

Abstract
Cell-to-cell transmission of retroviruses, such as human T lymphotropic virus type 1 (HTLV-1), is well documented, but the roles of viral regulatory or other nonstructural proteins in the modulation of T cell adhesion are incompletely understood. In this study we tested the role of the HTLV-1 accessory protein, p12I, on LFA-1-mediated cell adhesion. p12I is critical for early HTLV-1 infection by causing the release of calcium from the endoplasmic reticulum to activate NFAT-mediated transcription. We tested the role of this novel viral protein in mediating LFA-1-dependent cell adhesion. Our data indicated that T cells expressing a mutant HTLV-1 provirus that does not produce p12I mRNA (ACH.p12I) exhibited reduced LFA-1-mediated adhesion compared with wild-type HTLV-1-expressing cells (ACH). Furthermore, the expression of p12I in Jurkat T cells using lentiviral vectors enhanced LFA-1-mediated cell adhesion, which was inhibited by the calcium chelator BAPTA-AM, the calcium channel blocker SK&F 96365, and calpeptin, an inhibitor of the calcium-dependent protease calpain. Similar to the intracellular calcium mobilizer, thapsigargin, the expression of p12I in Jurkat T cells induced cell surface clustering of LFA-1 without changing the level of integrin expression. Our data are the first to indicate that HTLV-1 p12I, in addition to enhancing T cell activation, promotes cell-to-cell spread by inducing LFA-1 clustering on T cells via calcium-dependent signaling.
Human T lymphotropic virus type 1 (HTLV-1),4 the first identified human retrovirus, infects ~15–25 million people worldwide (1) and causes adult T cell leukemia/lymphoma and other lymphocyte-mediated diseases (2–5). The primary targets for HTLV-1 infection are CD4+ T cells, although CD8+ T cells, B cells, and macrophages also can be infected (4, 6). HTLV-1 is naturally transmitted in a highly cell-associated manner via beast milk, semen, and blood (2, 7, 8). Thus, cell-to-cell contact is required for efficient HTLV-1 transmission both in vivo (9) and in vitro (10, 11). Furthermore, HTLV-1 transmission occurs directly through cell-to-cell junctions (virological synapse) that mimic immunological synapses (12, 13). HTLV-1-infected cells have reorganized microtubule-organizing centers (MTOC) near points of cell contacts (14). At these sites, structural proteins, such as Gag, along with the HTLV-1 genome aggregate and transfer from infected to target lymphocytes. Interestingly, microtubule re-organization is triggered by Abs to ICAM-1 (CD54) or the IL-2R (CD25) (14, 15). Although HTLV-1 Tax contributes to microtubule reorganization (12–15), it is unclear whether other viral proteins, known to be important in the early events of infection, modulate cell-to-cell contact to accomplish viral transmission.
The HTLV-1 genome encodes structural and enzymatic (Gag, Pol, and Env), regulatory (Tax and Rex), and nonstructural or accessory (p12I, p27I, p13II, and p30II) proteins (16–20). HTLV-1 nonstructural proteins are important in viral replication and viral spread in vivo (18, 20–23). A highly conserved pX open reading frame I-encoded protein, p12I, is expressed during the natural viral infection (24, 25) and is critical for the establishment of HTLV-1 infection in nonactivated T cells in vitro and in animal models (26, 27). p12I localizes to the endoplasmic reticulum (ER) and Golgi apparatus and releases calcium from ER stores to trigger NFAT-mediated transcription, leading to IL-2 production and T cell activation (21, 28–30).
One of the key events of T cell activation is modulation of the expression of adhesion molecules such as LFA-1, which mediates lymphocyte adherence to the vascular wall and lymphocyte migration into tissues and contributes to the immunological synapse (31). LFA-1 is a heterodimeric transmembrane molecule composed of a unique α subunit (αL; CD11a) and a β2 subunit (CD18) (32, 33). Activation of LFA-1 is tightly controlled by inside-out signaling, which is induced by ligation of the TCR or certain chemokine receptors (32–35). LFA-1-mediated T cell adhesion results from either increasing the affinity for its ligand, ICAM-1, or clustering of LFA-1 (avidity) on the cell membrane (32–35). Although recent studies suggested that talin, Rap1, and RapL proteins are required for regulation of LFA-1 expression (31, 36), the detailed mechanism of signaling leading to increased affinity is unclear (34). Under experimental conditions, divalent cations, such as Mn2+ or Mg2+, and activating Abs can induce affinity activation of LFA-1 (34). The signaling pathways that result in increased integrin lateral mobility and clustering include proteins such as Vav-1 (guanine exchange factor), adhesion and degranualtion-promoting adaptor protein, Src kinase-associated protein of 55kDa (adaptor protein), and Rap1 (small GTPase) (34, 37–40). Protein kinase C activation by phorbol esters can also trigger integrin clustering by interacting with the β2 subunit (34, 40). Furthermore, integrin clustering can be induced by elevated cytoplasmic calcium via TCR activation or treatment with calcium mobilizers, such as ionomycin and thapsigargin (TG) (41). Calcium-mediated LFA-1 activation also requires the calcium-dependent protease calpain (41). Interestingly, the HTLV-1 accessory protein p12I mimics the function of TG and induces calcium release from ER stores to activate T cells (29).
In this study we tested whether the expression of HTLV-1 p12I could activate LFA-1-mediated T cell adhesion in a calcium-dependent manner. LFA-1-mediated adhesion of wild-type HTLV-1 (ACH) T cells was greater than that of T cells expressing a proviral clone lacking p12I (ACH.p12). The expression of p12I in Jurkat T cells also enhanced LFA-1-mediated cell adhesion, which was inhibited by the calcium chelator BAPTA-AM, the calcium channel blocker SK&F 96365, and calpeptin, an inhibitor of the calcium-dependent protease calpain. Similar to the effects of TG, the expression of p12I in Jurkat T cells induced cell surface clustering of LFA-1 without changing LFA-1 expression. Our data indicate that p12I induces LFA-1 clustering on T cells via calcium-dependent signaling, which would promote spread of HTLV-1 to target T cells.
