

Summary
Cell death plays an important role both in shaping the developing nervous system and in neurological disease and traumatic injury. In spite of their name, death receptors can trigger either cell death or survival and growth. Recent studies implicate five death receptors — Fas/CD95, TNFR1 (tumor necrosis factor receptor-1), p75NTR (p75 neurotrophin receptor), DR4 and DR5 (death receptors-4 and -5) — in different aspects of neural development or degeneration. Their roles may be neuroprotective in models of Parkinson’s disease, or pro-apoptotic in ALS and stroke. Such different outcomes likely reflect the diversity of transcriptional and post-translational signaling pathways downstream of death receptors in neurons and glia.
Introduction
The term “death receptor” refers to a group of eight members of the TNFR superfamily of single-pass transmembrane proteins [1]. The death receptors are the only members of this family whose cytoplasmic tail contains a conserved 80-amino acid sequence referred to as the “death domain”. In the appropriate cellular context, activation of a death receptor leads to formation of a death-inducing signaling complex (DISC) at the death domain and subsequent triggering of downstream cascades leading to death of the cell which expresses the receptor [1,2•]. Physiologically, death receptor activation can occur in cis, by transmembrane ligand expressed in the same cell as the receptor, or in trans, by ligand expressed at the surface of other cells or released into the extracellular milieu.
This review will focus on five death receptors and their ligands for which new roles in the nervous system have been reported over the last two years. FasL/CD95L triggers cell death through its receptor Fas/CD95 by recruiting the adapter protein FADD (Fas-associated death domain) and pro-caspase-8. This ligand-receptor couple will be referred to simply as FasL and Fas in the body of the review. TNFα (tumor necrosis factor alpha) acts as a soluble homotrimer through two TNFR family members: TNFR1, which has a death domain, and TNFR2, which does not. Death signaling by TNFR1 involves recruitment of adapter signaling molecules TRADD (TNFR-associated death domain), FADD and RIP (receptor interacting protein). Death receptors-4 and -5 (DR4 and DR5) share the same ligand - TRAIL (TNF-related apoptosis inducing ligand) - and signal similarly to Fas. Lastly, p75NTR can signal cell death when bound by NGF (nerve growth factor) or other ligands. Like the other 30 members of the TNFR family, death receptors also play multiple roles in cell differentiation, growth and proliferation [2•]. We will highlight examples of these as a reminder that expression of a death receptor in a given tissue does not necessarily imply a role in degeneration or death.
Death receptors in nervous system development
Death receptors and their ligands are expressed in the developing nervous system, and many recent reports confirm their ability to trigger death of embryonic neurons and glia in vitro [3-15]. However, with the exception of earlier reports concerning p75NTR, there is still relatively little evidence that death receptors play a role in nervous system development in vivo. This may be because they mostly regulate phenomena other than cell death at this stage. The spontaneous mouse mutants lpr and gld, which are partial loss-of-function mutants for Fas and FasL, respectively, have normal numbers of cortical, hippocampal and motor neurons at birth [16,17•]. However, they do show reduced dendritic branching of hippocampal and cortical neurons during the period at which synaptogenesis is occurring [17•]. At similar stages, mice deficient in TNFα, which is normally produced by glia, show defects in synaptic scaling and ocular dominance plasticity in the visual system [18,19]. However, a full understanding of the overall role of death receptors in nervous system development will require a more systematic approach in null mutant mice.
Involvement of death receptors in disease mechanism and therapeutic strategies in ALS
There is now a considerable body of data pointing to a role for death receptors in pathological situations. This provides new opportunities for therapeutic intervention, focused on the death receptors themselves, their ligands or intracellular relays. Many pharmacological compounds, recombinant proteins and genetic strategies have therefore been designed and tested in rodent models of disease. Recent reports are summarized in Table 1.
Table 1. Pharmacological and genetic interventions in death signalling pathways in the nervous system in vivo.
The Table summarizes recent in vivo studies targeting the nervous system, grouping them by target — ligand, receptor or effector — and by the pathology that they most closely model. Genetic gain- or loss-of-function or pharmacological approaches have provided important validation (or invalidation) of mechanisms identified in vitro, and can also serve as proof-of-principle for subsequent therapeutic strategies. The principal outcomes of these interventions are described in the text. Abbreviations: MPTP (1-methyl-4-phenylpyridinium), SNA (sciatic nerve avulsion), (MCAO (middle cerebral artery occlusion), lpr (lymphoproliferative), gld (generalized lymphoproliferative disease), JEV (Japanese Encephalitis Virus).
