

Abstract
Curcumin, a phenolic compound from the curry spice turmeric, exhibits a wide range of activities in eukaryotic cells, including antiviral effects that are at present incompletely characterized. Curcumin is known to inhibit the histone acetyltransferase activity of the transcriptional coactivator proteins p300 and CBP, which are recruited to the immediate early (IE) gene promoters of herpes simplex virus type 1 (HSV-1) by the viral transactivator protein VP16. We tested the hypothesis that curcumin, by inhibiting these coactivators, would block viral infection and gene expression. In cell culture assays, curcumin significantly decreased HSV-1 infectivity and IE gene expression. Entry of viral DNA to the host cell nucleus and binding of VP16 to IE gene promoters was not affected by curcumin, but recruitment of RNA polymerase II to those promoters was significantly diminished. However, these effects were observed using lower curcumin concentrations than those required to substantially inhibit global H3 acetylation. No changes were observed in histone H3 occupancy or acetylation at viral IE gene promoters. Furthermore, p300 and CBP recruitment to IE gene promoters was not affected by the presence of curcumin. Finally, disruption of p300 expression using a short hairpin RNA did not affect viral IE gene expression. These results suggest that curcumin affects VP16-mediated recruitment of RNA polymerase II to IE gene promoters by a mechanism independent of p300/CBP histone acetyltransferase activity.
Keywords: Curcumin, chromatin, coactivator, transcriptional activation, lytic infection, VP16
Introduction
Curcumin (diferuloylmethane) is the major component of the curry spice turmeric (Curcuma longa Linn.). Curcumin can affect the metabolism of cells and organisms in a number of ways, including apoptosis, cell signaling, inflammation and carcinogenesis [reviewed in (Joe, Vijaykumar, and Lokesh, 2004; Sharma, Gescher, and Steward, 2005)]. Various antiviral effects of curcumin have been described, but the biochemical mechanisms of those effects have been incompletely defined (Bourne et al., 1999; Burke et al., 1995; Li et al., 1993; Mazumder et al., 1995; Sui et al., 1993). Curcumin reportedly inhibits transcription and replication of the human immunodeficiency virus (HIV-1) by blocking Tat-mediated transactivation or by diminishing viral protease and integrase activity (Burke et al., 1995; Li et al., 1993; Mazumder et al., 1995; Sui et al., 1993). Virions of herpes simplex virus type 2 (HSV-2) were rendered less infectious by exposure to curcumin prior to infection of HeLa cells or of mouse genitalia (Bourne et al., 1999). In some cases, the antiviral effects of curcumin arise from inhibition of a cellular process or a transcription factor. For example, curcumin has been shown to suppress transcription activation by the host protein AP-1 (Balasubramanian and Eckert, 2006) leading to diminished HTLV-1 and HPV-mediated cellular transformation (Divya and Pillai, 2006; Prusty and Das, 2005; Tomita et al., 2006).
The packaging of eukaryotic DNA in the form of chromatin presents a significant impediment to the transcriptional machinery (Li, Carey, and Workman, 2007). This barrier can be overcome by the action of coactivator proteins that typically comprise multi-protein complexes with either of two types of enzymatic activities. Some coactivators covalently modify histones by acetylation, methylation, phosphorylation, ubiquitinylation, prolyl isomerization or ADP-ribosylation (Iizuka and Smith, 2003; Jenuwein and Allis, 2001; Lo et al., 2004; Mellor, 2006; Nelson, Santos-Rosa, and Kouzarides, 2006; Turner, 2000). Other coactivators hydrolyze ATP in the process of remodeling the position of nucleosomes along DNA or in removing nucleosomes from DNA (Johnson, Adkins, and Georgel, 2005; Smith and Peterson, 2005). Curcumin inhibits the histone acetyltransferase (HAT) activity of the closely-related transcriptional coactivator proteins p300 and CBP in vitro (IC50 of 25 μM) and in vivo (Balasubramanyam et al., 2004; Marcu et al., 2006). This inhibition might disrupt expression of genes dependent on those coactivators.
Herpes simplex virus type 1 (HSV-1) is a large DNA virus with a linear double stranded genome of 152 kb. Upon entry by fusion of its envelope with the host cell plasma membrane, the viral capsid is transported to the nuclear pore and the viral genome is released into the nucleus which starts the temporally regulated gene expression cascade involving transcription of immediate-early (IE or α), early (E or β) and late (L or γ) genes. IE gene transcription is strongly stimulated by the virion-borne trans-activator VP16 (Campbell, Palfreyman, and Preston, 1984; Walker, Greaves, and O'Hare, 1993). VP16 comprises a core domain (residues 1-410) and an acidic activation domain (AD) (residues 413-490), which has often been used for creating novel transactivators in heterologous expression systems (Sadowski et al., 1988; Triezenberg, 1995). VP16 is recruited to TAATGARAT motifs on IE gene promoters through interactions of its core domain with two host proteins, HCF and Oct-1, forming the VP16-induced complex (VIC) (Wysocka and Herr, 2003). VP16, mainly by its activation domain, then interacts with various general transcription factors and recruits RNA polymerase II (RNAP II) machinery (Goodrich et al., 1993; Ingles et al., 1991; Klemm et al., 1995; Lin et al., 1991; Uesugi et al., 1997; Xiao et al., 1994). Although the HSV-1 genome has long been suggested to be non-nucleosomal during lytic infection (Leinbach and Summers, 1980; Lentine and Bachenheimer, 1990; Muggeridge and Fraser, 1986; Muller et al., 1980), we and others have recently reported that histone H3 is associated with the HSV-1 genome and that histone tail modifications such as methylation and acetylation can be detected on transcriptionally active viral promoters and ORFs (Herrera and Triezenberg, 2004; Huang et al., 2006; Kent et al., 2004). We have also shown that p300 and CBP HATs are recruited to IE gene promoters in a manner dependent on the VP16 AD (Herrera and Triezenberg, 2004), suggesting that histone acetylation by p300/CBP on the viral genome might be important for IE gene expression.
