: familial | c.498_502 delTGTGG and 39bp ins | Ex5 | NA | NA | NA | NA | |||
| P16 (M) sporadic | c.573_574 insG | Ex. 6 | Frameshift at codon 192; stop at 216 | NA | inability to flex right distal thumb phalanx | responsive SD | NA | ||
| Splice-site mutation | P17 (M) de novo | f, m, b: normal seq | Donor splice site IVS2 +2t→g | Intr2 | NA | none | responsive SD | RBC trx | |
| P18 (M) sporadic | Donor splice site IVS3 +1g→t | Intr3 | NA | none | responsive SD | RBC trx |
Abbreviations are as follows: P, proband; f, father; m, mother; s, sister; b, brother; d, daughter; ins, insertion; del, deletion; Ex, exon; In, intron; seq, sequence; mo, month; y, years; ND, not done; NA, not available; SD, steroid dependent; RBC trx, red blood cell transfusions; ASD, atrial septal defect; VSD, ventricular septal defect.
Small patent ductus arteriosus, mild mitral valve prolapse, mild mitral regurgitation.
Double outlet right ventricle, pulmonary stenosis, left pulmonary artery stenosis, patent ductus arteriosus.
macrocytic anemia diagnosed at age 11, DBA not diagnosed until years later.
Table 3.
RPL11 Mutations in 13 Probands and Five Family Members from a Total of 196 Families Studied
| Mutation Type | Proband's ID (Gender) Inheritance | Family Members | DNA Mutation | Exon/Intron | Predicted Amino Acid Change | Age at Diagosis | Malformation Status | Response at First Steroid Therapy | Present Therapy |
|---|---|---|---|---|---|---|---|---|---|
| Nonsense mutation | P19 (F) de novo | f, m: normal seq | c.223C→T | Ex 3 | R75Stop | 2 mo | triphalangeal thumbs | responsive SD | steroid therapy |
| Deletion/Insertion | P20 (F) familial | c.60_61 delCT | Ex2 | Frameshift at codon 20; stop at 53 | 9 mo | VSD, narrow pulmonary artery | responsive | RBC trx | |
| m: familial | c.60_61 delCT | Ex2 | Frameshift at codon 20; stop at 53 | NA | triphalangeal thumb | no steroid trial | RBC trx | ||
| gm: familial | c.60_61 delCT | Ex2 | Frameshift at codon 20; stop at 53 | NA | triphalangeal thumb | NA | NA | ||
| P21 (F) sporadic | c.94_97 delAGAC | Ex2 | Frameshift at codon 32; stop at 32 | 1 mo | none | responsive SD | steroid therapy | ||
| P22 (M) sporadic | c.160_161 insA | Ex3 | Frameshift at codon 54; stop at 66 | NA | abnormal thumbs | responsive SD | RBC trx | ||
| P23 (F) de novo | f, m: normal seq | c.290 delA | Ex4 | Frameshift at codon 97; stop 14 aa behind wt stop | 1 mo | short neck | unresponsive | stem cell transplant | |
| P24 (M) de novo | f, m: normal seq | c.291_292 insA | Ex4 | Frameshift at codon 97; stop at 120 | NA | horseshoe kidney | responsive SD | steroid therapy | |
| P25 (M) familial | c.314_315 delTT | Ex4 | Frameshift at codon 105; stop at 119 | NA | none | responsive | no therapy | ||
| son | c.314_315 delTT | Ex4 | Frameshift at codon 105; stop at 119 | NA | small extra thumbs | no therapy | no therapy | ||
| P26 (F) familial | c.482_484 delAGG | Ex5 | 161Edel | 2 mo | none | responsive to high doses | RBC trx | ||
| m: familial | c.482_484 delAGG | Ex5 | 161Edel | NA | none | no therapy | no therapy | ||
| Splice-site mutation | P27 (F) de novo | f, m: normal seq | Donor splice site IVS1 +2t→c | In1 | 3 mo | none | unresponsive | RBC trx | |
| P28 (M) de novo | f, m, b: normal seq | Acceptor splice site IVS2 -1g→a | In2 | 3 mo | flat thenar muscle, small jaw | responsive SD | steroid therapy | ||
| P29 (M) sporadic | Acceptor splice site IVS2 -1g→a | In2 | 2 y | Tetralogy of Fallot, bilateral grade 3 vesicoureteral reflex | responsive SD | steroid therapy | |||
| P30 (F) de novo | f, m, b: normal seq | Donor splice site IVS4 +1g→t | In4 | 2.5 mo | hypoplastic thumb, flat thenar muscle | responsive/ stopped due to growth retardation | RBC trx | ||
| P31 (M) familial | Donor splice site IVS4 +1g→a | In4 | 12 mo | small extra thumbs, Tetralogy of Fallot | responsive SD | steroid therapy | |||
| m: familial | Donor splice site IVS4 +1g→a | In4 | 21 y | left thumb: additional short phalanx | no steroid therapy | RBC trx (2x/yr) |
Abbreviations are as follows: P, proband; f, father; m, mother; gm, grandmother; s, sister; b, brother; ins, insertion; del, deletion; Ex, exon; In, intron; seq, sequence; mo, month; y, years; NA, not available; SD, steroid dependent; RBC trx, red blood cell transfusions; VSD, ventricular septal defect.
Table 4.