Materials and Methods
Cell lines
ACH.2 and ACH.p12.4 cell lines were generated from the outgrowth of immortalized PBMC previously transfected with molecular clones of HTLV-1 (ACH and ACH.p12I) (29, 42). The ACH.p12I plasmid has a mutation resulting in complete ablation of p12I mRNA expression without affecting the expression of other viral genes (26). ACH.2p and ACH.p12.4p cells were infected cell lines created by coculturing naive human PBMC with lethally gamma-irradiated (10,000 rad) ACH.2 and ACH.p12.4 cells, respectively. Target PBMC were preactivated for 4 days with human IL-2 (10 U/ml) and PHA (2 μg/ml). ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cells were maintained in RPMI 1640 supplemented with 15% FBS, L-glutamine (0.3 mg/ml), penicillin (100 U/ml), streptomycin (100 μg/ml), and rIL-2 (10 U/ml). Viral p19 Ag production from the ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cell lines was measured in cell culture supernatants by ELISA in quadruplicate samples (Zeptomatrix) according to the manufacturer’s protocol. Jurkat T cells (clone E6-1; American Type Culture Collection; catalog no. TIB-152) were maintained in RPMI 1640 (Invitrogen Life Technologies) supplemented with 10% FBS, 100 μg of streptomycin-penicillin per ml, and 2 mM L-glutamine. For lentivirus production, the 293T cell line (American Type Culture Collection), which stably expresses the SV40 T Ag, was maintained in DMEM (Invitrogen Life Technologies) supplemented with 10% FBS, 100 μg/ml streptomycin-penicillin, and 2 mM L-glutamine.
Recombinant lentivirus production and infection of Jurkat T cells
Recombinant lentiviruses were prepared as previously described (21). A lentivisus vector containing a posttranscriptional regulatory element of woodchuck hepatitis virus, pWPT-IRES-GFP (empty plasmid), was generated by cloning an internal ribosome entry site (IRES) sequence (pHR′CMV/Tax1/enhanced GFP (eGFP); G. Feuer, State University of New York, Syracuse, NY) into pWPT-GFP (D. Trono, University of Geneva, Geneva, Switzerland). pWPT-p12I-hemagglutinin (p12IHA)-IRES-GFP was generated by cloning p12IHA from pME-p12I (G. Franchini, National Cancer Institute, National Institutes of Health, Bethesda, MD) into pWPT-IRES-GFP. To generate recombinant virus, 293T cells (5 × 106) were transfected with 2 μg of pHCMV-G, 10 μg of pCMVΔR8.2, and 10 μg of pWPT-p12IHA-IRES-GFP or pWPT-IRES-GFP using calcium phosphate. Supernatants were collected 24, 48, and 72 h after transfection and filtered through a 0.2-μm pore size filter. To obtain viral pellets, supernatants were centrifuged at 6500 × g for 16 h at 4°C, then pellets were suspended in DMEM overnight at 4°C. The concentrated virus was aliquoted and stored at −80°C. After determination of viral titer, Jurkat T cells were infected with recombinant virus at a multiplicity of infection of 3 in the presence of 8 μg/ml polybrene (Sigma-Aldrich) and spin-infected at 2700 rpm for 1 h at 25°C. The expression of eGFP and p12I was confirmed by flow cytometry, Western blot, or RT-PCR (21). For detection of p12I mRNA, cellular RNA was isolated using RNAqueous (Ambion). RNA was converted to cDNA using the RT system (Promega) and was amplified with AmpliTaq DNA polymerase (PerkinElmer) using CCTCTTTCTCCCGCTCTTTT (forward) and GGCCAAGCTAGC GTAATCTG (reverse) primers. Transduced Jurkat T cells (control and p12I-expressing cells) were used for experiments 10 – 60 days after infection, when expression of eGFP was >85% as determined by flow cytometric analysis.
Cell adhesion to immobilized ICAM-1
Cell adhesion assays were performed as described previously (41, 43). Briefly, recombinant human ICAM-1/Fc chimera (R&D Systems) in PBS (0.5 μg/well) was used for coating 96-well tissue culture plates overnight at 4°C. Nonspecific binding was blocked with BSA in PBS for 1 h at room temperature, followed by three washes in PBS and one wash with RPMI 1640 or HEPES buffer (20 mM HEPES, 140 mM NaCl, and 2 mg/ml glucose (pH 7.4)) for MgCl2/EGTA stimulation. Cells (2 × 105 in 50 μl of RPMI 1640 or HEPES buffer) were added to each well in the presence or the absence of 50 μl of integrin stimulatory or inhibitory reagents (2× final concentrations) as indicated in Results and figure legends. Ionomycin (Sigma-Aldrich), TG (Calbiochem), PMA (Sigma-Aldrich), and MgCl2-EGTA were used as stimuli to test LFA-1-mediated adhesion. The intracellular calcium chelator BAPTA-AM (Invitrogen Life Technologies), the calcium release-activated calcium channel blocker SK&F 96365 (Calbiochem), and the calpain inhibitor calpeptin (Calbiochem) were used for LFA-1 inhibition. The plates were incubated on ice for 20 min, centrifuged for 1 min at 30 × g, and incubated for 30 min at 37°C. The plates were washed carefully with prewarmed RPMI 1640 or HEPES buffer three times. The percentage of attached cells was calculated by viable cell staining with tetrazolium dye (CellTiter 96 Cell Proliferation Assay; Promega). Statistical differences between groups were determined using the Wilcoxon Mann-Whitney test, because we assumed that differences between the two groups were not normally distributed. A value of p < 0.05 was considered significant.
Flow cytometry and soluble ICAM-1Fc binding assay
Flow cytometric analysis of the expression of cell surface markers CD11a and CD18 was performed as previously described (42). R-PE-conjugated anti-CD11a and FITC-conjugated anti-CD18 Abs were used according to the manufacturer’s recommendations (Southern Biotechnology Associates). For soluble ICAM-1 Fc (sICAM-1) binding assay, cells were washed with RPMI 1640 or HEPES buffer and resuspended in 50 μl of stimulator (2× final concentrations) containing medium or HEPES buffer. Cells were mixed with 50 μl of recombinant human ICAM-1 Fc chimera (R&D Systems) at a final concentration 5 μg/ml and incubated for 30 min at 37°C. Subsequently, the cells were washed twice in ice-cold 0.2% BSA and 0.2% sodium azide in PBS and incubated with R-PE-conjugated goat anti-human IgG Fc-specific Ab (Jackson ImmunoResearch Laboratories) for 20 min on ice. Cells were washed twice in ice-cold 0.2% BSA and 0.2% sodium azide in PBS and fixed in 1% paraformaldehyde. The fluorescence of cells was measured by FACScan (BD Biosciences) analysis.
