

Abstract
Background
Traumatic experiences may lead to debilitating psychiatric disorders including acute stress disorder and post-traumatic stress disorder. Current treatments for these conditions are largely ineffective; therefore, novel therapies are needed. A cardinal symptom of these pathologies is the re-experiencing of the trauma through intrusive memories and nightmares. Studies in animal models indicate that memories can be weakened by interfering with the post-retrieval re-stabilization process known as memory reconsolidation. We previously reported that, in rats, intra-amygdala injection of the glucocorticoid receptor antagonist RU38486 disrupts the reconsolidation of a traumatic memory. Here we tested parameters important for designing novel clinical protocols targeting the reconsolidation of a traumatic memory with RU38486.
Methods
Using rat inhibitory avoidance, we tested the efficacy of post-retrieval systemic administration of RU38486 on subsequent memory retention and evaluated several key preclinical parameters.
Results
Systemic administration of RU38486 before or after retrieval persistently weakens IA memory retention in a dose-dependent manner, and memory does not re-emerge following footshock reminders. The efficacy of treatment is a function of the intensity of the initial trauma, and intense traumatic memories can be disrupted by changing the time and number of interventions. Furthermore, one or two treatments are sufficient to maximally disrupt the memory. The treatment selectively targets the reactivated memory without interfering with the retention of another non-reactivated memory.
Conclusions
RU38486 is a potential novel treatment for psychiatric disorders linked to traumatic memories. Our data provide the parameters for designing promising clinical trials for the treatment of flashback-type symptoms of PTSD.
Introduction
Traumatic experiences such as military combat, accidents, disasters, sexual assaults or terrifying events result in strong emotional reactions and debilitating disturbances of the emotional state that can last for several weeks (1). This symptomatology is known as acute stress disorder (ASD). ASD generally recedes and the traumatized individual returns to an asymptomatic state within a few weeks. However, in some cases, ASD persists and develops into post-traumatic stress disorder (PTSD), a psychiatric disorder associated with pervasive and debilitating symptoms of intense emotional distress that results in functional impairment. In the United States, approximately 8% of the population develops PTSD after a significant traumatic event (2). Unfortunately, the treatments currently available for PTSD, which include psychotherapeutic and pharmacologic types of approaches, rarely exceed a 60% rate of success, and fewer than 20-30% of patients achieve full remission (3, 4). Hence, there is an urgent need for identifying novel, efficacious treatments.
The most characteristic symptom of both ASD and PTSD is the re-experiencing syndrome during which the patient continuously relives the initial trauma (1). This re-experiencing can take on different forms such as nightmares, hallucinations, intrusive memories and emotional crises. It has been postulated that recalling and re-experiencing intrusive, repetitive, and vivid emotionally laden memories of a trauma are pivotal to the development of PTSD. These memories are thought to develop through classical fear conditioning processes (5, 6). In fact, fear conditioning appears to be critically involved in the development of a variety of anxiety disorders, PTSD and panic disorders (7-9).
Although they do not precisely reproduce the entire PTSD pathology, animal models of fear conditioning can be used to replicate the fear response of human traumatic memories (10-12).
In rodents, established memories, including fear conditioning, can be disrupted if interfering events or pharmacological treatments are presented or administered in a timely fashion following their retrieval (13-15). This indicates that, following retrieval, a traumatic memory becomes temporarily labile and undergoes a re-stabilization process in order to be maintained. This process is known as memory reconsolidation (13, 14). Thus, interfering with the reconsolidation of a traumatic memory may provide an opportunity for preventing or alleviating PTSD (14, 16).
To date, several types of compounds have been found to interfere with the reconsolidation of fear memories in animal models. These include, among others, inhibitors of protein synthesis (13, 17-19), inhibitors of mitogen-activated protein kinase (MAPK) (20), inhibitors of mammalian target of rapamycin (mTOR) (21) and antagonists of N-methyl-D-aspartate (NMDA) (22), adrenergic (23, 24), and glucocorticoid receptors (25, 26). Most of the animal-based studies that have investigated these compounds have evaluated the contribution of specific brain regions to the reconsolidation process by stereotactically targeting specific intracerebral areas. However, in order to design clinical protocols that aim to disrupt the reconsolidation of traumatic memories, it is critical that we establish the effect of systemic treatments. For example, it is crucial to know the time course and dosage parameters that permit the most powerful disruption of traumatic memory reconsolidation. In addition, it is important to determine the degree of specificity of the intervention.
