

Abstract
Methamphetamine (METH) damages dopamine (DA) nerve endings by a process that has been linked to microglial activation but the signaling pathways that mediate this response have not yet been delineated. Cardona et al. [Nat. Neurosci. 9 (2006), 917] recently identified the microglial-specific fractalkine receptor (CX3CR1) as an important mediator of MPTP-induced neurodegeneration of DA neurons. Because the CNS damage caused by METH and MPTP is highly selective for the DA neuronal system in mouse models of neurotoxicity, we hypothesized that the CX3CR1 plays a role in METH-induced neurotoxicity and microglial activation. Mice in which the CX3CR1 gene has been deleted and replaced with a cDNA encoding enhanced green fluorescent protein (eGFP) were treated with METH and examined for striatal neurotoxicity. METH depleted DA, caused microglial activation, and increased body temperature in CX3CR1 knockout mice to the same extent and over the same time course seen in wild-type controls. The effects of METH in CX3CR1 knockout mice were not gender-dependent and did not extend beyond the striatum. Striatal microglia expressing eGFP constitutively show morphological changes after METH that are characteristic of activation. This response was restricted to the striatum and contrasted sharply with unresponsive eGFP-microglia in surrounding brain areas that are not damaged by METH. We conclude from these studies that CX3CR1 signaling does not modulate METH neurotoxicity or microglial activation. Furthermore, it appears that striatal-resident microglia respond to METH with an activation cascade and then return to a surveying state without undergoing apoptosis or migration.
Keywords: dopamine, fractalkine receptor, methamphetamine, microglial activation, neurotoxicity
Microglial activation has been implicated as a contributing factor in numerous neurodegenerative disorders to include Alzheimer's disease (Streit 2004) and Parkinson's disease (Klegeris et al. 2007), to list but a few. In addition, drugs such as methamphetamine (METH) and MPTP, which cause damage to the dopamine (DA) neuronal system, are also thought to exert at least part of their neurotoxicity via microglial activation (LaVoie et al. 2004; Thomas et al. 2004b; Kim et al. 2007). One microglial-associated receptor was recently implicated in MPTP-induced neurotoxicity when it was shown that mice lacking the receptor for fractalkine (CX3CR1) were hypersensitive to the damaging effects of MPTP on DA neurons of the substantia nigra (Cardona et al. 2006). The CX3CR1 is expressed predominantly in microglial cells in brain (Nishiyori et al. 1998; Maciejewski-Lenoir et al. 1999; Mizuno et al. 2003) and its activation by fractalkine (CX3CL1) released from neurons serves to limit or minimize microglial activation (Zujovic et al. 2000; Mizuno et al. 2003). The null mutation in the CX3CR1 gene has acute as well as long-term negative consequences on neurons, highlighting the important role of the CX3CR1 signaling system in maintaining neuronal homeostasis (Meucci et al. 2000) and in resisting drug- and disease-induced damage.
Methamphetamine intoxication results in a constellation of gene expression changes in brain areas known to be damaged by this drug of abuse and many of the genes altered by METH can be traced to microglial cells (Thomas et al. 2004a). It is becoming evident that microglial activation is a very early response in the neurotoxic cascade associated with METH (LaVoie et al. 2004; Thomas et al. 2004b; Thomas and Kuhn 2005a) and blockade of this reaction with drugs like MK-801 and dextromethorphan protect against METH-induced nerve ending damage (Thomas and Kuhn 2005b).
In light of the important role of CX3CR1 in dampening microglial activation in general (Zujovic et al. 2000; Mizuno et al. 2003), and in protecting against MPTP-induced DA neuronal damage (Cardona et al. 2006), we hypothesized that METH would cause heightened DA nerve ending damage in mice lacking the CX3CR1. Because CX3CR1 knockout mice express the gene for enhanced green fluorescent protein (eGFP) in place of the CX3CR1, these mice have constitutively fluorescent microglial cells, allowing visualization of the morphological response to METH. We report presently that METH provokes microglial activation and DA nerve ending damage in the striatum of CX3CR1 knockout mice to the same extent seen in wild-type mice. In addition, the remarkable specificity of METH-induced microglial activation in brain is highlighted by the drug-induced changes in microglial status in damaged areas (i.e., striatum) and contrasts sharply with adjacent sites not damaged by METH (i.e., cortex). These results suggest that METH does not provoke microglial activation via the CX3CR1 signaling pathway and they also highlight some fundamentally different mechanisms of action of METH and MPTP.
Materials and methods
Materials
(+) Methamphetamine hydrochloride, pentobarbital, horseradish peroxidase (HRP)-conjugated Isolectin B4 (from Griffonia simplicifolia), 3,3′-diaminobenzidine, paraformaldehyde, Triton X-100, dopamine, methanol, EDTA, all buffers and HPLC reagents were purchased from Sigma-Aldrich (St. Louis, MO). CitriSolv and Permount were products of Fisher Scientific (Pittsburgh, PA). Alexa Fluor 568 conjugated ILB4 was obtained from Invitrogen (Carlsbad, CA).