Since curcumin inhibits p300/CBP HAT activity, we wanted to test whether curcumin could act as a potential anti-herpetic compound by inhibiting IE gene expression. In this report, we show that curcumin treatment of HeLa cells at non-toxic levels slows HSV-1 replication and decreases the ability of cells to support infection by HSV-1. This effect is mediated at least in part by inhibition of IE gene expression, since curcumin reduced the recruitment of RNAP II to IE gene promoters but not to cellular control promoters. However, the concentrations of curcumin required for these effects on viral gene expression were far less than the concentrations required to substantially inhibit global H3 acetylation. Moreover, the occupancy of histone H3 on IE gene promoters and the acetylation of H3 at those promoters did not change significantly in the presence of curcumin. Furthermore, no effect on viral gene expression was observed when p300 expression was disrupted by a short hairpin RNA. These results suggest that curcumin might inhibit recruitment of RNAP II machinery through a mechanism independent of the effect of curcumin on p300 or CBP HAT activity.
Results
Curcumin diminishes HSV-1 infectivity and slows HSV-1 replication
A previous report indicated that curcumin could reduce the infectivity of HSV-2 virions when the chemical was added to virus preparations prior to infection (Bourne et al., 1999). This observation suggests that curcumin has a biochemical effect on the structure or integrity of the virion itself. Given the evidence that curcumin can also inhibit a range of cellular processes on which viral gene expression and replication might depend, we tested whether the ability of cells to support HSV-1 infection and replication was altered by the presence of curcumin. To address whether curcumin inhibits infection by HSV-1, we performed plaque assays in cells that were treated with curcumin. The presence of curcumin reduced the number of plaques on Vero cell monolayers by approximately three-fold (Fig. 1A). The plaque size, which reflects multiple rounds of infection, was also affected by curcumin. After three days, the mean plaque size in the presence of curcumin was 0.62 (± 0.10) mm, whereas the mean plaque size in untreated cells was 0.73 (± 0.13) mm. The plaque size distributions in treated and untreated cells (Fig. 1B) are significantly different (p ≤ 0.00005 by Wilcoxon rank-sum test). The plaque morphology was also different in treated and untreated cells (Fig. 1C). In curcumin-treated cells, although the cytopathic effects of late-stage HSV infection were obvious in the middle of the plaque, the plaque was not cleared as in untreated cells. This effect might also be an indication that the lytic infectious cycle is slowed in the presence of curcumin. These results suggest that curcumin reduces the ability of cells to support HSV infection, and that curcumin slows but does not totally block HSV-1 replication.
Figure 1. Effects of curcumin on HSV-1 plaque formation and viral replication.
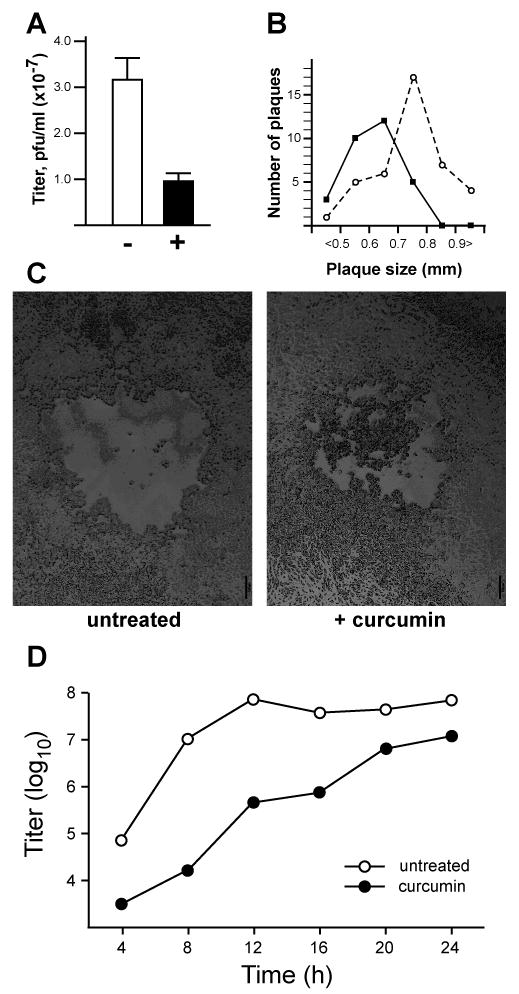
(A) Plaque assays were performed in Vero cells pretreated (+) with 10 μM curcumin or untreated (-) for 2 hours and infected with serial dilutions of a stock preparation of HSV-1. 10 μM curcumin was also present in the viral inoculums and overlay agar media of treated samples. Error bars represent the standard deviation among triplicate assays at two viral dilutions. (B) Plaque sizes in the absence (open circles) or presence (closed circles) of curcumin were measured by averaging randomly chosen vertical and horizontal diameters from plaques. The numbers of plaques within various size ranges (from less than 0.5 mm to greater than 0.9 mm) are shown. (C) Representative plaques from plaque assays performed in the absence (left panel) and presence (right panel) of 10 μM curcumin. Scale bar: 100 μm. (D) Single-step growth curves were performed in Vero cells infected with HSV-1 at an MOI of 1 pfu/cell. Virus titers in samples collected at 4 h intervals were assayed in triplicate on Vero cells. Closed circles represent curcumin-treated samples; open circles represent parallel cultures of untreated cells.
We then tested further the effect of curcumin on viral replication kinetics and production of infectious virions. Fig. 1D shows the results of a single-step growth curve assay indicating that production of infectious virus was diminished in cells treated with curcumin. In mock-treated Vero cells, production of infectious progeny virus reached a maximum of approximately 108 plaque-forming units per ml (pfu/ml) by 12 hours post-infection (hpi). In the presence of curcumin, progeny virus titers were lower throughout the infection period, but did increase steadily to a maximum of approximately 107 pfu/ml. We conclude that treatment of cells with curcumin decreases the ability of cells to support HSV-1 infection and slows but does not block the viral replication cycle.