| Molecule/Gene | Pathology | Model | Approach | Delivery | Reference | |
|---|---|---|---|---|---|---|
| Ligand | ||||||
| TNFa | TNFa | ALS | SOD1 G93A, G37R mice | gene KO | [26•] | |
| TNFa | Parkinson | MPTP neurotoxicity | gene KO | [50] | ||
| TNFa | synaptic scaling | Neuroplasticity | gene KO | [18,19] | ||
| TNFa binding protein |
ALS | wobbler mice | recombinant protein | subcutaneous | [27] | |
| TNFa and FasL | Thalidomide, lenalidomide |
ALS | SOD1 G93A mice | pharmacological | oral | [29•] |
| MMP-9 | ALS | SOD1 G93A mice | gene KO | [37,43] | ||
| TNFa & IFNγ | IFNγ receptor type 1 |
Alzheimer | APP mice | gene KO | [44] | |
| FasL | FasL | ALS | SOD1 G93A mice | gld mutation | [40] | |
| FasL | motor neuron injury |
SNA | gld mutation | [46] | ||
| FasL | branching | Neural development |
gld and lpr mutations |
[17•] | ||
| TIMP-3 | stroke | MCAO | gene KO | [15] | ||
| soluble Fas receptor |
spinal cord injury | recombinant protein | intrathecal | [45] | ||
| Receptor complex | ||||||
| TNFR | TNFR1/2 | Parkinson | MPTP neurotoxicity | gene KO | [56] | |
| TNFR1/2 | neuronal injury | Trimethyltin | gene KO | [55•] | ||
| TRADD siRNA | Encephalitis | JEV virus | gene silencing | intracerebral | [13] | |
| Fas | Fas siRNA | ALS | SOD1 G93A mice | gene silencing | intrathecal | [36•] |
| FLIP(L) | stroke | MCAO | transgenic expression |
[47] | ||
| Fas | motor neuron injury |
Axotomy | lpr mutation | [16] | ||
| Fas | Parkinson | MPTP neurotoxicity |
lpr and gld mutations |
[49] | ||
| FADD? | Lithium | ALS | SOD1 G93A mice, human patients |
pharmacological | oral, intraperitoneal |
[39•,41•] |
| p75NTR | p75NTR | Alzheimer | Aβ neurotoxicity | gene KO | [12]. | |
| Effector | ||||||
| DAXX | Daxx dominant negative |
ALS | SOD1 G93A mice | transgenic expression |
[9, 10] | |
| NO synthase | nNOS, iNOS | motor neuron injury |
SNA | gene KO | [46] | |
| Free radicals | Neu2000 | ALS | SOD1 G93A mice | pharmacological | oral | [39•] |
Currently the best evidence of a role for death receptors in neurological diseases comes from the study of amyotrophic lateral sclerosis (ALS). In patients with ALS, degeneration and death of motor neurons in the spinal cord lead to progressive and fatal muscle paralysis. Much of our understanding of the underlying mechanisms comes from studies of transgenic mice overexpressing toxic mutant forms of SOD1 (Cu, Zn-superoxide dismutase-1), which cause ALS in a subset of patients with a familial form of the disease. Mutant SOD1 triggers neurodegeneration in a cell-autonomous fashion in motor neurons, but also drives disease progression through non-autonomous mechanisms involving microglia and astrocytes [20-22]. Three death receptors — TNFα, p75NTR and Fas/CD95 — have been implicated in cell autonomous and/or non-autonomous aspects of the disease.
Upregulation of TNFα and TNF receptors is observed in parallel with the appearance of symptoms in SOD1 mice [23,24] and increased levels of TNFα are found in plasma of ALS patients [25]. However, genetic ablation of TNFα has no beneficial effect on survival, motor axon degeneration or gliosis in either of two different lines of mutant SOD1 mice [26•]. Although application of recombinant TNF-binding protein-1 (rhTBP-1) had protective effects in wobbler mice - another model of motor neuron degeneration - the treatment did not in fact reduce TNFα levels in the CNS [27]. The beneficial effects of treatments as diverse as thalidomide, folic acid, insulin-like growth factor-1, and metal chelators are all correlated in SOD1 mice with reductions in TNFα levels [28-31]. However, it does not seem likely that they all act through TNFα/TNFR directly to prevent degeneration; the reductions more probably reflect a decrease in inflammation secondary to neuroprotection by other mechanisms. Given the lack of evidence for a direct role in motor neuron death, an interesting possibility is that TNFα may trigger the redistribution of axonal mitochondria known to occur in ALS motor neurons in vivo [32].