Confocal microscopy to detect LFA-1 distribution
For detection of LFA-1 distribution on the surface of T cells by confocal microscopy, Jurkat T cells were attached to coverslips coated with a 0.01% solution of poly-L-lysine (Sigma-Aldrich). Cells were incubated for 30 min at 37°C with or without stimulation, then fixed with 0.5% paraformaldehyde for 10 min. After washing once with PBS and three times with RPMI 1640, cells were stained with R-PE-conjugated CD11a Ab (Southern Biotechnology Associates) for 30 min at 37°C, washed once with RPMI 1640, mounted, and analyzed using the Leica TCS SP2 AOBS confocal system. Leica confocal software (Leica Lite) was used to quantify the fluorescence signal on cell membranes representing LFA-1 clustering as described previously (41). Student’s t test was used for statistical analysis of differences between two groups.
Results
LFA-1-mediated adhesion in HTLV-1-immortalized T cells lacking p12I expression
To examine the role of p12I in LFA-1-mediated cell adhesion, we first conducted adhesion assays using wild-type HTLV-1 (ACH cell line; ACH.2 and ACH.2p) and T cells lacking p12I expression (ACH. p12I; ACH.p12.4 and ACH.p12.4p). We generated newly infected ACH.2p and ACH.p12.4p cells by coculturing preactivated naive human PBMC with lethally irradiated ACH.2 and ACH.p12.4, respectively, to rule out the influence of specific clonal effects during in vitro culture. Virus replication, measured by viral p19 Ag ELISA, was similar among ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cells (Fig. 1A). These results are consistent with previous studies that indicated that p12I is not required for HTLV-1 replication in activated T cells or immortalized T cell lines (26, 44).
FIGURE 1.

LFA-1-mediated adhesion is reduced in ACH.p12I cell lines. A, p19 Ag production from ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cell lines was measured by ELISA. Supernatants were collected 24 days after infection. Values are the mean ± SEM of triplicate samples and represent two independent experiments. B, Cells were treated with TG (5 μM), PMA (50 ng/ml), or Mg2+/EGTA (5 mM/1 mM) and incubated on ICAM-1 Fc-coated, 96-well plates for 30 min at 37°C. The proportion of adhesion was measured as a percentage of the total cells added per well. Values are the mean ± SEM of triplicate samples and represent four independent experiments. Significantly reduced adhesion was observed in TG-, PMA-, and Mg2+/EGTA-treated ACH.p12.4 and ACH.p12.4p cell lines (by Wilcoxon Mann-Whitney test, p < 0.05).
LFA-1-mediated T cell binding can result either from increased affinity for ICAM-1 or clustering of LFA-1 on the cell surface (32–35). LFA-1 clustering can be activated by TCR signaling, phorbol ester stimulation, or increased intracellular calcium levels (32–35). TG- and PMA-induced LFA-1-mediated adhesion of the wild-type ACH cell lines, ACH.2 and ACH.2p, was 2-fold greater compared with that of unstimulated cells (Fig. 1B). T cells lacking p12I expression (ACH.p12.4 and ACH.p12.4p) did not respond to the LFA-1 clustering reagents, TG or PMA (Fig. 1B). Mg2+/EGTA treatment, which resulted in an increase in LFA-1 affinity in vitro, increased ACH.2 and ACH.2p cell adhesion up to 50 – 60%, but did not induce significant adhesion in ACH.p12.4 and ACH.p12.4p cells (Fig. 1B). The failure of ACH.p12.4 and ACH.p12.4p cells to respond to reagents that cause LFA-1 clustering and enhance T cell adhesion could be explained by these cells having either lowered LFA-1 expression or decreased binding affinity.
ACH.p12I cells express LFA-1, but have decreased binding of sICAM-1
To examine whether lowered LFA-1-mediated adhesion in ACH.p12 cells was due to changes in LFA-1 expression, we performed flow cytometric analysis using anti-CD11a and anti-CD18 Abs. There was no significant difference in integrin expression on the cell surface among ACH.2, ACH.2p, ACH.p12.4, and ACH.p12.4p (Fig. 2A).
FIGURE 2.
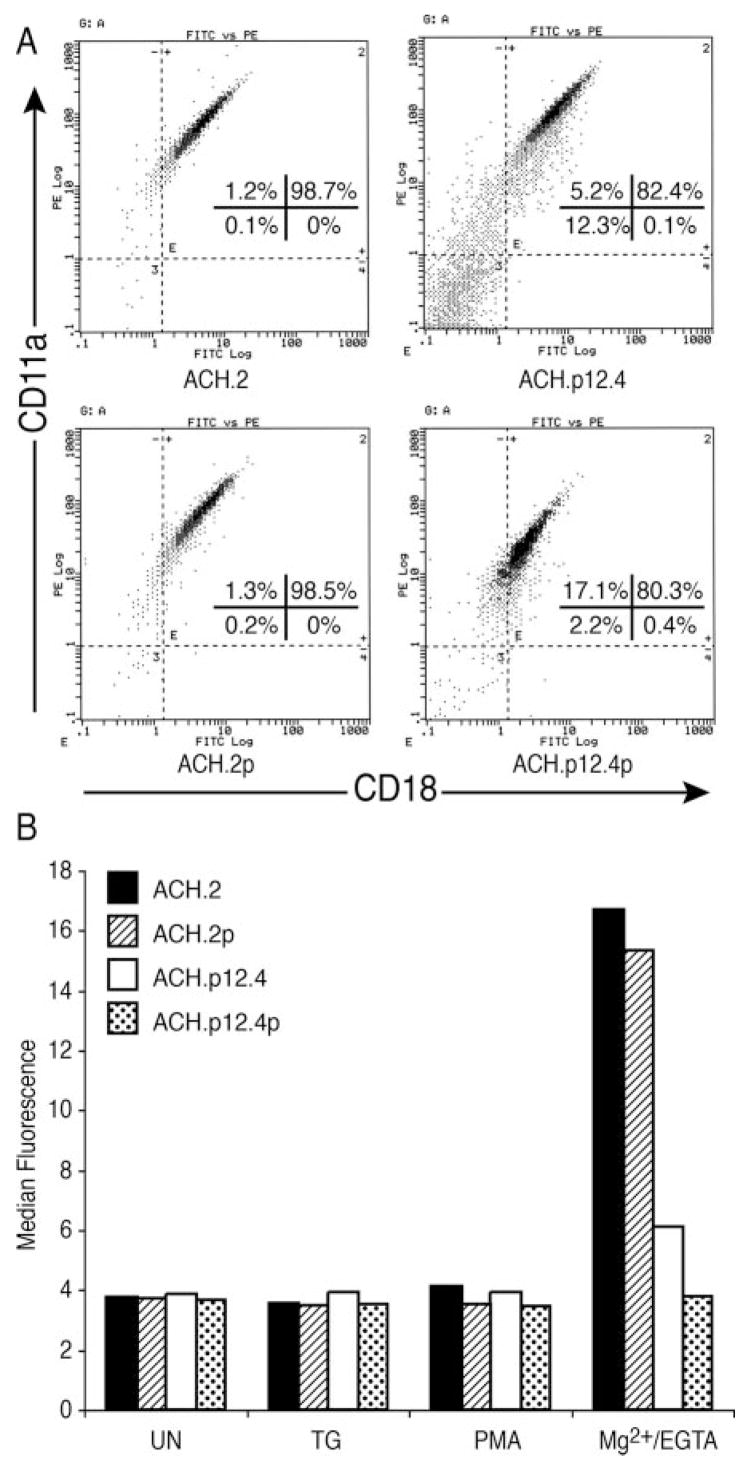