Curcumin inhibits IE gene expression without affecting the delivery of the viral genome to the nucleus
We have previously reported that the transcriptional coactivator and histone acetyltransferase proteins p300 and CBP are recruited to IE gene promoters in a manner dependent on the VP16 transcriptional activation domain (Herrera and Triezenberg, 2004). Curcumin can inhibit the HAT activity of p300 and CBP but not of other mammalian coactivators such as PCAF (Balasubramanyam et al., 2004). We hypothesized that if the HAT activities of p300 and CBP are important for IE gene expression and if curcumin inhibits that activity, then curcumin would inhibit IE gene expression. Indeed, in HeLa cells treated with nontoxic levels of curcumin and infected at a multiplicity of 1 pfu/cell, the steady-state mRNA levels for the IE genes ICP4 and ICP27 were significantly reduced (Fig. 2A), whereas levels of CBP transcripts (Fig.2A) and p300 transcripts (data not shown) were unchanged. This result suggests that curcumin inhibits VP16-dependent transcription activation, potentially by inhibiting p300/CBP HAT activity. Since curcumin accumulates in various cellular membranes (Jaruga et al., 1998), we were concerned that it might interfere with viral entry or delivery of the viral genome to the nucleus, which would result in diminished IE gene expression. This concern was alleviated by assaying viral DNA levels in the nuclear fractions of curcumin-treated and mock-treated cells. Quantitative PCR results shown in Fig. 2B show no difference in viral DNA levels between treated and untreated cells. We conclude that curcumin treatment does not disrupt the delivery of viral genomes to the infected cell nucleus, and infer that curcumin has a more direct role in inhibiting IE gene transcription.
Figure 2. Effects of curcumin on HSV-1 IE gene expression.

(A) HeLa cells were pretreated with 20 μM curcumin (+, black bars) or DMSO only (-, open bars) for 3 hours and then infected with HSV-1 at an MOI of 1 pfu/cell. Total RNA was isolated at 2 hpi and gene expression was analyzed by Q-RT-PCR, normalized against 18S rRNA levels. Mean values from three experiments for each gene tested are shown relative to values obtained from untreated cells; error bars represent standard deviations. (B) HeLa cells were infected with HSV-1 in the presence (+, black bars) or absence (-, open bars) of 20 μM curcumin. Aliquots of nuclear extracts (as used in ChIP assays, Fig. 3) were analyzed by Q-PCR. The amount of viral DNA (represented by the ICP27 gene promoter) was normalized against cellular DNA (represented by the U3 snRNA promoter). Error bars represent the standard deviation among five independent experiments.
Curcumin decreases RNAP II occupancy on IE gene promoters and ORFs but does not affect recruitment of VP16
One way that curcumin might block IE gene expression is by decreasing the recruitment of VP16 to IE gene promoters. Alternatively, curcumin might affect recruitment of RNAP II without affecting VP16 occupancy. To test these hypotheses, we performed chromatin immunoprecipitation (ChIP) assays to probe the presence of VP16 and RNAP II at IE genes in HeLa cells infected at an moi of 5 pfu/cell. Curcumin had no substantial effect on the recruitment of VP16 to the ICP0 or ICP27 promoters (Fig. 3A). The U3 snRNA gene promoter served as a negative control, showing that VP16 occupancy as detected by ChIP assays was specific to viral IE promoters, as previously shown (Herrera and Triezenberg, 2004). In contrast, curcumin caused a significant decrease in the occupancy of RNAP II on both the promoter and transcribed region (ORF) of ICP27 (Fig. 3B). Similar decreases in RNAP II occupancy were observed for the ICP0 gene promoter (data not shown). The modest effect of curcumin on the presence of RNAPII on the U3 snRNA promoter was not observed in other replicates of this experiment (data not shown). We conclude that curcumin decreases VP16-dependent recruitment of RNAP II and hence IE gene expression.
Figure 3. Curcumin decreases occupancy of RNA polymerase II but not of VP16 at IE genes.

HeLa cells treated with 20 μM curcumin (+, black bars) or DMSO only (-, open bars) were infected with HSV-1 at an MOI of 5 pfu/cell. Chromatin immunoprecipitation from sonicated nuclear extracts was performed using antibodies against VP16 (A) or RNA pol II (B). Samples were analyzed by Q-PCR using primers specific for the ICP0 promoter, ICP27 promoter, ICP27 ORF or the cellular U3 snRNA promoter. Aliquots of samples prior to immunoprecipitation (1%, 0.3%, 0.1%, 0.04% input) were used to create a standard curve of input DNA. This curve was used to determine the relative amounts of a given DNA fragment in immunoprecipitated or control (no antibody) samples. The values for no-antibody controls were subtracted from the values for corresponding IP samples. Error bars represent the range between technical duplicates for a representative experiment.
Curcumin has no significant effect on H3, AcH3, p300 and CBP occupancy on IE gene promoters and ORFs
One of the potential ways that curcumin might inhibit RNAP II recruitment is by inhibiting p300/CBP HAT activity (Balasubramanyam et al., 2004; Marcu et al., 2006), thus permitting formation of transcriptionally-repressive chromatin on viral DNA. We first tested whether the levels of curcumin (20 μM) that inhibit viral gene expression also affect total acetylation levels of histone H3. An immunoblot using an antibody specific to acetylated Lys9 and Lys14 of H3 revealed no significant change in total histone H3 acetylation after three hours, five hours, or overnight incubation in 20 μM curcumin (Fig. 4A). Those times correspond to beginning and end of viral infection, respectively, for the viral gene expression analysis indicated in Fig. 2A. A decrease in histone H3 acetylation was observed only after overnight incubation in the presence of higher curcumin concentrations (Fig. 4A). We infer that, under the conditions used in viral gene expression assays, curcumin does not significantly inhibit p300/CBP HAT activity.
Figure 4. Curcumin does not affect histone H3, p300, or CBP occupancy, nor H3 acetylation, at IE genes.