Sequence Changes in RPS7, RPS17, RPL36, RPS15, and RPS27A in DBA Patients
| Mutated Gene | Proband's ID (Gender) Inheritance | Family Members | DNA Mutation | Exon/Intron | Predicted Amino Acid Change | Age at Diagosis | Malformation Status | Response at First Steroid Therapy | Present Therapy |
|---|---|---|---|---|---|---|---|---|---|
| RPS7 | P32 (M) sporadic | s – normal sequence | Donor splice site IVS3+1g>a | In3 | NA | none | responsive SD | steroid therapy | |
| RPS17 | P33 (M) de novo | f, m, s – normal sequence | c. 200_201 delGA | Ex3 | Frameshift at codon 67; stop at 86 | 4 mo | none | responsive SD | steroid therapy |
| RPL36 | P34 (M) sporadic | c. 250_251 delGA | Ex3 | Frameshift at codon 84; stop 27 nt behind wt stop codon | 2 mo | left thumb abnormality | responsive SD | steroid therapy | |
| RPS15 | P35 (F) sporadic | c.208A→G | Ex3 | Met70Val | 1 d | truncus arteriosis type 1, VSD, colobomata, absent right radius, ulna and thumb, absent left ulna, small left thumb and 5th finger, unilateral absent fibulae and club foot | unresponsive | bone marrow transplant | |
| RPS27A | P36 (F) | m, b – normal sequence | c.169T→C | Ex4 | Ser57Pro | 15 mo | none | no therapy | no therapy |
| familial | f familial | c.169T→C | Ex4 | Ser57Pro | no DBA diagnosis | none | no therapy | no therapy |
Abbreviations are as follows: P, proband; f, father; m, mother; s, sister; b, brother; del, deletion; Ex, exon; In, intron; nt, nucleotide; wt, wild-type; mo, month; d, day; NA, not available; SD, steroid-dependent; VSD, ventricular septal defect.
To summarize, we identified sequence changes in RPL5 in 18 of 196 probands and in six additional family members, for a total of 24 individuals with RPL5 sequence changes. Seventeen of the identified changes cause premature termination either by nonsense or splice-site mutations or deletions and/or insertions of 1–5 nucleotides causing frameshifts. The 18th is a missense change, 418G→A, resulting in G140S substitution (Tables 1 and 2). All of these changes are unique, (i.e., not previously described in the literature or databases), and 13 of them were found in only a single kindred, whereas one was seen in two and another in three apparently unrelated families. Thirteen sequence changes were found in RPL11 among 196 DBA probands, and RPL11 sequence changes were also identified in five other family members. These included acceptor or donor splice-site mutations, deletions or insertions of 1–4 nucleotides causing a frameshift, and one nonsense mutation. All of these changes are also unique, and 11 of them are distinct from each other, because two changes were seen in two unrelated probands (Tables 1 and 3). Parental DNA was available in seven probands with RPL5 and in seven with RPL11 mutations. De novo sequence changes were identified in four and six probands with sporadic disease, respectively, further supporting the assessment that these sequence changes are probably pathogenic mutations. The single sequence changes in the five other RP genes, RPS7, RPS17, RPL36, RPS15, and RPS27A, in DBA patients are summarized in Table 4. These are a donor splice-site mutation in intron 2 of RPS7, a deletion of 2 nt causing frameshifts in RPS17 and RPL36, as well as two missense changes in RPS15 and RPS27A. Interestingly, only one RPS17 mutation was identified among 24 tested patients14 and only one sequence change was found in our patient cohort, demonstrating that mutations in RPS17 are rare events. None of the identified sequence changes was found on the NCBI SNP lists, and none was identified in at least 300 control chromosomes from a control population of similar, largely European origin. Since we consider it possible that the three missense changes in RPL5, RPS15, and RPS27A could be rare or private nonpathogenic genetic variants, DNA samples carrying these sequence changes are currently being screened for mutations in the remaining 50 RP genes.
Remarkably, the RPL5 combined deletion-insertion (c.498_502delTGTGG and c. 498, a 39 bp insertion) in exon 5 was identified in a family with anemia, triphalangeal thumbs, cleft lip, and a heart abnormality (P15; Table 2), a class of symptoms previously described as Aase syndrome (MIM 105650).31 Strikingly, review of the available medical data on 20 patients with mutations in RPL5 revealed that the majority of them (14/20) have physical malformations, including craniofacial, thumb, and heart anomalies (Tables 2 and 5). Similarly, a majority of patients with RPL11 mutations (12/18) presented with physical malformations, whereas among 76 reported DBA patients with RPS19 mutations, 35 (46%) presented with physical malformations, including short stature6,10,12,32 (Tables 3 and 5). In general, regardless of genotype, congenital malformations were described in 30%–50% of DBA patients.4,6,9,10 Remarkably, nine out of 14 patients with RPL5 mutations and physical abnormalities have cleft lip and/or palate or cleft soft palate, isolated or in combination with other facial malformations—such as micrognathia, hypertelorism, or mandibular hypoplasia with retrognathia—and/or with other physical abnormalities, such as heart or thumb anomalies (Table 5). In contrast, none of the 12 patients with RPL11 mutations and malformations have craniofacial abnormalities (p = 0.007, Fisher's exact test [FET]). Moreover, none of the 35 reported patients with RPS19 mutations and malformations6,10,12,32 presented with cleft lip and/or palate (p = 9.745 × 10−7 for RPL5 versus RPS19, FET) . Furthermore, among a group of 21 DBA patients with craniofacial abnormalities, reported by the Diamond-Blackfan Anemia Registry (DBAR) of North America, no RPS19 mutations were found, which strongly suggested that the DBA phenotype associated with cleft lip and/or palate is caused by gene(s) other than RPS19.33 A report from DBAR revealed that 5.7% of DBA patients present with cleft lip and/or palate4 and some cases were reported with additional microtia and/or ear abnormalities.34 In contrast, in the general population, 0.1%–0.2% of children are born with cleft lip and/or palate.35 Therefore, craniofacial clefting is clearly associated with DBA, particularly with mutations in RPL5. Because about half of DBA patients have unknown underlying genetic causes of DBA, it is likely that mutations in other DBA gene(s) yet to be discovered also cause craniofacial abnormalities. Importantly, it is possible that some DBA patients who present with craniofacial malformations and with only subtle (such as macrocytosis and/or elevated eADA) or no hematological abnormalities are underdiagnosed. RPL5 screening for mutations in these patients could potentially help establish the diagnosis of DBA.