Animals
Mice in which the CX3CR1 gene was deleted and replaced with a cDNA of eGFP (Jung et al. 2000) were purchased as breeding pairs from The Jackson Laboratory (Bar Harbor, ME; strain B6.129P-Cx3cr1tm1Litt/J). Mice homozygous for the deletion of the CX3CR1 gene were used to establish a colony of male and female offspring for use in these studies. The recommended wild-type control for these mice is the C57BL/6 strain. Male and female wild-type C57BL/6 mice were purchased from Harlan (Indianapolis, IN). The expression of the CX3CL1 gene was confirmed in wild-type controls and CX3CR1 knockout strains and the absence of expression of the CX3CR1 gene was confirmed in CX3CR1 knockout strain by RT-PCR (data not shown). Mice expressing eGFP under the control of a chicken beta-actin promoter and cytomegalovirus enhancer were also obtained from The Jackson Laboratory (strain C57BL/6-Tg(ACTbEGFP)1Osb/J). These mice express eGFP in all tissues with the exception of erythrocytes and hair (Okabe et al. 1997), and were used as bone marrow donors as described below. All mice were housed five per cage in small shoebox cages in a light and temperature controlled room and weighed 20-25 g at the time of experimentation. Mice had free access to food and water. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Pharmacological and physiological procedures
Mice were treated with a binge neurotoxic regimen of METH comprised of four injections of 5 mg/kg i.p. with a 2 h interval between each injection. This METH regimen is known to cause extensive microglial activation and DA nerve ending damage (Thomas et al. 2004b). Controls received i.p. injections of physiological saline on the same schedule used for METH. Mice were sacrificed at various times after the METH regimen to assess the status of striatal DA and microglial activation (specified below for each experiment). Body temperature was monitored by telemetry using IPTT-300 implantable temperature transponders from Bio Medic Data Systems, Inc. (Seaford, DE). Temperatures were recorded every 20 min non-invasively using the DAS-5001 console system from Bio Medic.
Lectin histochemical staining of microglia
Microglial activation was assessed by staining fixed brain sections with HRP-conjugated Isolectin B4 (ILB4) as developed by Streit (1990) and as previously described in our studies with METH (Thomas et al. 2004b). At the time of sacrifice, mice were deeply anesthetized with pentobarbital (120 mg/kg) and perfused transcardially with ice-cold 4% paraformaldehyde in PBS. Brains were removed and stored overnight in fixative at 4°C. Sections of 50 μm thickness were cut throughout the rostral/caudal axis of the striatum (+ 1.2 through - 0.1 mm with respect to Bregma). Sections were floated into phosphate buffered saline (PBS) containing 3% H2O2 for 30 min, washed once in PBS + 0.1% Triton X-100, then incubated in fresh PBS + 0.1% Triton X-100 for an additional 30 min. Microglia were labeled with HRP-conjugated ILB4 (10 μg/mL in PBS + 0.1% Triton X-100) overnight at 4°C. Excess ILB4 was removed by three washes with PBS + 0.1% Triton X-100 (5 min each) followed by a single wash in PBS before exposure to 3,3′-diaminobenzidine substrate (0.1 mg/mL) in PBS for 25 min. Following a single wash in PBS, sections were transferred to glass slides, air dried and dehydrated through a series of graded ethanol washes. Sections were incubated in Citrisolv for 5 min then coverslipped under Permount. For all pharmacological studies presented below, brain sections from drug-treated mice were processed simultaneously with controls to normalize staining among treatment groups. Microglial reactivity was viewed under the light microscope and the number of stained cells observed after various treatments was counted using NIH Image. Cell counts were sampled from a 0.38 mm2 area of the striatum by a person blinded to the treatment conditions. Cells were counted from three independent sections from all like-treated mice, bilaterally, generating an average count for each treated subject.
Fluorescence imaging of microglia
In some experiments, sections from CX3CR1 knockout mice were incubated with Alexa Fluor 568 conjugated ILB4 to assess activation of the eGFP-expressing microglia. Sections were incubated in PBS + 0.1% Triton X-100 for 30 min at 22°C and then overnight at 4°C with Alexa Fluor 568 conjugated ILB4 (10 μg/mL) in PBS + 0.1% Triton X-100. Excess label was removed by three washes in PBS + 0.1% Triton X-100 and one wash in PBS. Sections were then wet mounted on microscope slides and coverslipped. Activated microglia labeled with Alexa Fluor 568 conjugated ILB4 and microglia constitutively expressing eGFP were viewed and photographed using an Olympus BX51 fluorescence microscope.
Striatal DA content
Depletion of striatal DA after METH treatment is widely used as an index of METH-induced toxicity to DA nerve endings. DA depletion from striatum faithfully reflects other measures of DA nerve ending damage caused by METH, such as reduced tyrosine hydroxylase immunoreactivity or reduced ligand binding to the DA transporter, especially at longer times after treatment as used presently. Striata were dissected from brain and stored frozen at -80°C. Tissues were weighed and sonicated in 10 vol of 0.16N perchloric acid at 4°C. Insoluble protein was removed by centrifugation and DA was determined by HPLC with electrochemical detection.