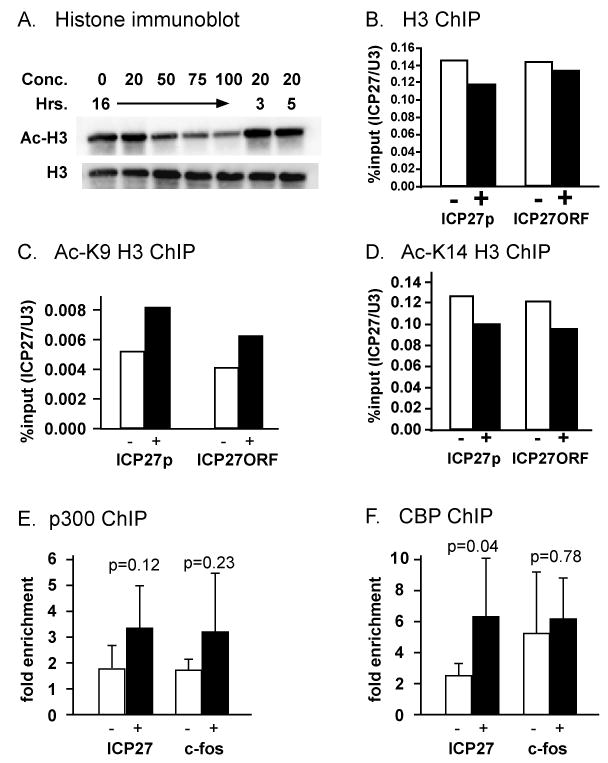
(A) HeLa cells were treated with curcumin (concentrations given in μM) for various times in the presence of 1.4 μM trichostatin A. Acid extracted histones were analyzed by immunoblotting using antibodies directed against H3 acetylated at Lys9 and Lys14 (top panel) or a carboxyl-terminal epitope of H3 (bottom panel). (B-F). Chromatin immunoprecipitation was performed as described for Fig. 3, but using antibodies against the H3 carboxyl-terminal domain (B), H3 acetylated at Lys9 (C), H3 acetylated at Lys14 (D), the coactivator p300 (E), or the coactivator CBP (F). The %input values in B-D were further normalized to U3 snRNA promoter as a means to control immunoprecipitation efficiency. Since the p300/CBP IP efficiencies were low, this data is represented as fold enrichment over no antibody control. Error bars represent the standard deviation among six (for ICP27 promoter) and four (for c-fos promoter) independent experiments (D, E). p-values in (D,E) were determined by Student-t-test.
We have previously shown that HSV-1 IE gene promoters are relatively devoid of histone H3 at 2 hpi, but that H3 and acetylated H3 can be found at IE gene ORFs and also at DE and L gene promoters at that time (Herrera and Triezenberg, 2004). However, during infection with a mutant virus (strain RP5) lacking the VP16 activation domain, H3 did associate with IE promoters, but acetylated H3 levels at IE ORF and DE or L gene promoters were reduced. On that basis, we hypothesized that p300/CBP HAT activity might be required for covalent modification and subsequent removal of histones from IE gene promoters, to allow recognition of those promoters by RNAP II. That model led to the prediction that curcumin, by inhibiting p300/CBP HAT activity, would diminish levels of acetylated histones at IE gene promoters and ORFs, and would increase the level of H3 at IE gene promoters, as observed during RP5 infection (Herrera and Triezenberg, 2004).
ChIP assays were performed using antibodies that recognize an H3 carboxyl-terminal epitope unaffected by known modifications or antibodies specific to H3 acetylated at either Lys9 or Lys14. Contrary to our hypothesis, we observed no significant increase in H3 occupancy (Fig. 4B) and no significant decrease in K9-acetylated H3 (Fig. 4C) or K14-acetylated H3 (Fig. 4D) occupancy on the ICP27 promoter or ORF. We conclude that the inhibition of IE gene expression caused by curcumin is not the result of changes in histone occupancy or acetylation at IE gene promoters, and thus the HAT activity of CBP and p300 is not likely to be critical for viral IE gene expression.
A recent report has suggested that relatively high concentrations of curcumin can inhibit the recruitment of p300 to promoter DNA fragments in vitro (Black et al., 2006). Thus, we considered the possibility that although their HAT activity per se might not be required, p300 and CBP might act as scaffolding proteins at IE gene promoters for the recruitment of other transcription factors. We used ChIP assays to test whether curcumin inhibits the recruitment of p300 and CBP to IE gene promoters. The results (Fig. 4E,F) indicate that curcumin did not block the recruitment of p300 or CBP to IE gene promoters nor to the c-fos promoter, which was previously shown to be dependent on p300/CBP (Janknecht and Nordheim, 1996; Ramirez et al., 1997). Instead, we observed a modest increase in recruitment of p300 and CBP at the ICP27 promoter. One potential explanation for this increase is that curcumin can inhibit the autoacetylation of p300 or CBP and hence inhibit the dissociation of CBP from the IE gene promoter, which would in turn prevent the recruitment of RNAP II. Taken together, these results suggest that curcumin affects recruitment of RNAP II to the HSV-1 genome but not to cellular control gene promoters, and that this effect is independent of the effects of curcumin on p300 and CBP HAT activity.
Diminished expression of p300 does not affect IE gene expression
In order to validate the conclusion that curcumin inhibits IE gene expression independent of p300 HAT activity, we have analyzed IE gene expression when p300 expression was diminished by RNA interference. Transfection of a plasmid expressing a short hairpin RNA (shRNA) specific for p300 resulted in efficient disruption of p300 expression assessed as either protein (Fig. 5A) or as mRNA (Fig. 5B). The disruption was specific for p300, since CBP expression was not affected (Fig. 5B). However, viral IE gene expression was not significantly affected (Fig. 5C), suggesting that p300 is not required for HSV-1 IE gene expression. This result also argues against the possible role of p300 as a scaffolding protein on IE gene promoters. Another possible explanation is that p300 and CBP may act redundantly on viral IE gene promoters and therefore knocking down only p300 does not affect IE gene expression. We are currently working on a more detailed analysis of coactivator disruption, which will be the subject of another manuscript. Our preliminary results suggest that neither p300 nor CBP are essential for IE gene expression (Kutluay, et al., manuscript in preparation).
Figure 5. Knocking down p300 expression does not affect HSV-1 IE gene expression.