Table 5.
Congenital Physical Abnormality in DBA Patients with RPL5, RPL11, and RPS19 Mutations
| Patients with Mutationsa | Patients with Malformations | Cleft Lip and/or Palateb | Thumb Abnormalityb | Heart Abnormalityb | Other Abnormalities, Including Short Statureb | Multiple Malformations | |
|---|---|---|---|---|---|---|---|
| RPL5 | 20 | 14 | 9c | 8 | 5 | 13 | 11 |
| RPL11 | 18 | 12 | 0 | 8 | 3 | 4 | 3 |
| RPS19d | 76 | 35 | 0 | 7 | 4 | 47 | 16 |
Total number of patients whose medical data are available.
Isolated or in association with other abnormalities.
Including two patients with soft cleft palate.
Based on 6,10,12,32.
In addition to craniofacial malformations in patients with RPL5 mutations, for patients whose medical records are available, eight out of 20 patients with mutated RPL5 and eight out of 18 patients with mutated RPL11 have thumb abnormalities, including triphalangeal or dysplastic thumbs, additional bilateral small thumbs, and bilateral long proximal thumb phalanges. In addition, two patients with RPL11 mutation have flat thenar muscles, one isolated and the other associated with thumb abnormality. In contrast, review of the literature reveals that various thumb abnormalities were reported in only seven out of 76 (9%) patients with RPS19 mutations6,10,12,32 (p = 0.0024 for RPL5 versus RPS19 and p = 0.0012 for RPL11 versus RPS19, FET). Moreover, congenital heart defects were found more often among patients with RPL5 mutations (5/20) compared with RPL11 (3/18) and RPS19 (4/76) (Table 5) (p = 0.017 for RPL5 versus RPS19, FET). Strikingly, the majority (11/20) of patients with RPL5 mutations presented with multiple, severe abnormalities, including craniofacial, heart, and/or thumb malformations. In contrast, patients with RPL11 and RPS19 mutations who presented with multiple physical abnormalities were uncommon: three patients out of 18 and 16 out of 76, respectively6,10,12,32 (Table 5) (p = 0.02 for RPL5 versus RPL11 and p = 0.0047 for RPL5 versus RPS19, FET). Elevated eADA was found in all eight patients with RPL5 mutations and in eight patients with RPL11 mutations for whom the results are available. Willig and colleagues6 described 11 families with RPS19 mutations and eADA measurements. They have found cosegregation of the RPS19 mutations and high eADA in four families, although partial cosegregation of the RPS19 mutations and elevated eADA were found in seven other families.
Our finding of an RPL5 mutation in the family first diagnosed with Aase syndrome,31 as well as other mutations of RPL5, RPL11, and, as described by Willigs and colleagues,6 RPS19 in DBA patients presenting with anemia and triphalangeal thumbs, confirms the notion that the Aase syndrome is a subtype of DBA and not a separate syndrome.
Interestingly, 16 out of 18 patients with RPL5 mutations whose treatment data were available required steroid treatment, and nine of them were responsive to the first steroid treatment, five were unresponsive, and one patient refused steroid treatment, choosing red blood cell transfusions. Similarly, 13 out of 17 patients with RPL11 mutations and available data of their medical treatment also underwent steroid therapy. Eleven of them responded well to the treatment, and two were unresponsive. According to data from the DBA Registry of North America, 79% of DBA patients have been initially responsive to steroid, 17% have been unresponsive, and 4% were never treated with steroids.4 In light of these data, steroid treatment of DBA patients with RPL5 and RPL11 mutations seems to have a similar outcome as that in the general DBA population. At present, 11 RPL5-mutation patients are dependent on red blood cell transfusions, two are steroid-dependent, one patient underwent stem cell transplantation, and three do not need any treatment. Seven RPL11-mutation patients are red blood cell transfusion-dependent, six are dependent on steroid treatment, one patient underwent stem cell transplantation, and three do not need any treatment.
Review of available medical data of 14 patients with RPL5 mutations revealed one patient with melanoma. Similarly, one patient out of 11 with RPL11 mutations and available medical data had uterine cancer. None of the patients with RPS7, RPS17, RPS15, RPL36, and RPS27A sequence changes suffered from cancer. Data from the DBA Registry of North America revealed eight DBA patients among 420 registered as having hematopoietic, nonhematopoietic, or mielodysplastic syndrome.4
DBA-associated ribosomal proteins RPS19, RPS24, and RPL35A were shown to be required for pre-rRNA maturation, a complex process that leads to the production of three rRNAs from a common precursor (Figure 1). To determine whether DBA mutations in RPL5 and RPL11 affected pre-rRNA maturation, we analyzed pre-rRNAs from lymphoblastoid cells established from DBA patients by northern blotting, with probes complementary to the internal transcribed spacer 2 (ITS2), and the signals were quantified by phosphoimager. Cells mutated in either of these genes displayed a conspicuous accumulation of 32S pre-rRNA when compared to cells derived from control individuals, this phenotype being more pronounced for mutations in RPL5 (Figure 2A). In parallel, we also observed a higher amount of 12S pre-rRNA (Figure 2A), as well as accumulation of smaller precursors of 5.8S rRNA, which were not detected in cells derived from the unaffected father of one of the patients with RPL11 mutation (Figure 2B). These results indicate defective maturation of ITS2, both at the initial endonucleolytic cleavage in the 32S pre-rRNA and during subsequent processing steps. The ITS1 probe also revealed a moderate increase in the levels of 30S and 18S-E pre-rRNAs (data not shown), consistent with several reports in yeast showing that perturbation of the pre-60S particle maturation affects maturation of the 18S rRNA.36
Figure 1.