ACH.p12I cells have decreased binding of sICAM-1 without alteration of LFA-1 expression. A, Flow cytometry specific for LFA-1 molecules CD11a and CD18 was performed on ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cell lines (106 cells/sample). B, Soluble ICAM-1Fc binding assay was performed on ACH.2, ACH.p12.4, ACH.2p, and ACH.p12.4p cell lines. Cells were treated with TG (5 μM), PMA (50 ng/ml), or Mg2+/EGTA(5 mM/1 mM) and incubated with sICAM-1Fc for 30 min at 37°C. Bound sICAM-1 was detected with R-PE-conjugated goat anti-human IgG Fc-specific Ab and analyzed by flow cytometry. Data are expressed as the mean fluorescence intensity. This figure is representative of two experiments.
T cell binding to sICAM-1 is increased only when LFA-1 affinity is enhanced by agonists such as Mg2+ stimulation. In contrast, LFA-1 clustering on T cells is stimulated by reagents that stimulate calcium release from cell stores, such as TG, or reagents that induce TCR signaling, such as PMA (41, 43). We therefore tested ACH wild-type and ACH.12 mutant T cells for their sI-CAM-1-binding properties using flow cytometry to measure LFA-1 affinity. As expected, none of the clustering stimulants (TG and PMA) exhibited any induction of sICAM-1 binding in any of the cell lines (Fig. 2B). Mg2+-stimulated wild-type ACH, ACH.2 and ACH.2p, cells had enhanced sICAM-1 binding compared with the ACH.p12I cell lines, ACH.p12.4 and ACH.p12.4p (Fig. 2B). These data were consistent with our Mg2+/EGTA experiments using immobilized ICAM-1 (Fig. 1B).
p12I expression in Jurkat T cells did not affect LFA-1 affinity
To test whether HTLV-1 p12I expression specifically influenced LFA-1-mediated T cell adhesion, we stably expressed p12I in Jurkat T cells using recombinant lentiviruses (21). All empty control and p12I-expressing Jurkat T cells were >85% GFP positive (Fig. 3A). The expression of HTLV-1 p12I also was confirmed by p12I mRNA amplification by RT-PCR (Fig. 3B) and immunoprecipitation using polyclonal HA-1 Ab (data not shown).
FIGURE 3.

p12I stable expression in Jurkat T cells did not alter LFA-1 affinity. A, Flow cytometric analysis for both GFP expression and sI-CAM-1 binding was performed in empty control and p12I-expressing Jurkat T cells. Both samples were >85% GFP positive, indicating high transduction efficiency. Soluble ICAM-1Fc binding was measured concurrently. Cells were treated with TG (5 μM), PMA (50 ng/ml), or Mg2+/EGTA (5 mM/1 mM) and incubated with sI-CAM-1 Fc for 30 min at 37°C. Bound sICAM-1 was detected with R-PE-conjugated, goat anti-human IgG Fc-specific Ab and analyzed by flow cytometry. B, RT-PCR was performed using total cellular RNA isolated from mock, empty control, and p12I-expressing Jurkat T cells 30 and 60 days after infection with lentiviral vectors.
Soluble ICAM binding was tested using both empty control and p12I Jurkat T cells. As expected, TG and PMA, which activate LFA-1 clustering, did not induce sICAM-1 binding (Fig. 3A). Mg2+/EGTA treatment, which stimulates LFA-1 affinity, was used to compare sICAM-1 binding in empty control and p12I Jurkat T cells. Our data indicated no significant difference in LFA-1 affinity between the two cell types (Fig. 3A). These data demonstrated that p12I expression does not affect LFA-1 affinity.
Expression of p12I in Jurkat T cells induced LFA-1 clustering
Calcium mobilizers, such as ionomycin and TG, enhance LFA-1-mediated T cell adhesion by affecting LFA-1 clustering (34, 41). Because HTLV-1 p12I functionally mimics TG by increasing intracellular calcium release from ER stores, we tested whether p12I expression might affect LFA-1 clustering. Jurkat T cells that stably expressed p12I exhibited enhanced cell adhesion to ICAM-1 molecules compared with empty vector-transduced control Jurkat T cells in all untreated and TG, ionomycin, PMA and Mg2+/EGTA treatment groups (Fig. 4A). Because p12I did not affect LFA-1 affinity (Fig. 3), the enhancement observed in Mg2+/EGTA-treated p12I Jurkat T cells was probably due to preclustered LFA-1 in p12I-expressing cells.
FIGURE 4.