Migration of hematopoietic cells into striatum after METH treatment
To investigate if hematopoietic cells in the peripheral circulation can serve as the source of striatal microglia after migration into brain, we analyzed donor cell engraftment into the CNS as previously described (Priller et al. 2001; Vallieres and Sawchenko 2003). Briefly, female C57BL/6 mice (n = 10) were exposed to a lethal dose of gamma radiation from a cesium source (10 gray total body irradiation in two equal doses 3 h apart). Approximately 24 h later, irradiated mice were injected via the tail vein with ~ 5 × 106 bone marrow cells harvested from the femurs of C57BL/6 female mice expressing eGFP in all tissues (Oka et al. 2003). After rescue by bone marrow transplant, mice were housed in sterilized caging. After a 10-day recovery period, mice (n = 5) were treated with the neurotoxic regimen of METH and controls (n = 5) received injections of saline on the same schedule. Brains were fixed by perfusion 2 days after METH as described above and examined for microglial activation and engraftment of eGFP expressing cells in striatum.
Data analysis
Individual treatment groups were compared to appropriate controls for DA and microglial counts with a one-way ANOVA followed by Tukey's Multiple Comparison Test in GraphPad Prism 5. The effects of drug treatments on core body temperature were analyzed by two-way ANOVA followed by Bonferroni's post-test. Differences were considered significant if p < 0.05.
Results
Wild-type and CX3CR1 knockout female mice were treated with a neurotoxic regimen of METH and the effects on striatal DA are presented in Fig. 1(a). METH reduced striatal DA content to approximately 25-30% of control at 2 days after treatment. This effect persisted for 7 days and 14 days, remaining at about 60-70% depletion. Wild-type and CX3CR1 knockout mice did not differ in their response to METH at any time point. Cardona et al. (2006) used male CX3CR1 knockout mice to show enhancement of MPTP neurotoxicity. In order to rule out a gender effect, we tested male CX3CR1 knockout mice for their response to METH. These data are presented in Fig. 1(b) and show that males from wild-type and CX3CR1 knockout strains were significantly depleted of striatal DA to the same extent 2 days, 7 days, and 14 days after METH treatment (i.e., depletion to about 20-30% of control) but did not differ from each other. METH-induced DA depletion at all time points and in both strains was significantly different from control (p < 0.001). It can also be seen that male mice (Fig. 1b) of both strains were slightly more responsive to METH toxicity than females (Fig. 1a). However, the only significant difference between males and females was the comparison between males at the 2-day time point of both strains to wild-type females at the 14 d time point (p < 0.05).
Fig. 1.
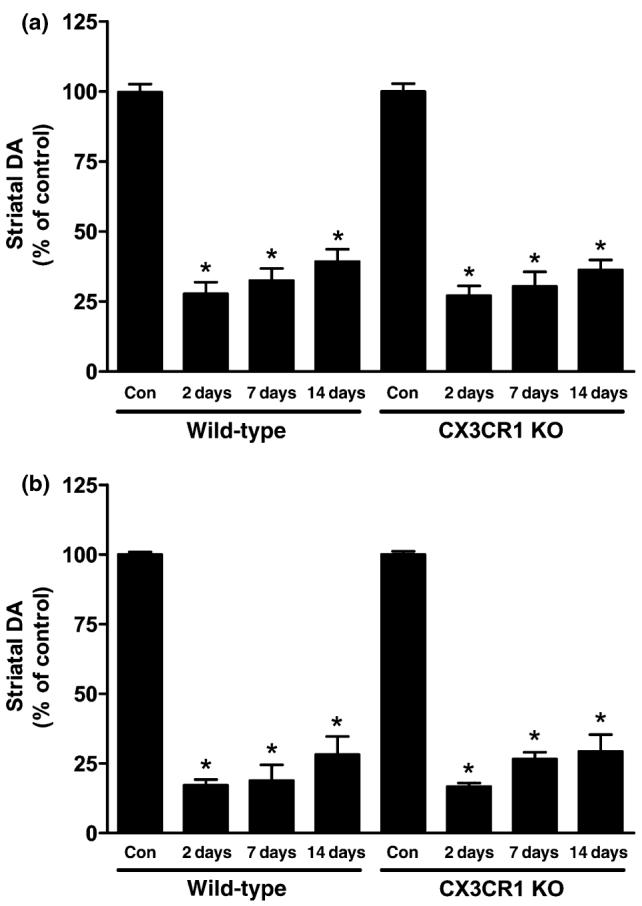
Effects of a neurotoxic METH regimen on striatal DA levels in wild-type and CX3CR1 knockout (KO) mice. Female (a) and male (b) mice (n = 5-8 per group) were treated with METH (4 × 5 mg/kg). Striatal DA levels were determined 2-day, 7-day, and 14-day post-METH. Results are presented as mean % of saline-treated controls ± SEM. DA levels in striatum of untreated mice were 17.5 ng/mg tissue (wet weight). Significant differences were determined via oneway ANOVA followed by Tukey's multiple comparison test, and are indicated as follows: *p < 0.001 relative to control (Con).
The effect of METH on striatal microglia was assessed 2 days after treatment, the time point at which METH-induced microglial activation is greatest (Thomas et al. 2004b), using lectin histochemical staining with ILB4. The results in Fig. 2 show that METH leads to extensive microglial activation in striatum in males and females of both strains. Counts of activated microglia indicated that the effect of METH was significant in female mice from both WT (p < 0.001) and CX3CR1 knockout strains (p < 0.001) as well as in male wild-type (p < 0.001) and CX3CR1 knockout mice (p < 0.001). A strain effect was not evident with regard to METH-induced microglial activation (p > 0.05). The extent of microglial activation seen in males trended toward a greater increase by comparison to females, but the overall effect of gender was not significant (p > 0.05).