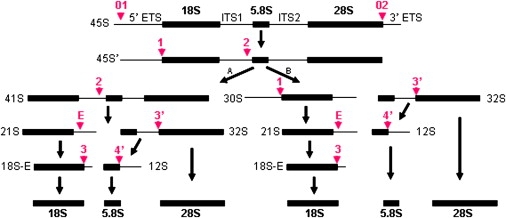
Pre-rRNA Processing in Human Cells, According to Hadjiolova49 and Choesmel22
The mature 18S, 5.8S, and 28S rRNAs are separated by ITS1 and ITS2 and flanked by external transcribed spacers, 5′-ETS and 3′-ETS. The red arrows and the numbers in red indicate the cleavage sites. “A” and “B” represent two pathways of the rRNA processing.
Figure 2.
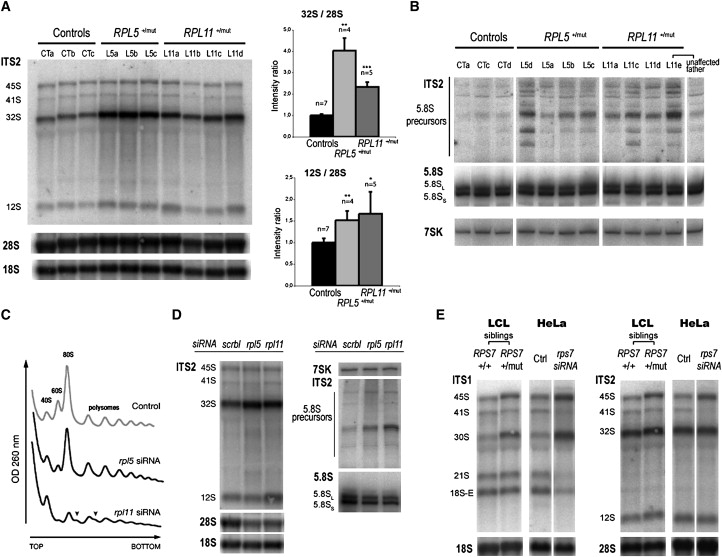
Impact of DBA Mutations in RPL5, RPL11, and RPS7 on Pre-rRNA Processing
(A) Northern blot analysis of total RNAs extracted from lymphoblastoid cells with mutations in RPL5 and RPL11. Precursor and mature rRNAs were detected with probes complementary to the ITS2, 18S rRNA, and 28S rRNA sequences. Bar graphs show signal quantification with a phosphoimager and include experiments from additional samples processed separately. Bars indicate variation of 32S/28S and 12S/28S intensity ratio relative to controls (means ± SD). Significance of the difference compared to controls was assessed with a Student's t test assuming nonequal variance (single asterisk indicates p ≤ 0.05; double asterisk indicates p ≤ 0.01; triple asterisk indicates p ≤ 0.001).
(B) Northern blot analysis of total RNAs fractionated on denaturing 6% polyacrylamide gel. After transfer, the blot was successively hybridized with probes complementary either to the junction between 5.8S rRNA and ITS2 (probe 5′-ITS2) or to 5.8S rRNA. 7SK RNA, a stable and abundant noncoding RNA involved the regulation of RNA polymerase II, was chosen as a loading control.
(C) Analysis on sucrose gradient of cytoplasmic ribosomes isolated from HeLa cells 48 hr after transfection with siRNAs directed against the mRNAs encoding RPL5 or RPL11. Arrowheads indicate polysomes containing half-mers. Control: transfection without siRNA. Expression of both siRNAs results in significant reduction of 60S particles. Differences in the extent of reduction are related to differing efficiencies of the siRNAs used.
(D) Northern blot analysis of total RNAs from cells treated as in (C) with probes complementary to the ITS2, the 18S rRNA and the 28S rRNA (1% agarose gel), or with probes complementary to the junction between the 5.8S rRNA and the ITS2 (probe 5′-ITS2) and to the 5.8S (6% polyacrylamide gel). The 7SK RNA is shown as a loading control for the polyacrylamide gel. Scrbl: scramble siRNA.