Reactive astrocytes in the spinal cord of SOD1 mice express NGF and can trigger motor neuron death in vitro in a p75NTR-dependent manner [33,34]. Moreover, NGF acting through p75NTR in the absence of nitric oxide triggers the death of cultured motor neurons from SOD1 mice, but not from controls [8]. However, earlier pharmacological studies produced conflicting results as to the role of p75NTR in disease progression in vivo. Genetic testing of the potential role of p75NTR in ALS mice is therefore a crucial next step.
Probably the strongest evidence for death receptor involvement in ALS concerns Fas/CD95. Motor neurons can be triggered to die in vitro by activation of a motor neuron-specific pathway downstream of Fas (called the Fas/NO pathway), which includes an amplification feedback loop Fas-NO-FasL-Fas [10•,35]. Motor neurons cultured from SOD1 mutant embryos are hyper-sensitive to Fas activation [10•,35,36•]. In presymptomatic SOD1 mice in vivo, activation or misexpression of all intermediates in the Fas/NO pathway, together with enhancement of Fas-FADD interactions, is observed [10•,24,36•-39•]. Importantly, therapeutic benefit has been observed following inhibition of Fas/FasL function. Intrathecal infusion of siRNAs against Fas in mutant SOD1 mice almost completely blocks the pathological activation of caspase-8, p38 and other elements of the Fas/NO pathway, protects motor neurons and prolongs lifespan [36•]. Partial loss-of-function mutations for FasL confer a modest extension of survival in the same mice [40]. It will be important to repeat these experiments using true null mutants for Fas and FasL. Intriguingly, lithium, a drug widely used in mood disorders, was also reported to block the activation of Fas, FADD and caspases-8 and -3 [39•] and to provide neuroprotection, not only in mutant SOD1 mice [39•,41•] but also in a small cohort of ALS patients [41•]. It remains to be determined whether these data can be replicated in larger controlled studies and whether lithium acts by inhibiting apoptosis or by activating autophagy [41•].
More indirect evidence of a role for Fas comes from studies of the matrix metalloproteinases (MMPs), which are involved in the proteolytic processing of TNFα and FasL from precursor to soluble forms. Inhibition of MMPs with the broad spectrum inhibitor Ro28-2653 had some positive effects in mutant SOD1 mice [42]. However, genetic knockout of MMP-9 attenuated disease in one study [37] but exacerbated symptoms in another [43]. Tissue inhibitors of metalloproteinases can signal through MMP inhibition to enhance stabilization and activation of death receptors such as Fas. One member of this family, TIMP-3, is strongly upregulated in degenerating motor neurons in spinal cords of SOD1 mice [38].
Overall, therefore, there is intriguing presumptive evidence that Fas signaling may play a role in ALS, but more data from genetic studies in mice and human patients are required to support this hypothesis.
Death receptors in other diseases and degenerative processes in the nervous system
In models of Alzheimer’s disease, death receptor signaling may potentially play a role both upstream and downstream of accumulation of the Aβ (amyloid-beta) peptide. TNFα stimulates expression of BACE1 (beta-site amyloid precursor protein-cleaving enzyme-1) and consequently production of Aβ [44]. In turn, cell death of septohippocampal neurons induced by Aβ in vitro and in vivo requires the presence of p75NTR [12]. Data from several different models of spinal cord injury, axotomy, deafferentation and ischemia provide strong evidence that Fas activation has a deleterious effect on survival of injured neurons [15,16,45-48]. In contrast, in the MPTP model of Parkinson’s disease, Fas activation protects dopaminergic neurons from MPTP toxicity, providing the first in vivo evidence that death receptors can act to enhance neuronal survival [49]. In the same MPTP model, TNFα has opposite effects depending on the cell context: it is protective for hippocampal neurons but triggers degeneration of dopaminergic axons [50]. These results exemplify the importance of cellular context in determining whether death receptor signaling is pro- or anti-apoptotic.
Signaling mechanisms downstream of death receptors in neural systems
The generality of the classical model involving activation of caspases and mitochondrial pro-apoptotic factors through formation of a DISC is currently questioned [2•]. It has been particularly challenged by two sets of results in the nervous system: (a) demonstrated requirements for novel signaling and transcriptional events in the execution of neuronal death; and (b) unexpected outcomes of death receptor activation such as cell survival and neurite outgrowth [2•]. These are summarized for dying and healthy neurons and glia in Figure 1.
Figure 1. Death receptor signaling pathways in neurons and glia.