Expression of p12I in Jurkat T cells induced LFA-1-mediated cell adhesion. Adhesion to immobilized ICAM-1 was performed using empty control and p12I-expressing Jurkat T cells. A, Cells were treated with TG (5 μM), ionomycin (0.5 μM), PMA (50 ng/ml), or Mg2+/EGTA (5 mM/1 mM) and incubated with ICAM-1 Fc for 30 min at 37°C. Adhesion was measured as a percentage of the total cells added per well. Values are the mean ± SEM of triplicate samples and represent three independent experiments. A value of p < 0.05 (by Wilcoxon Mann-Whitney U test) was considered significant. B–E, Cells were treated with increasing concentrations of TG (B), ionomycin (C), PMA (D), and Mg2+ (E; with constant 1 mM EGTA) and incubated for the adhesion assay. The fold increase was calculated by dividing the percentages of adhered cells in the presence of each treatment with that of untreated cell adhesion. This figure is representative of two experiments.
To clarify whether p12I expression affected clustering of or conformational changes in LFA-1, we measured the fold increase in cell adhesion over that in untreated cells in the empty control and p12I Jurkat T cells that were treated with increasing concentrations of TG, ionomycin, PMA, or Mg2+(Fig. 4, B–E). As expected, the calcium mobilizers, TG and ionomycin, induced increased cell adhesion in a dose-dependent manner at lower concentrations and led to decreased cell adhesion at higher concentrations in both cell types (Fig. 4, B and C). Peak adhesion by TG and ionomycin was observed between 2.5 and 5 μM and 0.5 and 1 μM, respectively, similar to a previous report (41). Another LFA-1 clustering agent, PMA, also induced a dose-dependent increase in adhesion to ICAM-1 at lower concentrations, but adhesion was saturated at higher concentrations in both cell types (Fig. 4D). At the lower concentrations, p12I Jurkat T cell adhesion was higher than that in empty control Jurkat T cells after TG, ionomycin, or PMA stimulation (Fig. 4, B–D), suggesting that p12I expression on Jurkat T cells reduced the threshold required for activation of LFA-1 clustering.
As expected, Mg2+ also induced a dose-dependent increase and saturation of cell adhesion in both cell types; however, the fold increase over untreated cells was not significantly different between empty control and p12I Jurkat T cells (Fig. 4E). The affinity of both cell types was increased to a similar level by Mg2+, suggesting that the expression of p12I did not affect LFA-1 affinity. These data indicated that enhanced LFA-1-mediated cell adhesion in p12I Jurkat T cells was due to enhanced LFA-1 clustering rather than increased affinity.
HTLV-1 p12I-mediated LFA-1 activation is calcium dependent
LFA-1 clustering requires signaling pathways leading to the elevation of calcium and activation of calpain, a calcium-dependent protease (41). To determine whether LFA-1-activation induced by p12I was dependent on calcium-mediated signaling pathways, we performed cell adhesion assays using inhibitors such as the intracellular calcium chelator BAPTA-AM, the calcium channel blocker SK&F 96365, and the calpain inhibitor calpeptin (Fig. 5). As expected, all inhibitors abolished TG- and PMA-induced cell adhesion in both cell types (Fig. 5). These results suggested that LFA-1 clustering, due to p12I, was dependent on both intracellular and extracellular calcium as well as calpain. In contrast to TG and PMA stimulation, cell adhesion induced by Mg2+/EGTA was not blocked completely by the inhibitors (Fig. 5). These data were consistent with a previous report that increases in Mg2+-induced affinity were not altered by calcium channel blockers or calpain inhibitors (41).
FIGURE 5.

HTLV-1 p12I-mediated LFA-1 activation is inhibited by calcium signal inhibitors. Empty control and p12I-expressing Jurkat T cells were pretreated with BAPTA-AM (50 μM), SK&F 96365 (100 μM), or calpeptin (100 μg/ml) for 30 min at 37°C, and an adhesion assay was performed in the presence or the absence of TG (5 μM), PMA (50 ng/ml), or Mg2+/EGTA (5 mM/1 mM) stimulation.
HTLV-1 p12I modulated surface distribution of LFA-1
To confirm that the membrane distribution of LFA-1 was altered in p12I-expressing Jurkat T cells, we measured LFA-1 expression by confocal microscopy using R-PE-conjugated CD11a. Untransduced mock Jurkat T cells were either untreated or treated with 1 μM TG as a control experiment. A higher intensity of LFA-1 fluorescence was frequently observed in TG-treated cells (Fig. 6A). Similar to TG treatment, p12I expression in Jurkat T cells had more than 2-fold enhanced fluorescence, indicating increased LFA-1 clustering on the cell membranes (Fig. 6B). Increased LFA-1 fluorescence in p12I-expressing Jurkat T cells was not due to increased cell surface expression of LFA-1. Cell surface expression of CD11a was measured by flow cytometry and was not significantly different between empty control and p12I-expressing Jurkat T cells (Fig. 6C). These data indicated that p12I expression altered LFA-1 cell surface distribution, favoring clustering of the adhesion molecule without changing total LFA-1 cell surface expression, and were consistent with our cell adhesion and sICAM-1 binding assays.
FIGURE 6.

Expression of p12I in Jurkat T cells modulated surface distribution of LFA-1 on the cell membrane. Untransduced mock Jurkat T cells (A) or transduced empty control and p12I expressing Jurkat T cells (B) were stained with R-PE-conjugated LFA-1 mAb and analyzed by confocal microscopy. Fluorescent signal exceeding 50 U is shown as a bright blue color. The signal of LFA-1 was quantified on the membrane of 30 cells. Mean fluorescence units (±SEM) were compared in the bar graphs on the right between TG-untreated and -treated groups (A) or between empty control and p12I-expressing Jurkat T cells (B). Significantly (by t test, p < 0.01) increased fluorescence in TG-treated and p12I-expressing Jurkat T cells was observed. C, Flow cytometric analysis of LFA-1 cell surface expression in GFP-expressing empty control and in empty control and p12I-expressing Jurkat T cells was performed.
Discussion
In this study we found that HTLV-1 p12I expression in T cells altered LFA-1-mediated T cell adhesion to ICAM-1. Abrogation of p12I expression in HTLV-1 infected cells (ACH.p12I) resulted in decreased LFA-1-mediated cell adhesion due to a reduced affinity of LFA-1 compared wild-type HTLV-1-infected cells. To identify the specific role of p12I in LFA-1-mediated cell adhesion, we stably expressed p12I in Jurkat T cells using lentiviral vectors. The expression of p12I increased LFA-1-mediated cell adhesion to ICAM-1 by inducing clustering of LFA-1 on Jurkat T cells. Our data are consistent with previous reports from our group and others (26–28, 45) that demonstrated the important role of HTLV-1 p12I in T cell activation and cell-to-cell transmission during the early stages of HTLV-1 infection.