(E) Northern blot analysis of pre-rRNA processing in lymphoblastoid cell lines (LCL) with mutation in RPS7 and in HeLa cells transfected with siRNAs targeting RPS7 mRNAs. The control RPS7+/+ lymphoblastoid cells are derived from an unaffected sibling of the RPS7+/mut patient. Ctrl: HeLa cells transfected without siRNAs. Methods: The 21 nt siRNA duplexes with a 3′ dTdT overhang, corresponding to L5 mRNA (5′-AAGGGAGCTGTGGATGGAGGC-3′)47 and to L11 mRNA (5′-AAGGTGCGGGAGTATGAGTTA-3′),48 were purchased from Eurogentec (Seraing, Belgium) and transfected via electrotransformation, as described previously.42 Detection of pre-rRNA on northern blots was performed as described previously50 with oligonucleotidic probes 18S, 28S, ITS1, ITS2b, and ITS2-d/e.42 For detection of ITS2, ITS2-b and ITS2-d/e probes were mixed in equal amounts. The remaining probes had the following sequences: 5′-ITS1, 5′-CCTCGCCCTCCGGGCTCCGTTAATGATC-3′, 5.8S, 5′-CAATGTGTCCTGCAATTCAC-3′; 5′-ITS2, 5′-GGGGCGATTGATCGGCAAGCGACGCTC-3′, 7SK (mix of two probes), 5′-CATGGAGCGGTGAGGGAGGA-3′, and 5′-GTGTCTGGAGTCTTGGAAGC-3′. For ribosome analysis on sucrose gradient, HeLa cells transfected with siRNAs for 48 hr were treated with 100 mg/ml cycloheximide (Sigma Aldrich, St. Louis, MO) for 10 min and fractionated, and the cytoplasmic fraction was analyzed on a 10%–50% sucrose gradient as described previously.42
HeLa cells transfected with siRNAs complementary to mRNAs encoding RPL5 and RPL11 had reduced levels of 28S and 5.8S when compared to controls. Consistently, analysis of cytoplasmic ribosomes on sucrose gradient showed low levels of free 60S subunit and formation of half-mers in the polysomes upon knockdown of RPL5 or RPL11 expression (Figure 2C), indicating that these proteins are essential for synthesis of the 60S subunit. Indeed, inhibition of RPL5 or RPL11 synthesis led to accumulation of 32S and 12S pre-rRNAs, together with shorter precursors to 5.8S (Figure 2D). Given the similarity of these pre-rRNA processing defects with those observed above in lymphoblastoid cells, we conclude that heterozygous mutation of RPL5 and RPL11 in DBA has a direct impact on pre-rRNA processing.
We applied a similar approach to evaluate the consequence of the mutation in RPS7 found in a single DBA patient. Lymphoblastoid cells established from this patient's cells displayed higher levels of 45S and 30S pre-rRNAs when compared to cells derived from an unaffected sibling (Figure 2E). Accordingly, knockdown of RPS7 synthesis in HeLa cells with siRNAs resulted in a strong defect in 5′-ETS processing. We observed accumulation of 45S and 30S pre-rRNAs and a marked decrease of the levels of the 41S, 21S, and 18S-E intermediates, whereas the amount of precursors to the large ribosomal subunit RNAs was unchanged (Figure 2E). This defective processing pattern is very reminiscent of that observed upon depletion of RPS24.25 As for mutations in RPL5 and RPL11, these results strongly suggest that mutation of RPS7 in this DBA patient directly affects maturation of the pre-rRNA.
RPL5, a 297 amino acid, 34 kDa protein component of the 60S large ribosomal subunit, localizes to the cytoplasm and the nucleolus. RPL5 is a nucleocytoplasmic shuttle protein that plays an important role in 5S rRNA intracellular transport during assembly of the large ribosomal subunit.37,38 Likewise, RPL11, a 178 amino acid, 21 kDa protein, is also a component of the large ribosomal subunit. RPL5 and RPL11 are closely connected during ribosome biogenesis; it has been shown, by the recent demonstration in yeast Saccharomyces cerevisiae, that they form a subcomplex with the 5S rRNA prior to incorporation into preribosomes.39 RPS19 deficiency was shown to affect 18S rRNA maturation and small-ribosomal-subunit formation in yeast and in human cells.22–24,40 We showed that mutations of RPS24 in human cells alter maturation of the 18S rRNA at an earlier stage than do mutations in RPS19.25 Similarly, alterations of pre-RNA processing and large-ribosomal-subunit synthesis were demonstrated in cell lines and in DBA cells with RPL35A deficiency.15
Here, we show that mutations in RPL5 and RPL11 in DBA cells lead to accumulation of the 32S and 12S intermediates of 28S rRNA and smaller precursors on the 5.8S rRNA maturation pathways. As shown by the analysis of cytoplasmic ribosomes on sucrose gradient, both RPL5 and RPL11 proteins are essential for rRNA maturation and 60S ribosomal subunit formation. RPL5 and RPL11 knockdown induces accumulation of precursors to 5.8S rRNA, whereas siRNA-mediated knockdown of RPL35A transcripts results in a decrease of these rRNA species,15 indicating that these RPs fulfill different functions in pre-rRNA processing, in agreement with recently published data.41 RPS7 mutation, in turn, affects the 5′-ETS cleavage, similar to RPS24 mutation. The role of RPS15 in nuclear export of the 40S subunit has been documented in mammalian cells42 and in Saccharomyces cerevisiae,21,43 whereas depletion of RPS31, the ortholog of RPS27A in yeast, is also necessary for production of the 40S ribosomal subunit.43 The findings of several ribosomal protein genes mutated in DBA, which subsequently cause aberrant rRNA maturation and small or large subunit formations, supports a hypothesis of a unifying model of DBA as a disorder of ribosome biogenesis that might stem from diverse defects in pre-rRNA processing.