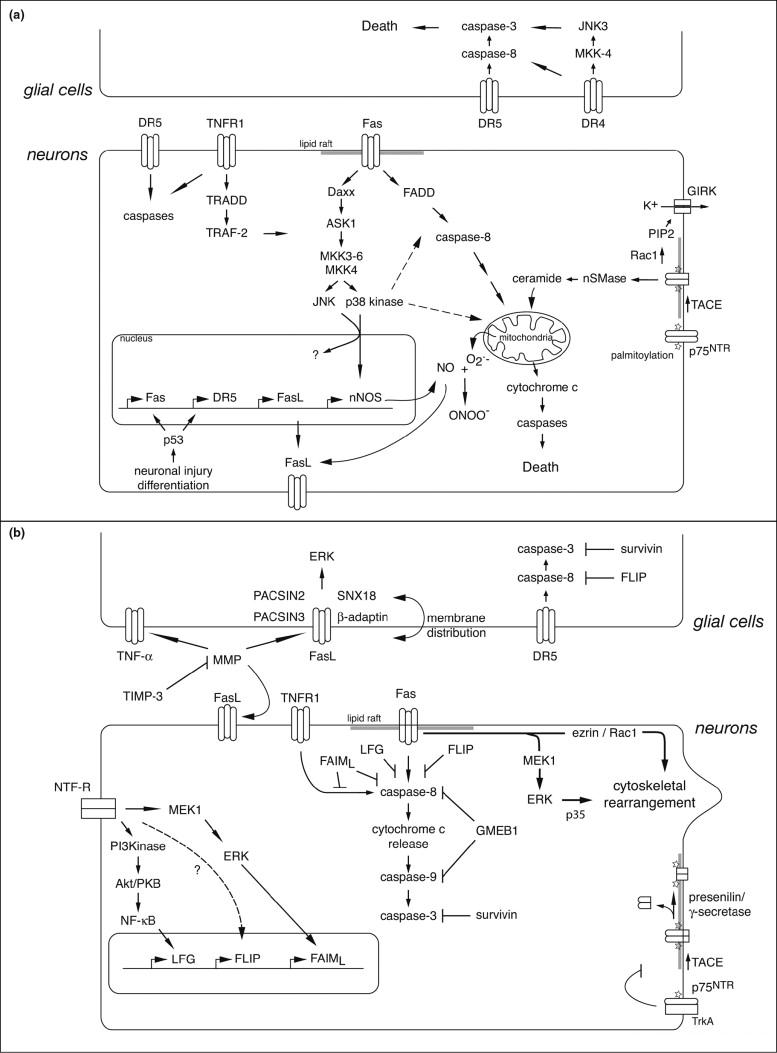
The two panels summarize novel aspects of death receptor signaling reviewed in the text. (A) Signaling events activated during cell death or degeneration. Once activated by their ligands (not shown, for clarity), death receptors cluster into multi-molecular assemblies in lipid rafts and trigger a series of post-translational and transcriptional events leading to caspase activation downstream of mitochondrial decision points. (B) Signaling events activated during cell survival or growth. Multiple control mechanisms act coordinately to prevent death receptor activation and inhibit downstream signaling. In addition, death receptors can stimulate signaling pathways directly associated with growth or survival.
Involvement of stress-activated protein (SAP) kinases represents a first layer of complexity superimposed on the prototypic death receptor pathway. Depending on the cellular context, different SAP kinases are involved. To kill motor neurons, Fas needs to selectively activate p38 kinase but not JNK (c-Jun N-terminal kinase) [35]. This in turn leads to obligate transcriptional upregulation of neuronal nitric oxide synthase [10•]. DR4 acts through JNK3 but not p38 kinase to trigger oligodendrocyte apoptosis [6], whereas TNFR1 in neuroblastoma cells signals through both JNK and p38 kinase [13]. p75NTR death signalling also shows cell type-dependent complexities. Production of ceramide by neutral sphingomyelinase is an obligatory step for caspase activation in NGF-treated motor neurons [8] whereas in sensory neurons, p75NTR elicits potassium efflux through phosphatidylinositol-4,5-biphosphate-dependent activation of G protein-coupled inward rectifier potassium (GIRK) channels [4•].
In spite of their name, death receptors can activate non-apoptotic pathways in appropriate cellular contexts, and are indeed required for cell survival and tissue regeneration in several systems [2•,49]. Fas-induced branching of cortical and hippocampal and sensory neurons does not require caspase activation [17•]. In agreement with this, Fas-triggered axonal outgrowth involves activation of the MEK1/ERK1/p35 pathway in sensory neurons and of the Ezrin-Rac1 pathway in cortical neurons [51].