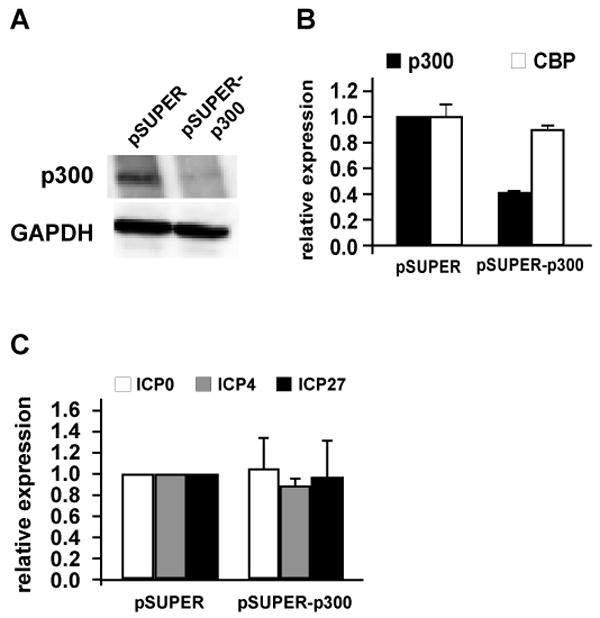
HeLa cells were transfected with 3 μg of pSUPER (empty) or pSUPER-p300 plasmid that expresses a shRNA targeting p300. 48 hours post-transfection cells were infected with HSV-1 at an MOI of 5 pfu/cell and total protein and RNA were isolated. p300 knockdown efficiency was analyzed by immunoblotting (A) and Q-RT-PCR (B). IE gene expression was analyzed by Q-RT-PCR as indicated in previous figure legends (C). Data represents the average of three independent experiments done at least in biological duplicates. Error bars represent the standard deviation.
Discussion
This study was performed to assess whether curcumin would act as an antiviral agent for HSV-1 infection, based on the hypothesis that curcumin could prevent IE gene expression by inhibiting the p300 and CBP HATs (Balasubramanyam et al., 2004) that are recruited to IE gene promoters during lytic infection (Herrera and Triezenberg, 2004). Our results demonstrate that curcumin-treated cells are indeed diminished in their ability to support HSV-1 infection and replication. The presence of curcumin did not block the delivery of the viral genome to the nucleus, indicating that binding, entry, and intracellular transit of the virus are not substantially affected by curcumin. We did observe a significant decrease in expression of viral IE genes, corresponding to diminished recruitment of RNAP II to IE gene promoters. These outcomes are all consistent with the hypothesis that curcumin specifically affects viral IE gene expression.
However, additional observations suggest that the antiviral effects of curcumin do not arise through the mechanism initially proposed. The effects of curcumin on IE gene expression were evident using concentrations and times much less than those required for significant inhibition of overall histone acetylation. We expected that curcumin, as an inhibitor of p300 and CBP HAT activity, would result in diminished histone acetylation and increased histone H3 occupancy at IE gene promoters, but those effects were not observed. Moreover, disruption of p300 protein expression had no significant effect on IE gene expression. We conclude that the HAT activity of p300 and CBP is not essential for histone clearance from HSV IE gene promoters or for IE gene expression, but that curcumin blocks other, yet unknown steps that are required for recruitment of RNAP II and subsequent transcription of IE genes. We cannot yet exclude the possibility that other HATs such as PCAF and hGCN5 may act redundantly on viral genes, masking any curcumin-dependent changes in histone acetylation (Chan and La Thangue, 2001).
Several potential mechanisms whereby curcumin blocks IE gene expression can be eliminated based on additional ChIP assays. The recruitment of the virion-borne activator protein VP16 to IE promoters was not affected by curcumin. Although a prior report suggested that curcumin can inhibit recruitment of p300 to target gene promoters by the chimeric activator protein Gal4-VP16 in an in vitro experiment (Black et al., 2006), we observed no effect of curcumin on p300 or CBP occupancy on IE gene promoters. One possible reason for this discrepancy might be the high concentrations of curcumin (300 μM) used in the in vitro system (Black et al., 2006), whereas we used 10-20 μM curcumin because of cytotoxicity observed at higher concentrations and long incubation periods.
Our observation that the occupancy of histone H3 and acetylated H3 on IE gene promoters and ORFs did not change upon curcumin treatment leads to the question of why the dramatic decrease in viral gene expression does not correspond to an increase in histone occupancy on the viral genome. Since histones are not packaged with the HSV-1 genome in virion particles (Gibson and Roizman, 1971), one possibility is that the level of histone deposition on the viral genome at 2 hours post-infection is still relatively low compared to cellular genes and hence the histones do not create a substantial barrier for transcription. Another possibility is that the VP16 activation domain recruits other coactivators such as the chromatin remodeling enzymes Brm and Brg-1 (Herrera and Triezenberg, 2004), independent of the HAT activities of CBP and p300, to keep the viral genome free of histones. In support of the latter hypothesis, VP16 AD has been recently shown to recruit the yeast remodeling complex SWI/SNF to mono- and dinucleosomal arrays to catalyze the eviction of nucleosomes by SWI/SNF (Gutierrez et al., 2007). These hypotheses will be tested in the context of viral infection in future experiments.
We note that the relatively modest effects of curcumin on the viral infection and replication (Fig. 1) do not seem to reflect the dramatic reduction in IE gene expression (Fig. 2A) and in RNAP II recruitment to IE promoters (Fig. 3B). We suspect that this discrepancy may be due to the decay of the biological activity of curcumin when exposed to light or to cell culture media (Blasius et al., 2004; Wang et al., 1997) in the longer experiments probing viral infection and replication, relative to the shorter experiments used for IE gene expression and ChIP assays.
Others have reported that virions of HSV-2 were rendered less infectious by exposure to curcumin prior to infection of HeLa cells or of mouse genitalia (Bourne et al., 1999). To date, no biochemical mechanism for this virucidal effect has been established. In contrast, the work presented here indicates that curcumin can inhibit viral gene expression and thus replication through mechanisms operative within the infected cells. This is especially evident in our observations that curcumin did not affect delivery of viral DNA to the nucleus, but had a dramatic effect on IE gene expression (Fig. 2). Nonetheless, we cannot fully exclude the possibility that the virucidal effects of curcumin might contribute to the diminished replication and plaque formation evident in Fig. 1.
Although curcumin has been widely studied in the context of cancer and apoptosis, the current literature does not clearly define the effects of curcumin on RNAPII machinery or transcription factors. One report has shown that curcumin inhibits certain cellular differentiation events by triggering protease-dependent degradation of the transcription factor AP-1 (Balasubramanian and Eckert, 2006). Although our results cannot directly exclude the possibility that VP16 is degraded similarly, our ChIP data suggests that more or less equivalent amounts of VP16 are immunoprecipitated with IE gene promoters and hence the mechanism whereby curcumin inhibits viral IE gene expression is unlikely to be mediated by degradation of VP16. The ChIP results also suggest that curcumin does not prevent the formation of the DNA-binding protein complex comprising VP16, Oct-1 and HCF. Since VP16 itself cannot bind to DNA with high affinity (Babb et al., 2001; Kristie and Sharp, 1990; Marsden et al., 1987) if curcumin was preventing the interaction between VP16, Oct-1 and HCF, then less VP16 would have been immunoprecipitated to IE gene promoters in the presence of curcumin.