Interestingly, RPL11 has been recently implicated in the negative regulation of c-Myc function. The c-Myc oncoprotein enhances ribosome biogenesis and promotes cell growth. Overexpression of c-Myc was shown to alter ribosome production and tumorigenesis, whereas overexpression of RPL11 inhibits these actions of c-Myc. In contrast, reduction of RPL11 expression increases c-Myc-induced transcription and cell proliferation.44 Both RPL5 and RPL11, as well as RPL23, were shown to inhibit MDM2-mediated p53 ubiquitination and degradation in response to ribosomal stress by restoring p53-mediated transactivation, accumulating p21 protein levels, and inducing a p53-dependent cell-cycle arrest.30,45–48 It will be interesting to evaluate the effects of RPL5 and RPL11 mutations of these pathways in future studies for assessment of their potential role(s) in apoptosis and in promoting tumorigenesis in patients with DBA.
In summary, we found that mutations in RPL5 and RPL11 are present in about 9% and 6.5% of our DBA proband cohort, respectively. After correcting for the fact that our study population was prescreened for RPS19, RPS24, and RPL35A mutations, we estimate that RPL5 and RPL11 mutations are present in about 6.6% and in 4.8% of the overall DBA population, respectively. Remarkably, RPL5 and RPL11 mutations are associated with physical malformations in about 70% and 67% of mutated patients, respectively, as compared to only 46% of patients with RPS19 mutations, none of whom have been reported to have cleft lip and/or cleft palate.6,10,12,32 Thus, to our knowledge, RPL5 is the first ribosomal protein gene to be associated with cleft lip and/or cleft palate abnormalities in DBA patients. Moreover, mutations in RPL5 appear to cause a more severe phenotype, compared to mutations in RPL11 and RPS19, whereas mutations in RPL11 are predominantly associated with thumb abnormalities. The data do not permit definitive determination of pathogenicity for the mutations that we found in RPL36, RPS15, and RPS27A, which will require further study by groups with access to additional DBA patient cohorts.
Acknowledgments
This work was supported by the Diamond-Blackfan Anemia Foundation (to H.T.G.), the Manton Foundation (to H.T.G and A.H.B.) and the French National Research Agency (Agence Nationale de la Recherche—RIBODBA project). DNA sequencing was performed by the Children's Hospital Boston Program in Genomics, Molecular Genetics Core, supported by the Developmental Disabilities Research Center (NIH P30 HD18655) and the Harvard Neuromuscular Disease Project (NIH P50 NS040828). The authors thank Kyriacos Markianos for his help with statistical analysis. Special thanks to Marie Arturi for stimulating DBA research collaboration and to physicians and DBA patients for participating in the study.
Web Resources
The URLs for data presented herein are as follows:
Entrez Gene, http://www.ncbi.nlm.nih.gov/sites/entrez and http://www.ncbi.nlm.nih.gov/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Primer3 Input, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
Accession Numbers
The GenBank accession numbers for human RPL5, RPL11, RPS7, RPS17, RPS15, RPL36, and RPS27A are NM_000969, NM_000975, NM_001011, NM_001021, NM_001018, NM_015414, and NM_002954, respectively.
References
- 1.Alter B.P., Young N.S. The bone marrow failure syndromes. In: Nathan D.G., Orkin H.S., editors. Hematology of Infancy and Childhood. Volume 1. Saunders; Philadelphia, PA: 1998. pp. 237–335. [Google Scholar]
- 2.Ball S.E., McGuckin C.P., Jenkins G., Gordon-Smith E.C. Diamond-Blackfan anaemia in the U.K.: Analysis of 80 cases from a 20-year birth cohort. Br. J. Haematol. 1996;94:645–653. doi: 10.1046/j.1365-2141.1996.d01-1839.x. [DOI] [PubMed] [Google Scholar]
- 3.Willig T.N., Niemeyer C.M., Leblanc T., Tiemann C., Robert A., Budde J., Lambiliotte A., Kohne E., Souillet G., Eber S. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Societe d'Hematologie et d'Immunologie Pediatrique (SHIP), Gesellshaft fur Padiatrische Onkologie und Hamatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr. Res. 1999;46:553–561. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Lipton J.M., Atsidaftos E., Zyskind I., Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: An update from the Diamond Blackfan Anemia Registry. Pediatr. Blood Cancer. 2006;46:558–564. doi: 10.1002/pbc.20642. [DOI] [PubMed] [Google Scholar]
- 5.Glader B.E., Backer K., Diamond L.K. Elevated erythrocyte adenosine deaminase activity in congenital hypoplastic anemia. N. Engl. J. Med. 1983;309:1486–1490. doi: 10.1056/NEJM198312153092404. [DOI] [PubMed] [Google Scholar]
- 6.Willig T.N., Draptchinskaia N., Dianzani I., Ball S., Niemeyer C., Ramenghi U., Orfali K., Gustavsson P., Garelli E., Brusco A. Mutations in ribosomal protein S19 gene and diamond blackfan anemia: Wide variations in phenotypic expression. Blood. 1999;94:4294–4306. [PubMed] [Google Scholar]