Fig. 2.

Effects of a neurotoxic METH regimen on microglial activation in wild-type and CX3CR1 KO mice. Mice (n = 3-5 per group) were treated with saline (a, c, e and g) or METH (b, d, f and h) and analyzed for microglial activation in the striatum after 48 h. Microglia counts were obtained as described in the Materials and Methods and are presented as means ± SEM: wild-type female control (a; 8 ± 1) and METH (b; 149 ± 13); wild-type male control (c; 11 ± 3) and METH (d; 151 ± 9); CX3CR1 KO female control (e; 7 ± 4) and METH (f; 143 ± 11); CX3CR1 KO male control (g; 12 ± 2) and METH (h; 157 ± 8). Significant differences were determined via one-way ANOVA followed by Tukey's multiple comparison test. All METH-treated conditions were statistically different relative to their respective controls (gender and strain; p < 001). Differences among controls (panels a, c, e. and g) or METH-treated conditions (panels b, d, f, h) were not significant (p > 0.05). Scale bar represents 150 μm.
We hypothesized that CX3CR1 knockout mice would be more responsive to METH neurotoxicity, as they are to MPTP (Cardona et al. 2006), but this was not the case (Figs 1 and 2). Therefore, we measured the core temperature response to METH in both strains to rule out the possibility that the CX3CR1 knockout mice differed from wild-type controls with regard to the development of hyperthermia. METH caused a significant increase in core body temperature in females (Fig. 3a) and males (Fig. 3b) of both strains (p < 0.0001), but the effect of strain was not significant in either gender (p > 0.05). It is also apparent from Fig. 3 that males show a different pattern of body temperature response by comparison to females. Males of both strains treated with METH show pronounced spikes in body temperature that begin just after each of the four METH injections and reach maximum over the next 60 min. Body temperatures fall over the ensuing 60 min. These sharp increases and decreases in body temperature become larger with each successive METH injection in males. This effect reached statistical significance in males of both strains at the 400 min (p < 0.001), 420 min (p < 0.001) and 440 min (p < 0.05) time points (Bonferroni's post-test). While females of both strains develop significant hyperthermia (Fig. 3a), the increases and decreases between METH injections are not quite as pronounced. The body temperature response of METH-treated males was significantly greater than that of females (p < 0.0001).
Fig. 3.
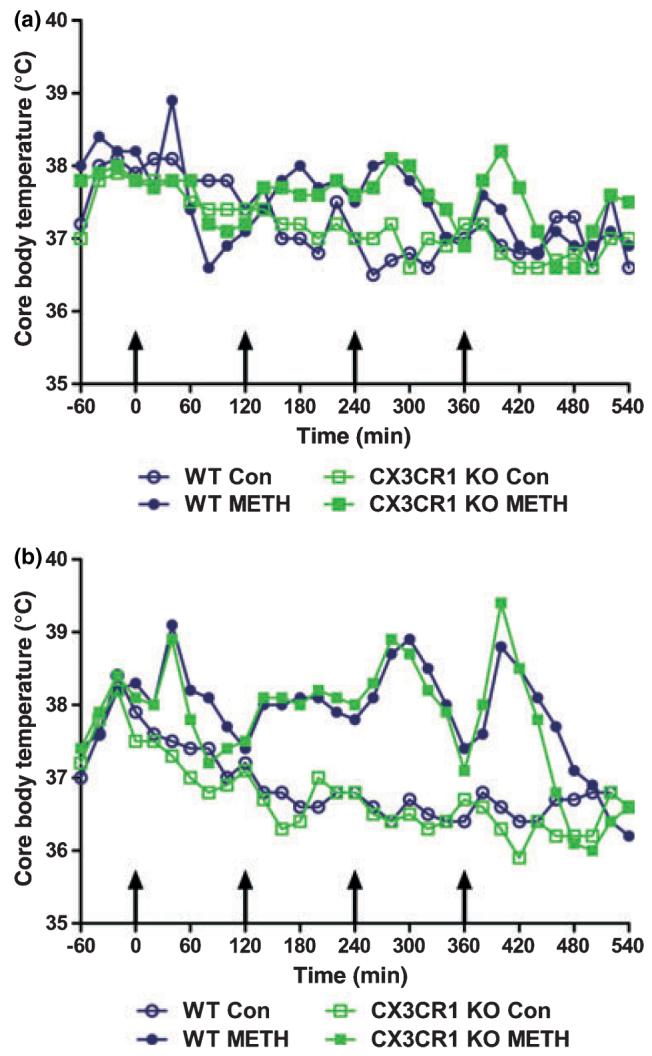
Effects of a neurotoxic METH regimen on the core body temperatures of wild-type and CX3CR1 KO mice. Female (a) and male (b) mice (n = 5 per group) were treated with saline or METH, and core body temperatures monitored by telemetry every 20 min for 10 h. Results are presented as mean body temperature (°C) for each group at the indicated time points. Saline and METH were administered at t = 0, 120, 240 and 360 min (indicated by arrows). SEM bars were omitted for the sake of clarity and were less than 10% of the mean in all groups.