Curcumin is a potent compound with various biological properties. We have shown that curcumin significantly affects HSV-1 IE gene expression which thereby diminishes the ability of the virus to launch the lytic infectious cycle. Whether curcumin can be used as an anti-HSV-1 therapeutic agent will likely be limited by its low bioavailability and high degradation rates when exposed to light. Moreover it is still not known how curcumin is metabolized in vivo and how these by-products might affect cellular processes. Therefore understanding the fate of curcumin in vivo is key to developing curcumin as an alternative drug for HSV-1 treatment. Hence, our results can be considered as an early step in elucidating the molecular basis of the antiviral activities of curcumin.
Methods
Cell lines, viruses and treatment
HeLa (ATCC, Cat.# CCL-2) and Vero (ATCC, Cat.# CCL-81) cells obtained from the American Type Culture Collection were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco). The KOS strain of HSV-1 was used to infect HeLa cells at a multiplicity of infection (MOI) of 1-5 pfu/cell, titered in Vero cells. Curcumin (Sigma, Cat. # C1836) was dissolved in dimethyl sulfoxide (DMSO). Trichostatin A (TSA) (Sigma, Cat. # T8552) was dissolved in ethanol. Cells in normal growth media were treated with curcumin as indicated in figure legends.
Plasmids and transfection
Construction of the pSUPER-p300 plasmid has been previously described (Ducat, Herrera, and Triezenberg, 2003). Briefly, a 64-mer doublestranded oligonucleotide targeting p300 was cloned into the pSUPER vector (OligoEngine) digested with Bgl II and Hind III. The oligonucleotide sequences are as follows, with bold font indicating the p300-specific region:
Forward primer: 5′- GAT CCC CGT CTT GGC ATG GTA CAA GAT TCA AGA GAT CTT GTA CCA TGC CAA GAC TTT TTG GAA A -3′
Reverse primer: 5′- AGC TTT TCC AAA AAG TCT TGG CAT GGT ACA AGA TCT CTT GAA TCT TGT ACC ATG CCA AGA CGG G -3′
HeLa cells were transfected with 3 μg of the indicated plasmids using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions.
Single-step growth curve
To determine growth curves of HSV-1 in the presence and absence of curcumin, Vero cells were plated at a density of 1 × 106 cells per 60 mm tissue culture plate. After 24 h, cells were pretreated for two hours with 20 μM curcumin prior to infection at a multiplicity of 1 pfu/cell. Duplicate samples were harvested at 4-h intervals by dislodging cells into the overlying medium with a disposable scraper. The cells were disrupted by sonication and insoluble cell debris was removed by centrifugation. The titer of each sample was determined in triplicate by plaque assay.
Plaque assay
Plaque assays were performed by infecting nearly confluent 60 mm plates of Vero cells in triplicate for each virus dilution. 100 μl of each dilution was added to plates and plates were rocked every 15 minutes for 1 hour. After 1 hour, cells were washed with DMEM lacking FBS and then overlaid with DMEM containing 2% FBS and 0.9% Sea Plaque Agarose. After 3 days, plates were stained with neutral red (0.2 mg/ml in PBS) and incubated one more hour for visualization of plaques. Plaque sizes were measured using a Nikon Eclipse TE300 microscope equipped with a Hamamatsu digital camera, C4742-95, using OpenLab software.
ChIP assay
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (Herrera and Triezenberg, 2004). Briefly, HeLa cells were fixed with 1% formaldehyde and were collected in a hypotonic buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5 % IGEPAL CA-630 with protease inhibitors). Nuclei were released by dounce homogenizer, collected and sonicated to obtain 200 to 500 bp DNA fragments. After preclearing with protein G-agarose beads, IPs were performed overnight at 4°C. Antigen-antibody complexes were precipitated by using protein G-agarose beads. Beads were washed and the protein DNA complexes were eluted with 100 μl of 50 mM Tris-HCl (pH 8.0)-10 mM EDTA-1% sodium dodecyl sulfate for 20 min at 65°C. Cross-links in DNA-protein complexes were reversed by overnight incubation at 65°C and then precipitated with ethanol. DNA was then further purified by Proteinase K digestion, phenol:chloroform (1:1) extraction and ethanol precipitation. DNA samples were resuspended in 75 μl of water and subjected to quantitative real-time PCR analysis.
ChIP assays were performed using antibodies or antisera directed against VP16 (Triezenberg, Kingsbury, and McKnight, 1988), RNA Pol II (8WG16; Covance), CBP (A-22; Santa Cruz Biotechnology), p300 (N-15; Santa Cruz Biotechnology), histone H3 acetylated at Lys9 (07-352, Upstate), histone H3 acetylated at Lys14 (07-353, Upstate) and a C-terminal epitope of histone H3 (ab1791; Abcam). Control IPs were performed using no antibodies.
RNA isolation, reverse transcription, Q-PCR
Total RNA was isolated by Trizol according to manufacturer's instructions. 1 ug RNA was reverse transcribed using the Reverse Transcription System (Promega). Quantitative real-time PCR analysis for gene expression and ChIP experiments was performed by SYBR Green assay using ABI 7500 real time PCR system (Applied Biosystems). The primer pair for the c-fos promoter was: 5′-GAACTGCGAAATGCTCACGAGATTA-3′ (forward) and 5′-AGTGTAAACGTCACGGGCTCAA-3′ (reverse). The primer pair for the c-fos ORF was: 5′-TCAACGCGCAGGACTTCTG-3′ (forward) and 5′-GCAGTGACCGTGGGAATGA-3′ (reverse). The primer pair for p300 was: 5′- CAATGAGATCCAAGGGGAGA-3′ (forward) and 5′- ATGCATCTTTCTTCCGCACT-3′ (reverse). The primer pair for CBP was: 5′-GTGCTGGCTGAGACCCTAAC-3′ (forward) and 5′- GGCTGTCCAAATGGACTTGT-3′ (reverse). Other primer pairs were previously described (Herrera and Triezenberg, 2004; Ottosen et al., 2006).