The important roles of LFA-1 and ICAM-1 in HTLV-1 cell-to-cell transmission were indicated in a recent study by Barnard et al. (14). By cross-linking with Abs to cell surface molecules, they identified that LFA-1 and ICAM-1 were involved in triggering MTOC polarization, which is characteristic of HTLV-1-induced virological synapses. Our data suggested that p12I may synergize to modify cell adhesion and contribute to MTOC polarization by increasing cell adhesion through clustered LFA-1. Interestingly, the transmission of other viruses is also enhanced by the LFA-1/ICAM-1 interaction. For example, measles virus infection induced increased LFA-1 expression on monocytes, resulting in enhanced viral transmission to endothelial cells (14, 46). Furthermore, LFA-1 activation of T cells increased their susceptibility to HIV-1 infection (47, 48).
The results of our study support our previous findings, which demonstrated that T cells expressing ACH. p12I have reduced viral infectivity when cocultured with quiescent T cells (27). ACH.p12I cells do not exhibit efficient cell-to-cell contact and therefore are disadvantaged for cell-to-cell transmission. This tenet would explain in part data indicating that HTLV-1 p12I is required for in vivo viral infectivity in rabbits (26). The early block of infection observed in this animal study could be explained by inadequate LFA-1 activation on HTLV-1-infected cells, which would inhibit the ability of HTLV-1 to spread to nonactivated target cells. Our findings are also consistent with reports that HTLV-1 infection is established by early viral spread to T cells, but subsequently maintained by clonal expansion of T cells (49–51). If the virus lacks the ability to rapidly spread from infected cells to its targets early in the infection, it is likely that HTLV-1 infection is eliminated by a robust immune response (52).
The observed affinity reduction in ACH.p12I cells might have resulted from alteration of inside-out signaling. Although signaling pathways that regulate LFA-1 affinity are incompletely defined compared with that of LFA-1 clustering modulation (34), transient affinity increases in LFA-1 and α1β4 are induced by chemokine stimulation (53, 54). In addition, the lymphocyte-specific protein tyrosine kinase, Lck, has been identified as a critical enzyme to maintain the high affinity status of integrin α1β4 (55). Interestingly, the expression of the lck gene was down-regulated by the HTLV-1 accessory protein p30II (56). Thus, a balance of HTLV-1 regulatory and accessory protein expression is probably required to ensure cell-to-cell transmission of the virus.
HTLV-1 p12I expression in Jurkat T cells induced LFA-1-mediated cell adhesion in a calcium-dependent manner. p12I mimics TG, which is an LFA-1 clustering agent, by releasing calcium from ER stores and increasing intracellular calcium concentration without activation of the TCR signaling pathway (29). We cannot rule out the possibility that p12I may activate LFA-1-mediated adhesion via chemokine secretion, which can increase affinity and induce clustering. We have reported that p12I enhanced IL-2 production through NFAT activation (28, 30, 45). IL-2 produced by HTLV-1-infected cells may activate LFA-1 on both infected and target cells in an autocrine or paracrine manner in the local tissue microenvironment in vivo (30, 45). The data from our study together with the findings of these previous studies suggest that p12I expression in HTLV-1-infected T lymphocytes lowers the threshold of calcium signaling events to promote T cell activation and cell-to-cell transmission.
In summary, our data indicate that HTLV-1 p12I expression induced LFA-1-mediated T cell adhesion to ICAM-1 by activating LFA-1 clustering in a calcium-dependent manner. Our data demonstrate that p12I enhances HTLV-1 cell-to-cell transmission during the early stages of infection by activating LFA-1-mediated adhesion and supporting formation of the HTLV-1 virological synapse.
Acknowledgments
We thank H. Hiraragi and M. Burkhard for their critical review of the manuscript, and G. Franchini for sharing valuable reagents.
Footnotes
This work was supported by National Institutes of Health Grants CA100730 and RR14324 (to M.L.) and CA70529 from the National Cancer Institute (awarded through the Ohio State University Comprehensive Cancer Center.
Abbreviations used in this paper: HTLV-1, T lymphotropic virus type 1; eGFP, enhanced GFP; ER, endoplasmic reticulum; HA, hemagglutinin; IRES, internal ribosome entry site; MTOC, microtubule-organizing center; sICAM-1, soluble ICAM-1; TG, thapsigargin.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005;24:6058–6068. doi: 10.1038/sj.onc.1208968. [DOI] [PubMed] [Google Scholar]
- 2.Mahieux R, Gessain A. HTLV-1 and associated adult T-cell leukemia/lymphoma. Rev Clin Exp Hematol. 2003;7:336–361. [PubMed] [Google Scholar]
- 3.Franchini G, Fukumoto R, Fullen JR. T-cell control by human T-cell leukemia/lymphoma virus type 1. Int J Hematol. 2003;78:280–296. doi: 10.1007/BF02983552. [DOI] [PubMed] [Google Scholar]
- 4.Kehn K, Berro R, de La FC, Strouss K, Ghedin E, Dadgar S, Bottazzi ME, Pumfery A, Kashanchi F. Mechanisms of HTLV-1 transformation. Front Biosci. 2004;9:2347–2372. doi: 10.2741/1401. [DOI] [PubMed] [Google Scholar]
- 5.Hall WW, Fujii M. Deregulation of cell-signaling pathways in HTLV-1 infection. Oncogene. 2005;24:5965–5975. doi: 10.1038/sj.onc.1208975. [DOI] [PubMed] [Google Scholar]
- 6.Manns A, Hisada M, Lagrenade L. Human T-lymphotropic virus type I infection. Lancet. 1999;353:1951–1958. doi: 10.1016/s0140-6736(98)09460-4. [DOI] [PubMed] [Google Scholar]
- 7.Weiss RA. Milk-borne transmission of HTLV-1. Jpn J Cancer Res. 1993;84 inside front cover. [PubMed] [Google Scholar]
- 8.Bangham CR. The immune control and cell-to-cell spread of human T-lymphotropic virus type 1. J Gen Virol. 2003;84:3177–3189. doi: 10.1099/vir.0.19334-0. [DOI] [PubMed] [Google Scholar]