Using inhibitory avoidance (IA) in rats, we have previously found that amygdala injections of the glucocorticoid receptor antagonist RU38486 immediately after retrieval persistently disrupt memory retention. This memory does not re-emerge after a reminder footshock, suggesting that the RU38486 treatment may indeed permanently weaken memory storage (26). These results, together with the fact that RU38486 is well tolerated in humans, suggest that this is a novel potential treatment for ASD and PTSD. In the present study, using the same paradigm as a preclinical model, we investigated the temporal and procedural modalities that might be used to optimally disrupt an established traumatic memory in humans. Thus, we determined the dose-response curve of systemic administration of RU38486, the optimal number of treatments and time of administration in relationship to the intensity of the traumatic memory and the specificity of this type of intervention.
Methods and Materials
Animals
Long Evans adult male rats (Harlan, Indianapolis, Indiana) weighing between 250 and 350 grams were housed individually on a 12 hours light/dark cycle with ad libitum access to food and water. All experiments were conducted during the light cycle between 9 am and 6 pm. All protocols complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Mount Sinai School of Medicine Animal Care Committees.
Pharmacological treatments
RU38486 (Sigma, St Louis, Missouri) was dissolved at the final concentrations in 100% 1,2-Propanediol (propylene glycol) and administered subcutaneously at a 1.0 ml/kg volume (27); vehicle therefore consisted of 100% propylene glycol. Cycloheximide (Sigma, St Louis, Missouri) was dissolved in 0.9% saline and administered subcutaneously (s.c.) at 2.2 mg/Kg (28, 29).
Inhibitory Avoidance
The IA procedure was used as previously described (30, 31). See supplement 1 material for detailed method.
Open-Field and Locomotor activity Tests
Open-field and locomotor activity were carried out according to Weaver et al. (32), as detailed in the supplement 3 material.
Two task behavioral paradigm (IA and FC)
IA was performed as described above. Auditory-cued fear conditioning (FC) was carried out as previously described (18, 33) with some modifications as described in the supplement 2 material.
Statistical Analysis
One- or two-way analysis of variance (ANOVA) followed by respectively Newman-Keuls or Bonferroni-protected post hoc t test were used. One-way ANOVA compared latencies across treatments and two-way ANOVAs compared latencies across treatment and test. For two groups comparisons, student t-tests were used. Significance was taken at p < 0.05.
Results
Post-retrieval systemic administration of RU38486 weakens the memory of a traumatic experience in a dose-dependent manner
We investigated whether peripherally injected RU38486 following retrieval affects IA retention in a dose-dependent manner. Rats were trained in IA with 0.6 mA. Forty-eight hours post-training, memory retention was tested (Test 1, Figure 1A). This test reactivated the memory. Immediately after, the animals received a s.c. injection of 3, 30, 60, or 120 mg/Kg RU38486 or vehicle. Two days after Test 1, the animals were tested (Test 2). At Test 2, rats that received 30, 60 or 120 mg/Kg (Figure 1C-E) but not 3 mg/Kg RU38486 (Figure 1B) exhibited significantly weaker retention compared to vehicle-treated controls (two-way ANOVA, F1,80 = 17, p < 0.0001 for 30 mg/Kg RU38486; F1,76 = 17.90, p < 0.0001 for 60 mg/Kg RU38486, F1,52 = 19.22, p < 0.0001 for 120mg/Kg RU38486 followed by Bonferroni post-hoc tests, Figure 1C-E). To assess whether the effect of treatment was contingent upon memory reactivation, we injected 30, 60 or 120 mg/Kg s.c. 48 hours after training in the absence of Test 1. Rats were tested 96 hours after training (Test 2). At Test 2, no significant effect of RU38486 treatment in the absence of reactivation compared to vehicle-injected controls was found (Figure 1C-E). However, a significant effect was found between RU38486-injected rats in the absence of reactivation compared to RU38486-injected rats that underwent reactivation (one-way ANOVAs followed by Newman-Keuls post-hoc tests). Hence, RU38486 disrupts the reconsolidation of an established memory – that is, only if administered after the memory is reactivated.
Figure 1.