Although these studies do not establish a clear role for the microglial CX3CR1 in METH-induced neurotoxicity and microglial activation, it was possible to take advantage of the fact that the CX3CR1 knockout strain expresses eGFP constitutively in CNS microglia to visualize their morphological response to METH. Figure 4 shows the effects of METH on striatal microglia 2 days after treatment as assessed by endogenous fluorescence of eGFP-expressing microglia. Because there was no qualitative difference between male and female mice in these studies, results from female mice are shown. It can be seen that METH provokes a profound change in microglia morphology. Microglia in METH-treated mice (Fig. 4b), by comparison to controls (Fig. 4a), showed a substantial retraction of their processes and the cell bodies assumed a larger, more amoeboid shape, both of which are characteristic of activation. METH did not increase the number of eGFP-expressing microglia in striatum (data not shown; see also legend to Fig. 2). Figure 4 also shows the remarkable anatomical specificity of METH-induced microglial activation. Microglia in brain areas bordering the striatum (e.g., somatosensory cortex and corpus callosum) known not to be damaged by METH did not show these morphological changes. By 7 days, the appearance of the activated microglia in striatum had returned to that shown by controls for both wild-type and CX3CR1 knockout strains and there was no evidence of microglial cell loss at any time (data not shown). Figure 5(c) and (d) show representative images of striatal microglia from CX3CR1 knockout mice treated with saline or METH, respectively. These higher magnification micrographs offer a fuller appreciation of the effect of METH on microglial status as revealed by the enlarged cell bodies and retracted processes shown in Fig. 5(d) as compared to Fig. 5(c).
Fig. 4.

Effects of a neurotoxic METH regimen on microglial morphology in CX3CR1 KO mice. 48 h post-METH, CX3CR1 KO mice were perfused and their microglia examined via fluorescence microscopy. Coronal sections through striatum from control (a, c) and METH (b, d) treated mice were viewed at 200× (a, b) and 600× (c, d) magnification.
Fig. 5.

ILB4 labels activated microglia in wild-type and CX3CR1 KO mice. Wild-type and CX3CR1 KO mice were perfused 48 h after METH treatment and microglia were examined via fluorescence microscopy. Alexa 568-conjugated ILB4 was used to probe for activated microglia. Coronal sections through the striatum of control and METH treated mice were viewed at 600× magnification.
Alexa Fluor 568 conjugated ILB4 was used to determine if microglia expressing eGFP constitutively and showing METH-induced morphological changes were also stained by the lectin, and the results are shown in Fig. 5. Because there was no qualitative difference between male and female mice in these studies, results from only female CX3CR1 knockout mice are presented. Figure 5(a) shows that eGFP-expressing microglia in untreated CX3CR1 knockout mice are characteristic of resting microglia, in agreement with the results in Fig. 4. These cells are not labeled by Alexa Fluor 568 conjugated ILB4 (Fig. 5b) in agreement with results shown in Fig. 2 using HRP-labeled ILB4 (see Fig. 2a, c, e, and g). The METH-induced shift in microglial morphology is shown in Fig. 5(d) and it can be seen that this cell is also extensively labeled with Alexa Fluor 568 conjugated ILB4 (panel e). The merged image (panel f) shows that the eGFP-expressing microglia in this field is positive for labeling with lectin, confirming its status as activated. Finally, to rule out the possibility that expression of the eGFP transgene in microglia altered their response to METH, striatal sections from wild-type females were labeled with Alexa Fluor 568 conjugated ILB4 and the results are shown in Fig. 5 as well. It can be seen in these panels that METH caused a significant increase in microglial activation and the effect was not different from what is seen when HRP-conjugated ILB4 is used to assess microglial activation (see Fig. 2).
To investigate the possibility that peripheral hematopoietic cells migrate into brain after METH treatment and serve as the source of striatal microglia, irradiated C57BL/6 mice were rescued with a bone marrow transplant from mice expressing eGFP in all tissues (i.e., `green mice'; Okabe et al. 1997). Two days after treatment with the standard neurotoxic METH regimen used above, when microglial activation is maximal (Thomas et al. 2004b), we did not find any evidence of eGFP-expressing cells in striatum. These mice responded to METH in the same manner as normal mice (e.g., developed hyperactivity) and ILB4 staining of fixed sections from striatum confirmed extensive microglial activation. Control mice (i.e., irradiated, bone marrow transplant, saline treated) were also devoid of eGFP cells in striatum 12 days after transplant. These negative data (i.e., blank fluorescence images) are not shown in the interest of conserving space.
Discussion
The chemokine CX3CL1 is a neuronal transmembrane glycoprotein that can be released proteolytically in soluble form (Cook et al. 2001). Receptors for CX3CL1 are found predominantly on CNS resident microglial cells and it is becoming evident that neuronal-microglial signaling is mediated by CX3CL1-CX3CR1 interactions (Harrison et al. 1998). Release of CX3CL1 from neurons has been identified as an early response to neurotoxic insult (Chapman et al. 2000; Zujovic et al. 2000) and activation of the CX3CR1 is associated with neuroprotection, probably as a result of reduced microglial activation (Mizuno et al. 2003). The consequences of reduced CX3CR1 function could be profound under conditions where microglial activation is thought to contribute to neuronal damage. This situation was illustrated dramatically by Cardona et al. (Cardona et al. 2006) who showed that DA neurons of the substantia nigra in CX3CR1 knockout mice were hyper-responsive to the damaging effects of MPTP. In light of the fact that MPTP and METH exert highly specific neurotoxic effects within the DA neuronal system, and considering that each drug causes extensive microglial activation, we hypothesized that CX3CR1 knockout mice would also be hyper-responsive to METH neurotoxicity.