- 7.Janov A.J., Leong T., Nathan D.G., Guinan E.C. Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine (Baltimore) 1996;75:77–78. doi: 10.1097/00005792-199603000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Vlachos A., Klein G.W., Lipton J.M. The Diamond Blackfan Anemia Registry: Tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. J. Pediatr. Hematol. Oncol. 2001;23:377–382. doi: 10.1097/00043426-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Campagnoli M.F., Garelli E., Quarello P., Carando A., Sv S.V., Nobili B., Dl D.L., Pecile V., Zecca M., Dufour C. Molecular basis of Diamond-Blackfan anemia: New findings from the Italian registry and a review of the literature. Haematologica. 2004;89:480–489. [PubMed] [Google Scholar]
- 10.Orfali K.A., Ohene-Abuakwa Y., Ball S.E. Diamond Blackfan anaemia in the UK: Clinical and genetic heterogeneity. Br. J. Haematol. 2004;125:243–252. doi: 10.1111/j.1365-2141.2004.04890.x. [DOI] [PubMed] [Google Scholar]
- 11.Draptchinskaia N., Gustavsson P., Andersson B., Pettersson M., Willig T.N., Dianzani I., Ball S., Tchernia G., Klar J., Matsson H. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 12.Ramenghi U., Campagnoli M.F., Garelli E., Carando A., Brusco A., Bagnara G.P., Strippoli P., Izzi G.C., Brandalise S., Riccardi R., Dianzani I. Diamond-Blackfan anemia: Report of seven further mutations in the RPS19 gene and evidence of mutation heterogeneity in the Italian population. Blood Cells Mol. Dis. 2000;26:417–422. doi: 10.1006/bcmd.2000.0324. [DOI] [PubMed] [Google Scholar]
- 13.Gazda H.T., Grabowska A., Merida-Long L.B., Latawiec E., Schneider H.E., Lipton J.M., Vlachos A., Atsidaftos E., Ball S.E., Orfali K.A. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 2006;79:1110–1118. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cmejla R., Cmejlova J., Handrkova H., Petrak J., Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum. Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 15.Farrar J.E., Nater M., Caywood E., McDevitt M.A., Kowalski J., Takemoto C.M., Talbot C.C., Jr., Meltzer P., Esposito D., Beggs A.H. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamaguchi I., Ooka A., Brun A., Richter J., Dahl N., Karlsson S. Gene transfer improves erythroid development in ribosomal protein S19-deficient Diamond-Blackfan anemia. Blood. 2002;100:2724–2731. doi: 10.1182/blood.V100.8.2724. [DOI] [PubMed] [Google Scholar]
- 17.Gazda H.T., Zhong R., Long L., Niewiadomska E., Lipton J.M., Ploszynska A., Zaucha J.M., Vlachos A., Atsidaftos E., Viskochil D.H. RNA and protein evidence for haplo-insufficiency in Diamond-Blackfan anaemia patients with RPS19 mutations. Br. J. Haematol. 2004;127:105–113. doi: 10.1111/j.1365-2141.2004.05152.x. [DOI] [PubMed] [Google Scholar]
- 18.Angelini M., Cannata S., Mercaldo V., Gibello L., Santoro C., Dianzani I., Loreni F. Missense mutations associated with Diamond-Blackfan anemia affect the assembly of ribosomal protein S19 into the ribosome. Hum. Mol. Genet. 2007;16:1720–1727. doi: 10.1093/hmg/ddm120. [DOI] [PubMed] [Google Scholar]
- 19.Gregory L.A., Aguissa-Toure A.H., Pinaud N., Legrand P., Gleizes P.E., Fribourg S. Molecular basis of Diamond-Blackfan anemia: Structure and function analysis of RPS19. Nucleic Acids Res. 2007;35:5913–5921. doi: 10.1093/nar/gkm626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuramitsu M., Hamaguchi I., Takuo M., Masumi A., Momose H., Takizawa K., Mochizuki M., Naito S., Yamaguchi K. Deficient RPS19 protein production induces cell cycle arrest in erythroid progenitor cells. Br. J. Haematol. 2008;140:348–359. doi: 10.1111/j.1365-2141.2007.06930.x. [DOI] [PubMed] [Google Scholar]
- 21.Leger-Silvestre I., Milkereit P., Ferreira-Cerca S., Saveanu C., Rousselle J.C., Choesmel V., Guinefoleau C., Gas N., Gleizes P.E. The ribosomal protein Rps15p is required for nuclear exit of the 40S subunit precursors in yeast. EMBO J. 2004;23:2336–2347. doi: 10.1038/sj.emboj.7600252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choesmel V., Bacqueville D., Rouquette J., Noaillac-Depeyre J., Fribourg S., Cretien A., Leblanc T., Tchernia G., Da Costa L., Gleizes P.E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109:1275–1283. doi: 10.1182/blood-2006-07-038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flygare J., Aspesi A., Bailey J.C., Miyake K., Caffrey J.M., Karlsson S., Ellis S.R. Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood. 2007;109:980–986. doi: 10.1182/blood-2006-07-038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Idol R.A., Robledo S., Du H.Y., Crimmins D.L., Wilson D.B., Ladenson J.H., Bessler M., Mason P.J. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol. Dis. 2007;39:35–43. doi: 10.1016/j.bcmd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Choesmel V., Fribourg S., Aguissa-Toure A.H., Pinaud N., Legrand P., Gazda H.T., Gleizes P.E. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum. Mol. Genet. 2008;17:1253–1263. doi: 10.1093/hmg/ddn015. [DOI] [PubMed] [Google Scholar]
- 26.Miyake K., Utsugisawa T., Flygare J., Kiefer T., Hamaguchi I., Richter J., Karlsson S. Ribosomal protein S19 deficiency leads to reduced proliferation and increased apoptosis but does not affect terminal erythroid differentiation in a cell line model of Diamond-Blackfan anemia. Stem Cells. 2008;26:323–329. doi: 10.1634/stemcells.2007-0569. [DOI] [PubMed] [Google Scholar]