Western blot analysis
Histones from HeLa cells were isolated by acid-extraction. In brief, nuclei were released by Triton extraction buffer (PBS containing 0.5% Triton X-100 (v/v) and PMSF) and collected. Histones were then extracted using 0.2 N hydrochloric acid. Acid soluble proteins were loaded onto a 15% sodium dodecyl sulfate-polyacrylamide gel. Proteins were transferred to nitrocellulose membrane, blocked at room temperature for 1 hr in 5% BSA/TTBS and proteins were identified using antibodies specific to histone H3 (ab1791, Abcam) or H3 acetylated at Lys9 and Lys14 (06-599, Upstate).
For the p300 western blot, total cell lysate was prepared from cells by RIPA buffer and loaded onto a 6% sodium dodecyl sulfate-polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane and blocked at 4°C overnight in 5% non-fat dry milk-TTBS. p300 was detected by a rabbit polyclonal antibody from Santa Cruz (SC-584). GAPDH was detected by a mouse monoclonal antibody from Chemicon (MAB374).
Acknowledgments
This work was supported by the Department of Biochemistry and Molecular Biology at Michigan State University and by the Van Andel Research Institute. SK was supported by a special fellowship from the College of Natural Science at Michigan State University and by a predoctoral fellowship from the American Heart Association. JD was supported in part by summer undergraduate research fellowships from the American Society for Microbiology. We thank Dan Ducat and Dr. Franciso Herrera for preparing the pSUPER-p300 construct. We also thank Dr. Eric Xu, Dr. Xu Lu, and Dr. Francisco Herrera for thoughtful comments on the manuscript.
References
- Babb R, Huang CC, Aufiero DJ, Herr W. DNA recognition by the herpes simplex virus transactivator VP16: a novel DNA-binding structure. Mol Cell Biol. 2001;21:4700–12. doi: 10.1128/MCB.21.14.4700-4712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Eckert RL. Curcumin suppresses AP1 transcription factor-dependent differentiation and activates apoptosis in human epidermal keratinocytes. J Biol Chem. 2006 doi: 10.1074/jbc.M606003200. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–71. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- Black JC, Choi JE, Lombardo SR, Carey M. A mechanism for coordinating chromatin modification and preinitiation complex assembly. Mol Cell. 2006;23:809–18. doi: 10.1016/j.molcel.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Blasius R, Duvoix A, Morceau F, Schnekenburger M, Delhalle S, Henry E, Dicato M, Diederich M. Curcumin stability and its effect on glutathione S-transferase P1-1 mRNA expression in K562 cells. Ann N Y Acad Sci. 2004;1030:442–8. doi: 10.1196/annals.1329.055. [DOI] [PubMed] [Google Scholar]
- Bourne KZ, Bourne N, Reising SF, Stanberry LR. Plant products as topical microbicide candidates: assessment of in vitro and in vivo activity against herpes simplex virus type 2. Antiviral Res. 1999;42:219–26. doi: 10.1016/s0166-3542(99)00020-0. [DOI] [PubMed] [Google Scholar]
- Burke TR, Jr, Fesen MR, Mazumder A, Wang J, Carothers AM, Grunberger D, Driscoll J, Kohn K, Pommier Y. Hydroxylated aromatic inhibitors of HIV-1 integrase. J Med Chem. 1995;38:4171–8. doi: 10.1021/jm00021a006. [DOI] [PubMed] [Google Scholar]
- Campbell ME, Palfreyman JW, Preston CM. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J Mol Biol. 1984;180:1–19. doi: 10.1016/0022-2836(84)90427-3. [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Divya CS, Pillai MR. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol Carcinog. 2006;45:320–32. doi: 10.1002/mc.20170. [DOI] [PubMed] [Google Scholar]
- Ducat DC, Herrera FJ, Triezenberg SJ. Overcoming obstacles in DNA sequencing of expression plasmids for short interfering RNAs. Biotechniques. 2003;34:1140–2. 1144. doi: 10.2144/03346bm04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W, Roizman B. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc Natl Acad Sci U S A. 1971;68:2818–21. doi: 10.1073/pnas.68.11.2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Hoey T, Thut CJ, Admon A, Tjian R. Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell. 1993;75:519–30. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- Gutierrez JL, Chandy M, Carrozza MJ, Workman JL. Activation domains drive nucleosome eviction by SWI/SNF. Embo J. 2007;26:730–40. doi: 10.1038/sj.emboj.7601524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–96. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, Zeng PY, Fraser NW, Berger SL. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J Virol. 2006;80:5740–6. doi: 10.1128/JVI.00169-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Smith MM. Functional consequences of histone modifications. Curr Opin Genet Dev. 2003;13:154–60. doi: 10.1016/s0959-437x(03)00020-0. [DOI] [PubMed] [Google Scholar]
- Ingles CJ, Shales M, Cress WD, Triezenberg SJ, Greenblatt J. Reduced binding of TFIID to transcriptionally compromised mutants of VP16. Nature. 1991;351:588–90. doi: 10.1038/351588a0. [DOI] [PubMed] [Google Scholar]
- Janknecht R, Nordheim A. Regulation of the c-fos promoter by the ternary complex factor Sap-1a and its coactivator CBP. Oncogene. 1996;12:1961–9. [PubMed] [Google Scholar]
- Jaruga E, Salvioli S, Dobrucki J, Chrul S, Bandorowicz-Pikula J, Sikora E, Franceschi C, Cossarizza A, Bartosz G. Apoptosis-like, reversible changes in plasma membrane asymmetry and permeability, and transient modifications in mitochondrial membrane potential induced by curcumin in rat thymocytes. FEBS Lett. 1998;433:287–93. doi: 10.1016/s0014-5793(98)00919-3. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Joe B, Vijaykumar M, Lokesh BR. Biological properties of curcumin-cellular and molecular mechanisms of action. Crit Rev Food Sci Nutr. 2004;44:97–111. doi: 10.1080/10408690490424702. [DOI] [PubMed] [Google Scholar]
- Johnson CN, Adkins NL, Georgel P. Chromatin remodeling complexes: ATP-dependent machines in action. Biochem Cell Biol. 2005;83:405–17. doi: 10.1139/o05-115. [DOI] [PubMed] [Google Scholar]
- Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004;78:10178–86. doi: 10.1128/JVI.78.18.10178-10186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RD, Goodrich JA, Zhou S, Tjian R. Molecular cloning and expression of the 32-kDa subunit of human TFIID reveals interactions with VP16 and TFIIB that mediate transcriptional activation. Proc Natl Acad Sci U S A. 1995;92:5788–92. doi: 10.1073/pnas.92.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Sharp PA. Interactions of the Oct-1 POU subdomains with specific DNA sequences and with the HSV alpha-trans-activator protein. Genes Dev. 1990;4:2383–96. doi: 10.1101/gad.4.12b.2383. [DOI] [PubMed] [Google Scholar]
- Leinbach SS, Summers WC. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J Gen Virol. 1980;51:45–59. doi: 10.1099/0022-1317-51-1-45. [DOI] [PubMed] [Google Scholar]
- Lentine AF, Bachenheimer SL. Intracellular organization of herpes simplex virus type 1 DNA assayed by staphylococcal nuclease sensitivity. Virus Res. 1990;16:275–92. doi: 10.1016/0168-1702(90)90053-e. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li CJ, Zhang LJ, Dezube BJ, Crumpacker CS, Pardee AB. Three inhibitors of type 1 human immunodeficiency virus long terminal repeat-directed gene expression and virus replication. Proc Natl Acad Sci U S A. 1993;90:1839–42. doi: 10.1073/pnas.90.5.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Ha I, Maldonado E, Reinberg D, Green MR. Binding of general transcription factor TFIIB to an acidic activating region. Nature. 1991;353:569–71. doi: 10.1038/353569a0. [DOI] [PubMed] [Google Scholar]
- Lo WS, Henry KW, Schwartz MF, Berger SL. Histone modification patterns during gene activation. Methods Enzymol. 2004;377:130–53. doi: 10.1016/S0076-6879(03)77007-4. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, Neckers L. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–74. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- Marsden HS, Campbell ME, Haarr L, Frame MC, Parris DS, Murphy M, Hope RG, Muller MT, Preston CM. The 65,000-Mr DNA-binding and virion trans-inducing proteins of herpes simplex virus type 1. J Virol. 1987;61:2428–37. doi: 10.1128/jvi.61.8.2428-2437.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder A, Raghavan K, Weinstein J, Kohn KW, Pommier Y. Inhibition of human immunodeficiency virus type-1 integrase by curcumin. Biochem Pharmacol. 1995;49:1165–70. doi: 10.1016/0006-2952(95)98514-a. [DOI] [PubMed] [Google Scholar]
- Mellor J. It takes a PHD to read the histone code. Cell. 2006;126:22–4. doi: 10.1016/j.cell.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Muggeridge MI, Fraser NW. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J Virol. 1986;59:764–7. doi: 10.1128/jvi.59.3.764-767.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Schroder CH, Zentgraf H, Franke WW. Coexistence of nucleosomal and various non-nucleosomal chromatin configurations in cells infected with herpes simplex virus. Eur J Cell Biol. 1980;23:197–203. [PubMed] [Google Scholar]
- Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–16. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Ottosen S, Herrera FJ, Doroghazi JR, Hull A, Mittal S, Lane WS, Triezenberg SJ. Phosphorylation of the VP16 transcriptional activator protein during herpes simplex virus infection and mutational analysis of putative phosphorylation sites. Virology. 2006;345:468–81. doi: 10.1016/j.virol.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusty BK, Das BC. Constitutive activation of transcription factor AP-1 in cervical cancer and suppression of human papillomavirus (HPV) transcription and AP-1 activity in HeLa cells by curcumin. Int J Cancer. 2005;113:951–60. doi: 10.1002/ijc.20668. [DOI] [PubMed] [Google Scholar]
- Ramirez S, Ait-Si-Ali S, Robin P, Trouche D, Harel-Bellan A. The CREB-binding protein (CBP) cooperates with the serum response factor for transactivation of the c-fos serum response element. J Biol Chem. 1997;272:31016–21. doi: 10.1074/jbc.272.49.31016. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–4. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41:1955–68. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Smith CL, Peterson CL. A conserved Swi2/Snf2 ATPase motif couples ATP hydrolysis to chromatin remodeling. Mol Cell Biol. 2005;25:5880–92. doi: 10.1128/MCB.25.14.5880-5892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Salto R, Li J, Craik C, Ortiz de Montellano PR. Inhibition of the HIV-1 and HIV-2 proteases by curcumin and curcumin boron complexes. Bioorg Med Chem. 1993;1:415–22. doi: 10.1016/s0968-0896(00)82152-5. [DOI] [PubMed] [Google Scholar]
- Tomita M, Kawakami H, Uchihara JN, Okudaira T, Masuda M, Takasu N, Matsuda T, Ohta T, Tanaka Y, Mori N. Curcumin suppresses constitutive activation of AP-1 by downregulation of JunD protein in HTLV-1-infected T-cell lines. Leuk Res. 2006;30:313–21. doi: 10.1016/j.leukres.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ. Structure and function of transcriptional activation domains. Curr Opin Genet Dev. 1995;5:190–6. doi: 10.1016/0959-437x(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ, Kingsbury RC, McKnight SL. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 1988;2:718–29. doi: 10.1101/gad.2.6.718. [DOI] [PubMed] [Google Scholar]
- Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–45. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–3. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- Walker S, Greaves R, O'Hare P. Transcriptional activation by the acidic domain of Vmw65 requires the integrity of the domain and involves additional determinants distinct from those necessary for TFIIB binding. Mol Cell Biol. 1993;13:5233–44. doi: 10.1128/mcb.13.9.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Pan MH, Cheng AL, Lin LI, Ho YS, Hsieh CY, Lin JK. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15:1867–76. doi: 10.1016/s0731-7085(96)02024-9. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier JL, Triezenberg SJ, Reinberg D, Flores O, Ingles CJ, et al. Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol Cell Biol. 1994;14:7013–24. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]