Control of death receptor signaling can occur at multiple levels. In neurons, both Fas and TNFR localize to lipid rafts, specialized microdomains of the plasma membrane, and such localization seems to be required for death signaling [5]. Neuronal cell death induced by p75NTR requires palmitoylation, translocation to lipid rafts and metalloproteinase cleavage of the extracellular domain [14]. In agreement with the emerging importance of subcellular compartmentalization of death signaling pathways, proteomic analysis of reverse signalling through FasL reveals interactors associated with endocytosis and trafficking [52]. There are diverse alternative mechanisms for control of the outcome of death receptor activation. Regulation of Fas or TRAIL-R2/DR5 can occur at the transcriptional level in a p53-dependent manner [15,37,46]. In cancer cells, activation of DR4 and DR5 is regulated by O-glycosylation and by decoy receptors [1,53]. Intracellular inhibitory or decoy proteins such as FLIP (FLICE inhibitory protein), LFG (lifeguard), FAIML (Fas apoptosis inhibitory molecule, long form), survivin or GMEB1 (glucocorticoid modulatory element binding protein-1) can prevent caspase activation downstream of death receptors and thereby confer resistance to death signals, providing a novel mode of action for neurotrophic factors [3,5,7,11•,54].
Conclusions
Our vision of death receptor signaling has radically changed over the last decade, as the many data pointing to “positive” signaling outcomes such as growth or survival have been progressively taken into account. Cellular context strongly affects the choice of outcome in the nervous system but potential molecular mechanisms to explain this — alternative co-receptors, interaction with other signaling pathways — need to be further investigated. Nevertheless, the classical role of death receptors as triggers of degeneration and cell death seems to come into play in several forms of neurodegeneration, particularly ALS and stroke. New genetic and pharmacological tools will allow these hypotheses to be more fully tested in vivo.
Acknowledgements
Work in the authors’ laboratories is supported by Spinal Muscular Atrophy Foundation, Claire and Leonard Tow Charitable Foundation, Project A.L.S., Wings over Wall Street, New York State Office of Science Technology and Academic Research, National Institutes of Neurological Disease and Stroke (C.E.H.); Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Association Française contre les Myopathies and European Union Network of Excellence “NeuroNE” (G.H., B.P., CR); Région PACA and Agence Nationale pour la Recherche (G.H.); and ALS Association (C.R, B.P, C.E.H.).
Abbreviations and references in which they are mentioned
- ASK-1
- ERK
extracellular signal-regulated kinase [6]
- FADD
- FAIM
Fas apoptotic inhibitory molecule [11•]
- GIRK
G-protein-coupled inwardly rectifying potassium [4•]
- GMEB1
glucocorticoid modulatory element-binding protein 1 [54]
- JNK
- LFG
- MEKK1
mitogen-activated protein kinase/ERK kinase 1(MEK1) kinase
- MKK
- MMP
- nNOS
- nSMase
neutral sphingomyelinase [8]
- NTF-R
neurotrophic factor receptors
- PACSIN
protein kinase C and casein kinase substrate in neurons [52]
- SNX18
sorting nexin 18 [52]
- PIP2
phosphatidylinositol 4,5 biphosphate [4•]
- TACE
TNFα converting enzyme
- TIMP
- TRADD
tumor necrosis factor receptor-associated death domain [13]
- TRAF
- TRAIL-R
TNF-related apoptosis-inducing ligand (TRAIL) receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References and annotations
- 1.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 2•.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, et al. The CD95 receptor: apoptosis revisited. Cell. 2007:129–447. doi: 10.1016/j.cell.2007.04.031.This collaborative review summarizes well the changes in thinking in the field about the relative weight to be given to the pro- and anti-apoptotic functions of death receptors. It focuses on the prototypic death receptor Fas/CD95, but many of its conclusions are undoubtedly relevant for other family members.
- 3.Beier CP, Wischhusen J, Gleichmann M, Gerhardt E, Pekanovic A, Krueger A, Taylor V, Suter U, Krammer PH, Endres M, et al. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci. 2005;25:6765–6774. doi: 10.1523/JNEUROSCI.1700-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Coulson EJ, May LM, Osborne SL, Reid K, Underwood CK, Meunier FA, Bartlett PF, Sah P. p75 neurotrophin receptor mediates neuronal cell death by activating GIRK channels through phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2008;28:315–324. doi: 10.1523/JNEUROSCI.2699-07.2008.Using cultured sensory neurons and HEK293 cells, the authors show that overexpression of p75NTR or a C-terminal fragment triggers direct activation of inwardly rectifying potassium channels by PIP2. These events, which are G protein-independent, lead to efflux of potassium ions, caspase activation and death. This result ties in well with earlier studies that demonstrated a role for K+ efflux in neuronal cell death.