Because previous work showing hypersensitivity of DA neurons to MPTP in CX3CR1 knockout mice focused on the substantia nigra and was carried out using male mice exclusively (Cardona et al. 2006), we included an assessment of several variables that could influence the extent of METH neurotoxicity in these mice to include post-treatment time of survival, gender, body temperature response, and brain-regional effects. Initially, we tested female mice and we were unable to uncover a difference in response to METH between wild-type and CX3CR1 knockout strains with respect to striatal DA depletion, microglial activation, or the development of hyperthermia. These analyses extended up to 14 days after treatment with a binge neurotoxic regimen of METH. Similarly, male CX3CR1 knockout mice could not be distinguished from wild-type controls with regard to the extent to which they developed striatal DA neurotoxicity, microglial activation, or hyperthermia after METH. While strain differences were not apparent, we did observe mild gender effects in that the body temperature responses of male mice were greater in magnitude than females, and the extent of neurotoxicity was slightly greater in males at selected times after METH treatment. The gender effects observed in this study were not as substantial as previously reported for METH (Miller et al. 1998) but our present studies used lower doses of drug (4 × 5 mg/kg vs. 4 × 10 mg/kg). It was also interesting to observe that while male mice showed larger swings in body temperature between METH injections and a slightly greater overall hyperthermic response, they did not uniformly show greater neurotoxicity and microglial activation than females. This result could reflect a ceiling effect, making it difficult for male mice of either strain to develop more severe neurotoxicity or hyperthermia under the present treatment conditions. Finally, we also assessed tyrosine hydroxylase protein content in the substantia nigra by immunoblotting and observed that METH did not alter its levels in either strain at any time point (data not shown). Those few studies that have demonstrated loss of tyrosine hydroxylase content or DA cell bodies in the substantia nigra after METH also used much higher drug doses than used presently (Sonsalla et al. 1996; Brown et al. 2006). A neurotoxic regimen of METH does not change striatal expression of either the CX3CR1 or the CX3CL1 gene, as assessed by microarray analysis (Thomas et al. 2004a), and we observed presently that the expression of neither gene was altered by METH in wild-type controls nor was expression of CX3CL1 altered in the CX3CR1 knockout strain (data not shown). A number of important variables were included for analysis presently to determine if gender, survival time after drug treatment, or brain-regional specificity would reveal a strain effect in CX3CR1 knockout mice. The results indicated that METH exerts DA nerve ending neurotoxicity, microglial activation, and hyperthermia in CX3CR1 knockout mice to exactly the same extent as seen in wild-type controls. Therefore, considering all data presented above, the most parsimonious conclusion asserts that METH-induced microglial activation and DA nerve terminal toxicity are not mediated by CX3CR1 signaling. It is known that METH toxicity can extend to striatal serotonin terminals as well (Boger et al. 2007) and a role for CX3CR1 signaling in METH-induced toxicity within the serotonin neuronal system is under investigation.
The present data stand in contrast to the findings of Cardona and colleagues (Cardona et al. 2006) who showed that CX3CR1 knockout mice were much more sensitive to the destructive effects of MPTP on DA cell bodies of the substantia nigra. Cardona et al. (2006) also established that this interesting mouse strain was hyper-responsive to other models of neurotoxicity because of dysregulation in their microglial response. However, it is evident that METH leads to activation of striatal microglia in a manner that is independent of the CX3CR1. Certain similarities between METH and MPTP may point to a differential involvement of the microglial CX3CR1 in drug-induced neurotoxicity. For example, blockade of microglial activation with minocycline protects against MPTP-induced damage to DA cell bodies (Du et al. 2001; Wu et al. 2002). Minocycline also prevents some elements of microglial activation caused by single doses of METH and MPTP but it does not protect against drug-induced damage to DA nerve terminals (Sriram et al. 2006). Taken together, these results suggest that CX3CR1 signaling may influence toxicity to a greater extent in DA cell bodies than in nerve terminals. Despite the fact that it was not possible to assign to the microglial CX3CR1 a role in METH-induced DA nerve ending neurotoxicity, it was possible to take advantage of the fact that the CX3CR1 gene was replaced in the knockout strain with the cDNA of eGFP, producing a mouse with constitutive expression of eGFP in all CNS microglia.
Various lectins, including ILB4 as applied presently, are used to label activated microglia in brain, but these probes do not normally label resting microglia (e.g., see Fig. 2). Striatal sections from CX3CR1 knockout mice with eGFP reporter expression in microglia are visually striking in that the microscopic field is abundantly populated with ramified (i.e., resting) microglia. At 2-day post-METH, the time at which microglial activation is maximal (Thomas et al. 2004b), microglia show a profound change in morphology. These microglia have assumed a more amoeboid shape with retracted processes and enlarged cell bodies, phenotypic changes that are characteristic of activation (Davalos et al. 2005; Nimmerjahn et al. 2005). The endogenously fluores-cent striatal microglia in untreated mice are not labeled by ILB4 but they are labeled at 2 days after METH treatment, further affirming that the lectin primarily recognizes activated microglia (Streit 1990). It does not appear that METH changes the number of microglia in striatum over the time-course studied presently (i.e., 2-14 days). Instead, the microglia cycle from resting to activated and then back to resting (or a status of `surveying' as proposed by Hanisch and Kettenmann (Hanisch and Kettenmann 2007)) as judged from the observed morphological changes after METH. In light of results showing that eGFP-expressing microglia are also labeled by ILB4 in the identical manner as wild-type mice treated with METH, it is safe to conclude that expression of the eGFP transgene in microglia, per se, has not hampered microglial reactivity.