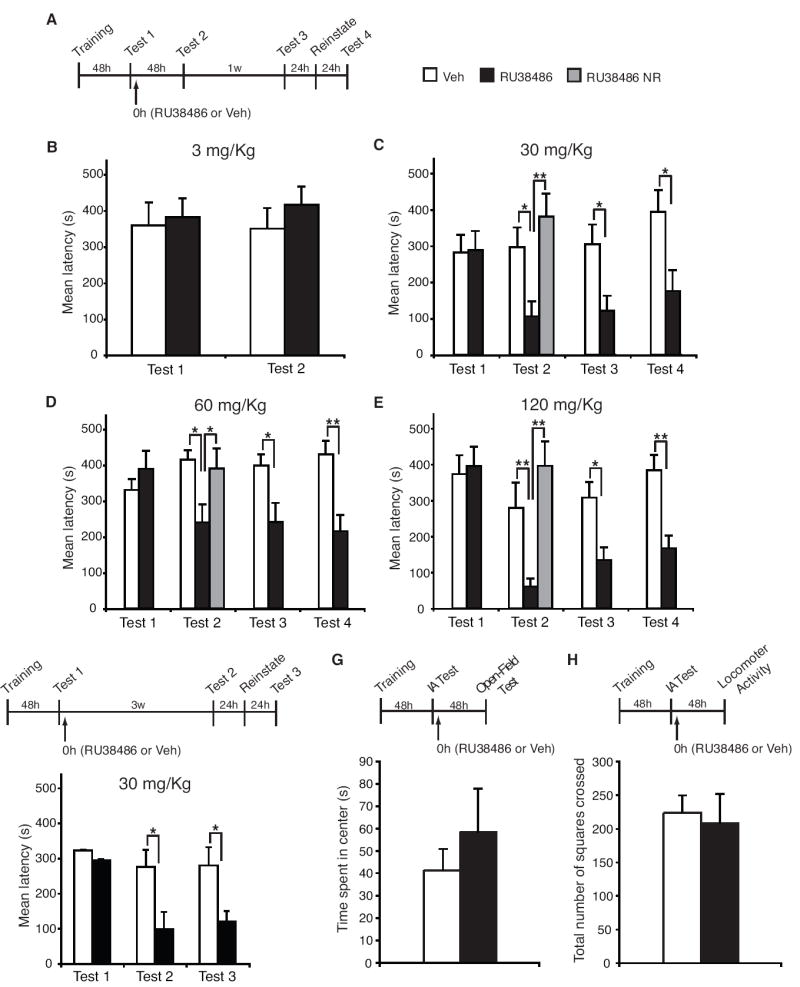
RU38486 administered following memory reactivation disrupts the retention of a traumatic memory in a dose-dependent manner. (A) IA training/testing and drug administration schedule. (B) Mean latencies ± s.e.m. of groups of rats systemically injected (↑) with 3 mg/Kg of RU38486 (n = 8) or vehicle (Veh, n = 7) immediately after Test 1 and re-tested 48 hours later (Test 2). (C) Mean latencies ± s.e.m. of groups of rats systemically injected with 30 mg/Kg of RU38486 (n = 11) or Veh (n = 11) immediately after Test 1 and re-tested 48 hours later (Test 2), 1 week later (Test 3) and after a reminder shock (Test 4). RU38486 NR: rats that received RU38486 in the absence of reactivation (n = 7). (**p < 0.01). (D) Identical to C, except that rats were injected with 60 mg/Kg of RU38486 (n = 9) or Veh (n = 12); NR, n = 7 (*p < 0.05; **p < 0.01). (E) Identical to C and D, except that animals were injected with 120 mg/Kg of RU38486 (n = 8) or Veh (n = 8); NR, n = 6 (*p < 0.05; **p < 0.01). (F) Identical to C, except that rats were tested 3 weeks after Test 1 (Test 2); RU38486 (n = 9); Veh (n = 8) (*p < 0.05). (G) Mean ± s.e.m. time spent exploring the inner arena. No significant difference was found between RU38486- and vehicle-treated groups (n=5 per group). (H) Mean ± s.e.m. locomotor activity in the arena (center). No significant difference was found between RU38486- and vehicle-treated groups (n=5 per group).
To establish whether the effect of RU38486 is long-lasting, the animals were re-tested one week after Test 2 (Test 3). The memory impairment persisted at all effective doses (Figure 1C-E, Bonferroni post-hoc). Since previous experiments have suggested that spontaneous memory recovery may occur over the course of several weeks (34), in a separate experiment, we tested whether the disruptive effect of RU38486 persisted for at least 3 weeks. Rats were trained and memory was reactivated by testing (Test1) as described in the previous experiments. Following reactivation, the animals were injected with either 30 mg/Kg of RU38486 or vehicle. Twenty-one days after reactivation they were tested (Test 2). A two-way ANOVA showed a significant effect of treatment (F1,45 = 17.2, p < 0.01) and test (F2,45 = 13.7, p < 0.05). Bonferroni post-hoc tests revealed a significant decrease in the latencies RU38486-injected rats compared to vehicle-injected controls at Test 2 (Figure 1F).