- 5.Fernandez M, Segura MF, Sole C, Colino A, Comella JX, Cena V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem. 2007;103:190–203. doi: 10.1111/j.1471-4159.2007.04767.x. [DOI] [PubMed] [Google Scholar]
- 6.Jurewicz A, Matysiak M, Andrzejak S, Selmaj K. TRAIL-induced death of human adult oligodendrocytes is mediated by JNK pathway. Glia. 2006;53:158–166. doi: 10.1002/glia.20249. [DOI] [PubMed] [Google Scholar]
- 7.Murata T, Tsuboi M, Hikita K, Kaneda N. Protective effects of neurotrophic factors on tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of murine adrenal chromaffin cell line tsAM5D. J Biol Chem. 2006;281:22503–22516. doi: 10.1074/jbc.M602579200. [DOI] [PubMed] [Google Scholar]
- 8.Pehar M, Vargas MR, Robinson KM, Cassina P, Diaz-Amarilla PJ, Hagen TM, Radi R, Barbeito L, Beckman JS. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–7785. doi: 10.1523/JNEUROSCI.0823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raoul C, Barthelemy C, Couzinet A, Hancock D, Pettmann B, Hueber AO. Expression of a dominant negative form of Daxx in vivo rescues motoneurons from Fas (CD95)-induced cell death. J Neurobiol. 2005;62:178–188. doi: 10.1002/neu.20086. [DOI] [PubMed] [Google Scholar]
- 10•.Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B, Henderson CE, Haase G. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci USA. 2006;103:6007–6012. doi: 10.1073/pnas.0508774103.The article demonstrates the existence of a feedback loop through which nitric oxide, a product of Fas activation in motor neurons, leads to upregulation of FasL and therefore further activation of the Fas pathway. Evidence for early activation of several intermediates in this feedback loop has been found in presymptomatic ALS model mice. It is tempting to speculate that low-level activation of such processes leads to a build-up of toxic intermediates that is only gradual, and which could therefore explain the late onset of ALS in patients and animal models.
- 11•.Segura MF, Sole C, Pascual M, Moubarak RS, Perez-Garcia MJ, Gozzelino R, Iglesias V, Badiola N, Bayascas JR, Llecha N, et al. The long form of Fas apoptotic inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J Neurosci. 2007;27:11228–11241. doi: 10.1523/JNEUROSCI.3462-07.2007.This study uses a combination of developmental expression studies, biochemical analysis of death receptor-adapter interactions and gain- and loss-of-function studies in cortical and motor neurons. The authors make a clear case for FAIML as an endogenous modulator of death receptor signaling in neurons
- 12.Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–3946. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swarup V, Das S, Ghosh S, Basu A. Tumor necrosis factor receptor-1-induced neuronal death by TRADD contributes to the pathogenesis of Japanese encephalitis. J Neurochem. 2007;103:771–783. doi: 10.1111/j.1471-4159.2007.04790.x. [DOI] [PubMed] [Google Scholar]
- 14.Underwood CK, Reid K, May LM, Bartlett PF, Coulson EJ. Palmitoylation of the C-terminal fragment of p75(NTR) regulates death signaling and is required for subsequent cleavage by gamma-secretase. Mol Cell Neurosci. 2008;37:346–358. doi: 10.1016/j.mcn.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA, Khokha R, Cunningham LA. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ. 2008;15:143–151. doi: 10.1038/sj.cdd.4402246. [DOI] [PubMed] [Google Scholar]
- 16.Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaun D, Henderson CE, Kato AC, Pettmann B, Hueber AO. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J Neurosci. 2003;23:8526–8531. doi: 10.1523/JNEUROSCI.23-24-08526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Zuliani C, Kleber S, Klussmann S, Wenger T, Kenzelmann M, Schreglmann N, Martinez A, del Rio JA, Soriano E, Vodrazka P, et al. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell Death Differ. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720.This is one of only a few studies to provide clear in vivo loss-of-function data for nonapoptotic roles for death receptors, in this case Fas/CD95. The results point to a role for Fas in axonal and dendritic branching and provide an intriguing potential explanation for the behavioral deficits observed in animals and patients with mutations in the fas/cd95 gene.
- 18.Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 20•.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511.See annotation after reference 33•.
- 21•.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885.See annotation after reference 33•.
- 22•.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876.See annotation after reference 33•.