- 9.Okochi K, Sato H, Hinuma Y. A retrospective study on transmission of adult T cell leukemia virus by blood transfusion: seroconversion in recipients. Vox Sang. 1984;46:245–253. doi: 10.1111/j.1423-0410.1984.tb00083.x. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto N, Okada M, Koyanagi Y, Kannagi M, Hinuma Y. Transformation of human leukocytes by cocultivation with an adult T cell leukemia virus producer cell line. Science. 1982;217:737–739. doi: 10.1126/science.6980467. [DOI] [PubMed] [Google Scholar]
- 11.Popovic M, Sarin PS, Robert-Gurroff M, Kalyanaraman VS, Mann D, Minowada J, Gallo RC. Isolation and transmission of human retro-virus (human T-cell leukemia virus) Science. 1983;219:856–859. doi: 10.1126/science.6600519. [DOI] [PubMed] [Google Scholar]
- 12.Dustin M. Viral spread through protoplasmic kiss. Nat Cell Biol. 2003;5:271–272. doi: 10.1038/ncb0403-271. [DOI] [PubMed] [Google Scholar]
- 13.Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 2003;299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 14.Barnard AL, Igakura T, Tanaka Y, Taylor GP, Bangham CR. Engagement of specific T cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood. 2005;106:988–995. doi: 10.1182/blood-2004-07-2850. [DOI] [PubMed] [Google Scholar]
- 15.Nejmeddine M, Barnard AL, Tanaka Y, Taylor GP, Bangham CR. Human T-lymphotropic virus, type 1, Tax protein triggers microtubule reorientation in the virological synapse. J Biol Chem. 2005;280:29653–29660. doi: 10.1074/jbc.M502639200. [DOI] [PubMed] [Google Scholar]
- 16.Pise-Masison CA, Jeong SJ, Brady JN. Human T cell leukemia virus type 1: the role of Tax in leukemogenesis. Arch Immunol Ther Exp. 2005;53:283–296. [PubMed] [Google Scholar]
- 17.Manel N, Battini JL, Taylor N, Sitbon M. HTLV-1 tropism and envelope receptor. Oncogene. 2005;24:6016–6025. doi: 10.1038/sj.onc.1208972. [DOI] [PubMed] [Google Scholar]
- 18.Bindhu M, Nair A, Lairmore MD. Role of accessory proteins of HTLV-1 in viral replication, T cell activation, and cellular gene expression. Front Biosci. 2005;10:17–37. doi: 10.2741/1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franchini G, Nicot C, Johnson JM. Seizing of T cells by human T-cell leukemia/lymphoma virus type 1. Adv Cancer Res. 2003;89:69–132. doi: 10.1016/s0065-230x(03)01003-0. [DOI] [PubMed] [Google Scholar]
- 20.Nicot C, Harrod RL, Ciminale V, Franchini G. Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene. 2005;24:6026–6034. doi: 10.1038/sj.onc.1208977. [DOI] [PubMed] [Google Scholar]
- 21.Nair A, Michael B, Hiraragi H, Fernandez S, Feuer G, Boris-Lawrie K, Lairmore M. Human T lymphotropic virus type 1 accessory protein p12I modulates calcium-mediated cellular gene expression and enhances p300 expression in T lymphocytes. AIDS Res Hum Retroviruses. 2005;21:273–284. doi: 10.1089/aid.2005.21.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hiraragi H, Michael B, Nair A, Silic-Benussi M, Ciminale V, Lairmore M. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis. J Virol. 2005;79:9449–9457. doi: 10.1128/JVI.79.15.9449-9457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Agostino DM, Silic-Benussi M, Hiraragi H, Lairmore MD, Ciminale V. The human T-cell leukemia virus type 1 p13(II) protein: effects on mitochondrial function and cell growth. Cell Death Differ. 2005;12(Suppl 1):905–915. doi: 10.1038/sj.cdd.4401576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dekaban GA, Peters AA, Mulloy JC, Johnson JM, Trovato R, Rivadeneira E, Franchini G. The HTLV-I orfI protein is recognized by serum antibodies from naturally infected humans and experimentally infected rabbits. Virology. 2000;274:86–93. doi: 10.1006/viro.2000.0406. [DOI] [PubMed] [Google Scholar]
- 25.Pique C, Uretavidal A, Gessain A, Chancerel B, Gout O, Tamouza R, Agis F, Dokhelar MC. Evidence for the chronic in vivo production of human T cell leukemia virus type I Rof and Tof proteins from cytotoxic T lymphocytes directed against viral peptides. J Exp Med. 2000;191:567–572. doi: 10.1084/jem.191.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins ND, Newbound GC, Albrecht B, Beard JL, Ratner L, Lairmore MD. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood. 1998;91:4701–4707. [PubMed] [Google Scholar]
- 27.Albrecht B, Collins ND, Burniston MT, Nisbet JW, Ratner L, Green PL, Lairmore MD. Human T-lymphotropic virus type 1 open reading frame I p12I is required for efficient viral infectivity in primary lymphocytes. J Virol. 2000;74:9828–9835. doi: 10.1128/jvi.74.21.9828-9835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht B, D’Souza CD, Ding W, Tridandapani S, Coggeshall KM, Lairmore MD. Activation of nuclear factor of activated T cells by human T-lymphotropic virus type 1 accessory protein p12I. J Virol. 2002;76:3493–3501. doi: 10.1128/JVI.76.7.3493-3501.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding W, Albrecht B, Kelley RE, Muthusamy N, Kim S, Altschuld RA, Lairmore MD. Human T lymphotropic virus type 1 p12I expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J Virol. 2002;76:10374–10382. doi: 10.1128/JVI.76.20.10374-10382.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding W, Kim SJ, Nair AM, Michael B, Boris-Lawrie K, Tripp A, Feuer G, Lairmore MD. Human T-cell lymphotropic virus type 1 p12I enhances interleukin-2 production during T-cell activation. J Virol. 2003;77:11027–11039. doi: 10.1128/JVI.77.20.11027-11039.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dustin ML, Bivona TG, Philips MR. Membranes as messengers in T cell adhesion signaling. Nat Immunol. 2004;5:363–372. doi: 10.1038/ni1057. [DOI] [PubMed] [Google Scholar]
- 32.Hogg N, Henderson R, Leitinger B, Mcdowall A, Porter J, Stanley P. Mechanisms contributing to the activity of integrins on leukocytes. Immunol Rev. 2002;186:164–171. doi: 10.1034/j.1600-065x.2002.18614.x. [DOI] [PubMed] [Google Scholar]