To determine whether a disrupted memory could be reinstated by a reminder footshock, one day after the test for persistence (Test 3 Figure 1C-E, and Test 2 Figure 1F) the rats received a 0.6 mA shock in a different context and were tested 24 hours later (Figure 1). Memory did not recover after this reminder shock in any of the RU38486-treated groups. A Bonferroni post-hoc test confirmed that, in all experiments, RU38486-treated animals maintained a significant memory loss compared to vehicle-injected controls. Additional control experiments confirmed that the same reminder footshock used in these experiments fully re-instated an extinguished IA memory (data not shown).
To evaluate whether changes in anxiety or overall activity contributed to the effect of RU38486 on memory retention, we determined, 48 hours after reactivation, the anxiety behavior in the open field and the locomotor activity of rats trained, reactivated and treated as described above. Student t-tests comparing vehicle to RU38486 injected animals revealed no difference in either open field or locomotor activity (Figures 1G and 1H), indicating that the treatment with RU38486 targets memory retention and not locomotor activity.
These results show that IA memory is persistently disrupted by systemic treatment of RU38486 in a dose- and reactivation-dependent manner and that 30 mg/Kg is sufficient for obtaining a maximal and persistent memory disruption.
One or two treatments of RU38486 are sufficient to maximally disrupt a traumatic memory
Here we examined how many post-reactivation RU38486 treatments are necessary to maximally disrupt the retention of a traumatic memory. Rats trained in IA using 0.6 mA underwent four subsequent tests, each separated by 24 hours (Tests 1-4, Figure 2). Following each test, animals received a s.c. injection of either RU38486 or vehicle. Each test served to both reactivate the fear memory and test the effect of the previous RU38486 treatment(s). Animals were tested two days later (Test 5). A two-way ANOVA found a significant effect of treatment (F1,65 = 55.48, p < 0.0001), test (F4,65 = 5.322, p < 0.001), and a treatment-test interaction (F4,65 = 2.712, p < 0.05). Bonferroni post-hoc tests revealed that a single treatment significantly and maximally disrupts the retention of an IA memory. Whereas the second treatment showed a small trend toward further disruption, subsequent treatments had no additive effects. Memory disruption was contingent upon reactivation, as, in fact, a one-way ANOVA followed by a Newman-Keuls post-hoc test showed no difference at Test 5 between post-retrieval vehicle-injected and RU38486-injected animals that did not receive reactivations. Conversely, post-reactivation RU38486-injected animals had significantly reduced latencies when compared to each of the other groups.
Figure 2.

One-two post-retrieval RU38486 treatments are sufficient to maximally disrupt the retention of a traumatic memory. Mean latencies ± s.e.m. of groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 8) or Veh (n = 7) immediately after Tests 1 through 4, and then re-tested 48 hours later (Test 5). (***p < 0.001). RU38486 NR: rats that received RU38486 in the absence of reactivation (n = 6). (*p < 0.05)
Thus, the effect of RU38486 is maximal after one or two consecutive post-retrieval administrations.
Pre-reactivation administration of RU38486 disrupts IA memory reconsolidation
Previous studies in animal models revealed that the time window of susceptibility to disruption is limited to only a few hours following reactivation (18, 35). For clinical applications, it is important to determine the effective temporal window of the pharmacological intervention. For example, if PTSD subjects receive the pharmacological treatment after a re-experiencing session (reactivation), it is possible that the time that elapses between the reactivation and the pharmacological action allows reconsolidation to occur, at least in part, thereby reducing the efficacy of treatment. Hence, as detailed in the supplementary material (supplement 4), we tested the effect of RU38486 administered before IA reactivation on both retrieval and subsequent memory retention. Results revealed that this intervention significantly disrupts the memory reconsolidation, while sparing retrieval (Figure 3).
Figure 3.