- 23.Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:2075–2087. doi: 10.1089/ars.2006.8.2075. [DOI] [PubMed] [Google Scholar]
- 24.Veglianese P, Lo Coco D, Cutrona M Bao, Magnoni R, Pennacchini D, Pozzi B, Gowing G, Julien JP, Tortarolo M, Bendotti C. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci. 2006;31:218–231. doi: 10.1016/j.mcn.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Cereda C, Baiocchi C, Bongioanni P, Cova E, Guareschi S, Metelli MR, Rossi B, Sbalsi I, Cuccia MC, Ceroni M. TNF and sTNFR1/2 plasma levels in ALS patients. J Neuroimmunol. 2008;194:123–131. doi: 10.1016/j.jneuroim.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 26•.Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006.Although the results could be classified as negative, this study provides a clear demonstration of the power of mouse genetics in vivo to support (or in this case weaken) hypotheses developed in vitro. In the present case, the absence of TNFa does not affect the ALS-like disease process in SOD1 mice, arguing against a role for TNFa either as a death inducer for motor neurons or as a pro-inflammatory agent.
- 27.Bigini P, Repici M, Cantarella G, Fumagalli E, Barbera S, Cagnotto A, De Luigi A, Tonelli R, Bernardini R, Borsello T, et al. Recombinant human TNF-binding protein-1 (rhTBP-1) treatment delays both symptoms progression and motor neuron loss in the wobbler mouse. Neurobiol Dis. 2008;29:465–476. doi: 10.1016/j.nbd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Dodge JC, Haidet AM, Yang W, Passini MA, Hester M, Clarke J, Roskelley EM, Treleaven CM, Rizo L, Martin H, et al. Delivery of AAV-IGF-1 to the CNS Extends Survival in ALS Mice Through Modification of Aberrant Glial Cell Activity. Mol Ther. 2008 doi: 10.1038/mt.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006.The authors show that thalidomide and its analog lenalidomide are both capable of inhibiting the expression of TNFα and FasL in motor neurons of SOD1 mutant mice. They also confer quite significant benefit in terms of lifespan of the ALS mice. It remains to be determined whether these two observations are linked, or whether thalidomide is acting through independent mechanisms to promote survival.
- 30.Petri S, Calingasan NY, Alsaied OA, Wille E, Kiaei M, Friedman JE, Baranova O, Chavez JC, Beal MF. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2007;102:991–1000. doi: 10.1111/j.1471-4159.2007.04604.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Chen S, Li L, Wang Q, Le W. Folic acid protects motor neurons against the increased homocysteine, inflammation and apoptosis in SOD1 G93A transgenic mice. Neuropharmacology. 2008;54:1112–1119. doi: 10.1016/j.neuropharm.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 32.Stommel EW, van Hoff RM, Graber DJ, Bercury KK, Langford GM, Harris BT. Tumor necrosis factor-alpha induces changes in mitochondrial cellular distribution in motor neurons. Neuroscience. 2007;146:1013–1019. doi: 10.1016/j.neuroscience.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 33•.Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x.Although not focused directly on death receptors, this paper and references 20•, 21• and 22• are significant because they clearly demonstrate the concept of active killing of motor neurons by glia in the mutant SOD1 mouse model of familial ALS.
- 34.Domeniconi M, Hempstead BL, Chao MV. Pro-NGF secreted by astrocytes promotes motor neuron cell death. Mol Cell Neurosci. 2007;34:271–279. doi: 10.1016/j.mcn.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 36•.Locatelli F, Corti S, Papadimitriou D, Fortunato F, Del Bo R, Donadoni C, Nizzardo M, Nardini M, Salani S, Ghezzi S, et al. Fas small interfering RNA reduces motoneuron death in amyotrophic lateral sclerosis mice. Ann Neurol. 2007;62:81–92. doi: 10.1002/ana.21152.The authors use intrathecal micropumps to administer siRNA to Fas to mutant SOD1 mice. They observe not only a complete return of the Fas/NO pathway to normal activation levels but also significant neuroprotection of motor neurons and prolongation of lifespan. The effects are particularly striking given that Fas siRNA was only administered for a limited time. These results are currently the strongest evidence for a role of Fas signaling in disease onset and progression in ALS. They will need to be confirmed using similar approaches in other ALS models, and by the use of mouse genetics to fully evaluate the protective effect of Fas/FasL depletion.