- 33.van KY, Figdor CG. Activation and inactivation of adhesion molecules. Curr Top Microbiol Immunol. 1993;184:235–256. doi: 10.1007/978-3-642-78253-4_19. [DOI] [PubMed] [Google Scholar]
- 34.Hogg N, Laschinger M, Giles K, Mcdowall A. T-cell integrins: more than just sticking points. J Cell Sci. 2003;116:4695–4705. doi: 10.1242/jcs.00876. [DOI] [PubMed] [Google Scholar]
- 35.Hogg N, Smith A, Mcdowall A, Giles K, Stanley P, Laschinger M, Henderson R. How T cells use LFA-1 to attach and migrate. Immunol Lett. 2004;92:51–54. doi: 10.1016/j.imlet.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Ardouin L, Bracke M, Mathiot A, Pagakis SN, Norton T, Hogg N, Tybulewicz VL. Vav1 transduces TCR signals required for LFA-1 function and cell polarization at the immunological synapse. Eur J Immunol. 2003;33:790–797. doi: 10.1002/eji.200323858. [DOI] [PubMed] [Google Scholar]
- 38.Griffiths EK, Krawczyk C, Kong YY, Raab M, Hyduk SJ, Bouchard D, Chan VS, Kozieradzki I, Oliveira-Dos-Santos AJ, Wakeham A, et al. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science. 2001;293:2260–2263. doi: 10.1126/science.1063397. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Moon EY, Azouz A, Wu X, Smith A, Schneider H, Hogg N, Rudd CE. SKAP-55 regulates integrin adhesion and formation of T cell-APC conjugates. Nat Immunol. 2003;4:366–374. doi: 10.1038/ni913. [DOI] [PubMed] [Google Scholar]
- 40.Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–1969. doi: 10.1128/mcb.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart MP, Mcdowall A, Hogg N. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J Cell Biol. 1998;140:699–707. doi: 10.1083/jcb.140.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins ND, D’Souza C, Albrecht B, Robek MD, Ratner L, Ding W, Green PL, Lairmore MD. Proliferation response to interleukin-2 and Jak/Stat activation of T cells immortalized by human T-cell lymphotropic virus type 1 is independent of open reading frame I expression. J Virol. 1999;73:9642–9649. doi: 10.1128/jvi.73.11.9642-9649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart MP, Cabanas C, Hogg N. T cell adhesion to intercellular adhesion molecule-1 (ICAM-1) is controlled by cell spreading and the activation of integrin LFA-1. J Immunol. 1996;156:1810–1817. [PubMed] [Google Scholar]
- 44.Robek MD, Wong FH, Ratner L. Human T-cell leukemia virus type 1 pX-I and pX-II open reading frames are dispensable for the immortalization of primary lymphocytes. J Virol. 1998;72:4458–4462. doi: 10.1128/jvi.72.5.4458-4462.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicot C, Mulloy JC, Ferrari MG, Johnson JM, Fu K, Fukumoto R, Trovato R, Fullen J, Leonard WJ, Franchini G. HTLV-1 p12I protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood. 2001;98:823–829. doi: 10.1182/blood.v98.3.823. [DOI] [PubMed] [Google Scholar]
- 46.Hummel KB, Bellini WJ, Offermann MK. Strain-specific differences in LFA-1 induction on measles virus-infected monocytes and adhesion and viral transmission to endothelial cells. J Virol. 1998;72:8403–8407. doi: 10.1128/jvi.72.10.8403-8407.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fortin JF, Cantin R, Tremblay MJ. T cells expressing activated LFA-1 are more susceptible to infection with human immunodeficiency virus type 1 particles bearing host-encoded ICAM-1. J Virol. 1998;72:2105–2112. doi: 10.1128/jvi.72.3.2105-2112.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arias RA, Munoz LD, Munoz-Fernandez MA. Transmission of HIV-1 infection between trophoblast placental cells and T-cells take place via an LFA-1-mediated cell to cell contact. Virology. 2003;307:266–277. doi: 10.1016/s0042-6822(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 49.Wattel E, Vartanian JP, Pannetier C, Wainhobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. 1995;69:2863–2868. doi: 10.1128/jvi.69.5.2863-2868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cavrois M, Leclercq I, Gout O, Gessain A, Wainhobson S, Wattel E. Persistent oligoclonal expansion of human T-cell leukemia virus type 1 infected circulating cells in patients with tropical spastic paraparesis/HTLV-1 associated myelopathy. Oncogene. 1998;17:77–82. doi: 10.1038/sj.onc.1201906. [DOI] [PubMed] [Google Scholar]
- 51.Mortreux F, Kazanji M, Gabet AS, de Thoisy B, Wattel E. Two-step nature of human T-cell leukemia virus type 1 replication in experimentally infected squirrel monkeys (Saimiri sciureus) J Virol. 2001;75:1083–1089. doi: 10.1128/JVI.75.2.1083-1089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosley AJ, Asquith B, Bangham CR. Cell-mediated immune response to human T-lymphotropic virus type I. Viral Immunol. 2005;18:293–305. doi: 10.1089/vim.2005.18.293. [DOI] [PubMed] [Google Scholar]
- 53.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate β2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 54.Chan JR, Hyduk SJ, Cybulsky MI. Chemoattractants induce a rapid and transient upregulation of monocyte α4 integrin affinity for vascular cell adhesion molecule 1 which mediates arrest: an early step in the process of emigration. J Exp Med. 2001;193:1149–1158. doi: 10.1084/jem.193.10.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feigelson SW, Grabovsky V, Winter E, Chen LL, Pepinsky RB, Yednock T, Yablonski D, Lobb R, Alon R. The Src kinase p56lck up-regulates VLA-4 integrin affinity: implications for rapid spontaneous and chemokine-triggered T cell adhesion to VCAM-1 and fibronectin. J Biol Chem. 2001;276:13891–13901. doi: 10.1074/jbc.M004939200. [DOI] [PubMed] [Google Scholar]
- 56.Michael B, Nair AM, Hiraragi H, Shen L, Feuer G, Boris-Lawrie K, Lairmore MD. Human T lymphotropic virus type-1 p30II alters cellular gene expression to selectively enhance signaling pathways that activate T lymphocytes. Retrovirology. 2004;1:39. doi: 10.1186/1742-4690-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]