- 27.Danilova N., Sakamoto K.M., Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008 doi: 10.1182/blood-2008-01-132290. published online May 30, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Bommer U.A., Stahl J., Henske A., Lutsch G., Bielka H. Identification of proteins of the 40 S ribosomal subunit involved in interaction with initiation factor eIF-2 in the quaternary initiation complex by means of monospecific antibodies. FEBS Lett. 1988;233:114–118. doi: 10.1016/0014-5793(88)81366-8. [DOI] [PubMed] [Google Scholar]
- 29.Heyn R., Kurczynski E., Schmickel R. The association of Blackfan-Diamond syndrome, physical abnormalities, and an abnormality of chromosome 1. J. Pediatr. 1974;85:531–533. doi: 10.1016/s0022-3476(74)80464-6. [DOI] [PubMed] [Google Scholar]
- 30.Jin A., Itahana K., O'Keefe K., Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aase J.M., Smith D.W. Congenital anemia and triphalangeal thumbs: A new syndrome. J. Pediatr. 1969;74:471–474. doi: 10.1016/s0022-3476(69)80208-8. [DOI] [PubMed] [Google Scholar]
- 32.Cmejla R., Blafkova J., Stopka T., Zavadil J., Pospisilova D., Mihal V., Petrtylova K., Jelinek J. Ribosomal protein S19 gene mutations in patients with diamond-blackfan anemia and identification of ribosomal protein S19 pseudogenes. Blood Cells Mol. Dis. 2000;26:124–132. doi: 10.1006/bcmd.2000.0286. [DOI] [PubMed] [Google Scholar]
- 33.Ellis S.R., Lipton J.M. Diamond Blackfan anemia: A disorder of red blood cell development. Curr. Top. Dev. Biol. 2008;82:217–241. doi: 10.1016/S0070-2153(07)00008-7. [DOI] [PubMed] [Google Scholar]
- 34.Gripp K.W., McDonald-McGinn D.M., La Rossa D., McGain D., Federman N., Vlachos A., Glader B.E., McKenzie S.E., Lipton J.M., Zackai E.H. Bilateral microtia and cleft palate in cousins with Diamond-Blackfan anemia. Am. J. Med. Genet. 2001;101:268–274. doi: 10.1002/ajmg.1329. [DOI] [PubMed] [Google Scholar]
- 35.Lidral A.C., Murray J.C. Genetic approaches to identify disease genes for birth defects with cleft lip/palate as a model. Birth Defects Res. Part A Clin. Mol. Teratol. 2004;70:893–901. doi: 10.1002/bdra.20096. [DOI] [PubMed] [Google Scholar]
- 36.Venema J., Tollervey D. Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 1999;33:261–311. doi: 10.1146/annurev.genet.33.1.261. [DOI] [PubMed] [Google Scholar]
- 37.Michael W.M., Dreyfuss G. Distinct domains in ribosomal protein L5 mediate 5 S rRNA binding and nucleolar localization. J. Biol. Chem. 1996;271:11571–11574. doi: 10.1074/jbc.271.19.11571. [DOI] [PubMed] [Google Scholar]
- 38.Rosorius O., Fries B., Stauber R.H., Hirschmann N., Bevec D., Hauber J. Human ribosomal protein L5 contains defined nuclear localization and export signals. J. Biol. Chem. 2000;275:12061–12068. doi: 10.1074/jbc.275.16.12061. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J., Harnpicharnchai P., Jakovljevic J., Tang L., Guo Y., Oeffinger M., Rout M.P., Hiley S.L., Hughes T., Woolford J.L., Jr. Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes Dev. 2007;21:2580–2592. doi: 10.1101/gad.1569307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leger-Silvestre I., Caffrey J.M., Dawaliby R., Alvarez-Arias D.A., Gas N., Bertolone S.J., Gleizes P.E., Ellis S.R. Specific Role for Yeast Homologs of the Diamond Blackfan Anemia-associated Rps19 Protein in Ribosome Synthesis. J. Biol. Chem. 2005;280:38177–38185. doi: 10.1074/jbc.M506916200. [DOI] [PubMed] [Google Scholar]
- 41.Robledo S., Idol R.A., Crimmins D.L., Ladenson J.H., Mason P.J., Bessler M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA. 2008;14:1918–1929. doi: 10.1261/rna.1132008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouquette J., Choesmel V., Gleizes P.E. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J. 2005;24:2862–2872. doi: 10.1038/sj.emboj.7600752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira-Cerca S., Poll G., Gleizes P.E., Tschochner H., Milkereit P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol. Cell. 2005;20:263–275. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Dai M.S., Arnold H., Sun X.X., Sears R., Lu H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007;26:3332–3345. doi: 10.1038/sj.emboj.7601776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lohrum M.A., Ludwig R.L., Kubbutat M.H., Hanlon M., Vousden K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y., Wolf G.W., Bhat K., Jin A., Allio T., Burkhart W.A., Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai M.S., Zeng S.X., Jin Y., Sun X.X., David L., Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun X.X., Dai M.S., Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 2007;282:8052–8059. doi: 10.1074/jbc.M610621200. [DOI] [PubMed] [Google Scholar]
- 49.Hadjiolova K.V., Nicoloso M., Mazan S., Hadjiolov A.A., Bachellerie J.P. Alternative pre-rRNA processing pathways in human cells and their alteration by cycloheximide inhibition of protein synthesis. Eur. J. Biochem. 1993;212:211–215. doi: 10.1111/j.1432-1033.1993.tb17652.x. [DOI] [PubMed] [Google Scholar]
- 50.Vanrobays E., Gelugne J.P., Gleizes P.E., Caizergues-Ferrer M. Late cytoplasmic maturation of the small ribosomal subunit requires RIO proteins in Saccharomyces cerevisiae. Mol. Cell. Biol. 2003;23:2083–2095. doi: 10.1128/MCB.23.6.2083-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]