Another advantage accruing from the use of mice constitutively expressing eGFP in CNS microglia is the opportunity to document the highly delimited anatomical locus of microglial activation resulting from METH intoxication. Activation of striatal microglia after METH was completely restricted to the caudate-putamen. Brain areas immediately bordering the striatum in all directions, including the somatosensory cortex and the nucleus accumbens, did not show morphological evidence of microglial activation. These results agree well with previous studies showing that microglial activation and DA nerve ending damage are highly correlated along the neuroanatomical axis (LaVoie et al. 2004; Thomas et al. 2004b) and add support to the notion that microglial activation is an integral component of the METH neurotoxic cascade in striatum.
The effects of METH on core body temperature did not differ between strains, but a mild gender effect was apparent. Males and females developed a significant hyperthermia after METH administration, but the pattern of response was different between genders, with males showing significantly greater effects. By measuring core body temperature via telemetry at 20 min intervals, the complexity of the effect of METH on body temperature becomes evident. The most common approach to body temperature determinations after METH injections in rodents involves the use of a rectal probe with measures taken hourly, probably to minimize the stress of repeated handling and insertion of the probe into the rectal cavity. Male and female mice alike show an interesting cyclic body temperature response to METH when assessed noninvasively by telemetry as shown in Fig. 3. In general, body temperatures begin to rise in the 60 min following the first injection of METH and then they fall over the next 60 min. Each subsequent METH injection prompts another rise and fall cycle during the 2 h period between injections. The fourth injection of the METH binge administration protocol leads to the greatest changes in body temperature. This pattern occurs in both genders but it is amplified in males. This difference between males and females in their body temperature response to METH was not reflected in the effects of drug on striatal DA neurochemistry and microglial activation where gender effects were not observed. Other investigators have shown that METH exerts greater DA nerve ending toxicity in males versus females, but most of these studies used higher doses of drug (Miller and O'Callaghan 1995; Hirata et al. 1996; Miller et al. 1998; Dluzen et al. 2003; Dluzen 2004; Dluzen and Mickley 2005). Thus, while it may be the case that the current METH treatment protocol created a ceiling effect in males, the present results emphasize the marked difference in body temperature response to METH when measured non-invasively via telemetry versus the use of a rectal probe.
It has been reported that hematopoietic cells can populate the brain and differentiate into neurons (Brazelton et al. 2000) or microglia (Eglitis and Mezey 1997; Priller et al. 2001; Hess et al. 2004; Simard and Rivest 2004), or they can retain their hematopoietic identity as macrophage (Vallieres and Sawchenko 2003). Microglial engraftment is enhanced by neuropathology as evidenced by the attraction of bone marrow-derived cells (i.e., eGFP-expressing) to the sites of neuronal damage (Priller et al. 2001). In light of these findings, we investigated the possibility that METH increases trafficking of hematopoietic cells into brain. METH treatment of irradiated mice rescued with bone marrow transplants from `green' mice did not reveal any evidence whatsoever of migration of eGFP-expressing cells into striatum. These mice did develop microglial activation 2 days after treatment that was identical to that seen in wild-type mice (see Fig. 2 above). Recruitment of monocyte/macrophage cells into the CNS is not impaired in mice lacking the gene for CX3CR1 (Huang et al. 2006). Therefore, it seems clear that the CX3CR1 knockout mouse model, in which METH-induced microglial activation is revealed through morphological and histochemical (i.e., ILB4 staining) analyses, augments the engraftment studies very well. Taken together, these results suggest that the eGFP-expressing microglia observed in the striatum of CX3CR1 mice are striatal-resident and METH does not appear to increase trafficking of macrophage- or monocyte-like cells from the periphery into the central compartment. This conclusion is entirely consistent with recent findings that microglia progenitor recruitment from the circulation in CNS degenerative diseases does not occur (Ajami et al. 2007; Mildner et al. 2007; Ransohoff 2007).
In summary, the present work reveals several new and interesting facets of striatal toxicity and microglial activation induced by neurotoxic doses of METH. First, neither the neurotoxicity nor the microglial activation caused by METH is mediated by CX3CR1 signaling as evidenced by the use of CX3CR1 knockout mice. Second, striatal microglia constitutively expressing eGFP show pronounced morphological changes over a time course that is the same as seen in wild-type controls treated with METH, and only those eGFP-microglia showing morphological evidence of activation are labeled by ILB4 histochemical staining. Third, striatal-resident microglia are activated soon after METH administration and it does not appear that macrophage- or monocyte-like cells traffic into the striatum from the circulation after drug treatment. Fourth, non-invasive measurement of the body temperature response to METH by telemetry reveals a highly complex and cyclic response to drug challenge that is amplified in males by comparison to females. The CX3CR1 knockout mouse model represents a valuable resource that will facilitate new lines of work aimed at the analysis of microglial responses to drug-, disease-, or injury-induced damage to the brain.