- 37.Kiaei M, Kipiani K, Calingasan NY, Wille E, Chen J, Heissig B, Rafii S, Lorenzl S, Beal MF. Matrix metalloproteinase-9 regulates TNF-alpha and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2007;205:74–81. doi: 10.1016/j.expneurol.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 38.Lee JK, Shin JH, Suh J, Choi IS, Ryu KS, Gwag BJ. Tissue inhibitor of metalloproteinases-3 (TIMP-3) expression is increased during serum deprivation-induced neuronal apoptosis in vitro and in the G93A mouse model of amyotrophic lateral sclerosis: a potential modulator of Fas-mediated apoptosis. Neurobiol Dis. 2008;30:174–185. doi: 10.1016/j.nbd.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 39•.Shin JH, Cho SI, Lim HR, Lee JK, Lee YA, Noh JS, Joo IS, Kim KW, Gwag BJ. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol Pharmacol. 2007;71:965–975. doi: 10.1124/mol.106.030676.This paper provides two types of evidence for a role for Fas receptor in neurodegeneration in ALS model mice. First, the authors show increased expression in SOD mice of Fas and its downstream signaling intermediates, and report that Fas-FADD interactions are intensified at one stage of the disease. In addition, they report that lithium can prevent neurodegeneration and lead to modest increases in lifespan of SOD1 mice. Lithium also strongly inhibits the expression of Fas and FADD, though it still needs to be determined whether this underlies its neuroprotective activity.
- 40.Petri S, Kiaei M, Wille E, Calingasan NY, Beal M Flint. Loss of Fas ligand-function improves survival in G93A-transgenic ALS mice. J Neurol Sci. 2006;251:44–49. doi: 10.1016/j.jns.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 41•.Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105.The data confirm and extend the report by ref. 39• of the beneficial effects of lithium in SOD1 mice. Strikingly, the authors also report significant benefit in a small cohort of patients with ALS. This article has stimulated considerable interest and some debate, and studies attempting to confirm its results in larger trials are currently underway. The authors favor the hypothesis that lithium is acting to promote autophagy; the article is cited here because of the link with Fas/FADD suggested by ref. 39•.
- 42.Lorenzl S, Narr S, Angele B, Krell HW, Gregorio J, Kiaei M, Pfister HW, Beal MF. The matrix metalloproteinases inhibitor Ro 28-2653 [correction of Ro 26-2853] extends survival in transgenic ALS mice. Exp Neurol. 2006;200:166–171. doi: 10.1016/j.expneurol.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 43.Dewil M, Schurmans C, Starckx S, Opdenakker G, Van Den Bosch L, Robberecht W. Role of matrix metalloproteinase-9 in a mouse model for amyotrophic lateral sclerosis. Neuroreport. 2005;16:321–324. doi: 10.1097/00001756-200503150-00003. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ackery A, Robins S, Fehlings MG. Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma. 2006;23:604–616. doi: 10.1089/neu.2006.23.604. [DOI] [PubMed] [Google Scholar]
- 46.Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–6459. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taoufik E, Valable S, Muller GJ, Roberts ML, Divoux D, Tinel A, Voulgari-Kokota A, Tseveleki V, Altruda F, Lassmann H, et al. FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J Neurosci. 2007;27:6633–6646. doi: 10.1523/JNEUROSCI.1091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landau AM, Luk KC, Jones ML, Siegrist-Johnstone R, Young YK, Kouassi E, Rymar VV, Dagher A, Sadikot AF, Desbarats J. Defective Fas expression exacerbates neurotoxicity in a model of Parkinson’s disease. J Exp Med. 2005;202:575–581. doi: 10.1084/jem.20050163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. Faseb J. 2006;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- 51.Ruan W, Lee CT, Desbarats J. A Novel Juxtamembrane Domain in TNF Receptor Superfamily Molecules Activates Rac1 and Controls Neurite Growth. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-02-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thornhill PB, Cohn JB, Drury G, Stanford WL, Bernstein A, Desbarats J. A proteomic screen reveals novel Fas ligand interacting proteins within nervous system Schwann cells. FEBS Lett. 2007;581:4455–4462. doi: 10.1016/j.febslet.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 53.Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 54.Tsuruma K, Nakagawa T, Morimoto N, Minami M, Hara H, Uehara T, Nomura Y. Glucocorticoid modulatory element-binding protein 1 binds to initiator procaspases and inhibits ischemia-induced apoptosis and neuronal injury. J Biol Chem. 2006;281:11397–11404. doi: 10.1074/jbc.M510597200. [DOI] [PubMed] [Google Scholar]
- 55•.Harry GJ, d’Hellencourt C Lefebvre, McPherson CA, Funk JA, Aoyama M, Wine RN. Tumor necrosis factor p55 and p75 receptors are involved in chemical-induced apoptosis of dentate granule neurons. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05382.x.This study investigates the role of TNF in neuronal damage induced by neuroinflammation. The toxin trimethyltin was known to induce a microglial response and an increase in TNFα, and is shown to induce cell death of hippocampal neurons. Genetic ablation of TNFR1 and/or TNFR2, or use of antibodies to TNFa, both provide significant protection against the effects of toxin treatment.
- 56.Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]