Acknowledgements
This work was supported by NIDA/NIH grants DA010756, DA017327, DA014692, and a VA Merit Award to DMK and by NIDA grant DA020680 and a VA MREP Award to DMT. We thank Dr. Joseph Kaplan, Department of Pediatrics, Wayne State University School of Medicine, for his invaluable assistance with the bone marrow transplant experiments.
Abbreviations used
- CX3CL1
fractalkine
- CX3CR1
fractalkine receptor
- HRP
horseradish peroxidase
- DA
dopamine
- eGFP
enhanced green fluorescent protein
- ILB4
Isolectin B4
- METH
methamphetamine
- PBS
phosphate-buffered saline
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self- renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H, Pacchioni AM, Granholm AC, McGinty JF. Long-term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line-derived neurotrophic factor heterozygous mice. J. Neurosci. 2007;27:8816–8825. doi: 10.1523/JNEUROSCI.1067-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- Brown JM, Gouty S, Iyer V, Rosenberger J, Cox BM. Differential protection against MPTP or methamphetamine toxicity in dopamine neurons by deletion of ppN/OFQ expression. J. Neurochem. 2006;98:495–505. doi: 10.1111/j.1471-4159.2006.03902.x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, Vassileva G, Lira SA. Generation and analysis of mice lacking the chemokine fractalkine. Mol. Cell. Biol. 2001;21:3159–3165. doi: 10.1128/MCB.21.9.3159-3165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Dluzen DE. The effect of gender and the neurotrophin, BDNF, upon methamphetamine-induced neurotoxicity of the nigrostriatal dopaminergic system in mice. Neurosci. Lett. 2004;359:135–138. doi: 10.1016/j.neulet.2004.01.061. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Mickley KR. Gender differences in modulatory effects of tamoxifen upon the nigrostriatal dopaminergic system. Pharmacol. Biochem. Behav. 2005;80:27–33. doi: 10.1016/j.pbb.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Tweed C, Anderson LI, Laping NJ. Gender differences in methamphetamine-induced mRNA associated with neurodegeneration in the mouse nigrostriatal dopaminergic system. Neuroendocrinology. 2003;77:232–238. doi: 10.1159/000070278. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc. Natl Acad. Sci. USA. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc. Natl Acad. Sci. USA. 1997;94:4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl Acad. Sci. USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, Ogawa M. Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 2004;186:134–144. doi: 10.1016/j.expneurol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Hirata H, Ladenheim B, Carlson E, Epstein C, Cadet JL. Autoradiographic evidence for methamphetamine-induced striatal dopaminergic loss in mouse brain: attenuation in CuZn-superoxide dismutase transgenic mice. Brain Res. 1996;714:95–103. doi: 10.1016/0006-8993(95)01502-7. [DOI] [PubMed] [Google Scholar]
- Huang D, Shi FD, Jung S, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Choi DH, Block ML, et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007;21:179–187. doi: 10.1096/fj.06-5865com. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer EG, McGeer PL. Therapeutic approaches to inflammation in neurodegenerative disease. Curr. Opin. Neurol. 2007;20:351–357. doi: 10.1097/WCO.0b013e3280adc943. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp. Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB. Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J. Immunol. 1999;163:1628–1635. [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl Acad. Sci. USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, et al. Microglia in the adult brain arise from Ly-6C(hi)CCR2(+) monocytes only under defined host conditions. Nat. Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Miller DB, O'Callaghan JP. The role of temperature, stress, and other factors in the neurotoxicity of the substituted amphetamines 3,4-methylenedioxymethamphetamine and fenfluramine. Mol. Neurobiol. 1995;11:177–192. doi: 10.1007/BF02740694. [DOI] [PubMed] [Google Scholar]
- Miller DB, Ali SF, O'Callaghan JP, Laws SC. The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann. N. Y. Acad. Sci. 1998;844:153–165. [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Oka T, Oka K, Kobayashi T, Sugimoto Y, Ichikawa A, Ushikubi F, Narumiya S, Saper CB. Characteristics of thermo-regulatory and febrile responses in mice deficient in prostaglandin EP1 and EP3 receptors. J. Physiol. 2003;551:945–954. doi: 10.1113/jphysiol.2003.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. `Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Priller J, Flugel A, Wehner T, et al. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Microgliosis: the questions shape the answers. Nat. Neurosci. 2007;10:1507–1509. doi: 10.1038/nn1207-1507. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738:172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O'Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J. Neurochem. 2006;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J. Histochem. Cytochem. 1990;38:1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and Alzheimer's disease pathogenesis. J. Neurosci. Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. Attenuated microglial activation mediates tolerance to the neurotoxic effects of methamphetamine. J. Neurochem. 2005a;92:790–797. doi: 10.1111/j.1471-4159.2004.02906.x. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005b;1050:190–198. doi: 10.1016/j.brainres.2005.05.049. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Liu X, Kuhn DM. Identification of differentially regulated transcripts in mouse striatum following methamphetamine treatment-an oligo-nucleotide microarray approach. J. Neurochem. 2004a;88:380–393. doi: 10.1046/j.1471-4159.2003.02182.x. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J. Pharmacol. Exp. Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Vallieres L, Sawchenko PE. Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J. Neurosci. 2003;23:5197–5207. doi: 10.1523/JNEUROSCI.23-12-05197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]