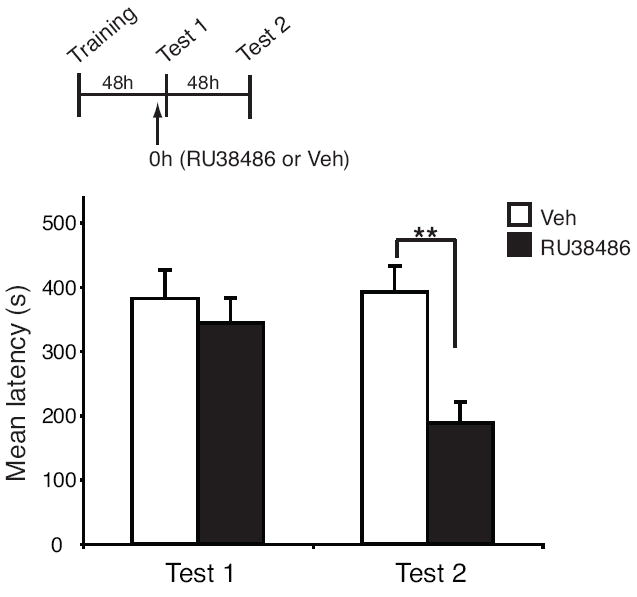
RU38486 disrupts a traumatic memory when administered prior to memory reactivation. Animals were trained using a 0.6 mA (mild) foot shock. Mean latencies ± s.e.m. of groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 13) or Veh (n = 11) 45 minutes prior to Test 1 and re-tested 48 hours later (Test 2). (**p < 0.01).
Post-retrieval RU38486 administration selectively disrupts the memory that has been reactivated but spares another non-reactivated memory
To determine whether RU38486 treatment selectively targets the reactivated memory or whether it may affect the retention of other previously established memories and perhaps those that engage overlapping memory systems, rats underwent training in two tasks, IA and auditory-cued fear conditioning (FC). In auditory FC, animals learn to associate a tone with a footshock, and both the consolidation and reconsolidation of this task, like IA, recruit the amygdala (36, 37). Subsequently, only one of the two experiences, either IA or FC, was reactivated and the effect of a systemic treatment with RU38486 was determined on the retentions of both IA and FC.
Rats were trained in IA and two days later in FC. Two days later, rats received either a reactivation test for IA (IA Test 1, Figure 4A) or a reactivation test for FC (FC Test 1, Figure 4B). Following reactivation, each group of rats were injected with either 30 mg/Kg RU38486 or vehicle. Two days later, they were tested for both IA and FC retentions. As shown in Figure 4, only the reactivated memory was selectively disrupted by RU38486 treatment, while the other memory remained intact.
Figure 4.

RU38486-mediated disruption of a traumatic memory is selective for the reactivated memory and does not interfere with the stability of another established memory. Groups of rats were trained in both IA and in FC. IA retention is expressed as mean latencies ± s.e.m., FC retention is expressed as mean % freezing ± s.e.m.. (A) Rats underwent reactivation of IA but not FC: Groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 8) or Veh (n = 6) immediately after IA reactivation (Test 1) and re-tested 48 hours later for both IA (IA Test 2; *p < 0.05) and FC (no effect) retentions. (B) Rats underwent reactivation of FC but not IA: Groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 8) or Veh (n = 8) immediately after FC Test 1 and tested 48 hours later for both IA (no effect) and FC (FC Test 2, *p < 0.05).
In the IA reactivation experiment (Figure 4A), a two-way ANOVA revealed a significant interaction (F1,24 = 4.792, p < 0.05) and Bonferroni post-hoc indicated a significant reduction in the IA retention of RU38486-treated animals compared to vehicle at Test 2. A student t-test revealed that, conversely, FC retention times did not differ between groups. When FC was reactivated, the treatment with RU38486 selectively disrupted the retention of FC but left IA memory intact (Figure 4B). A two-way ANOVA demonstrated a significant effect of treatment (F1,28 = 6.918, p < 0.05). A Bonferroni post-hoc test indicated a significant impairment for FC retention at Test 2 in the RU38486-treated animals compared to vehicle-injected controls whereas IA latencies remained unaffected (student t-test).
Together, these data indicate that RU38486 selectively disrupts the reactivated memory.
The effect of post-retrieval administration of RU38486 on memory retention is a function of the intensity of the initial traumatic experience
To determine whether RU38486 disrupts the reconsolidation of a more intense traumatic fear memory, we trained rats in IA with either 0.9 (moderate) or 1.2 mA (high) footshock intensity. Forty-eight hours after training, the animals were tested (Test 1, Figure 5A). Immediately after, they received an injection of either 30 mg/Kg RU38486 or vehicle and two days later were tested (Test 2).
Figure 5.

The efficacy of post-retrieval RU38486 treatmment is a function of the intensity of the initial trauma. (A) IA training/testing and drug administration schedule. (B) Animals were trained using a 0.9 mA (moderate) foot shock. Mean latencies ± s.e.m. of groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 8) or Veh (n = 10) immediately after Test 1 and re-tested 48 hours later (Test 2), 1 week later (Test 3) and after a reminder shock (Test 4). NR, n = 7. (***p < 0.001). (C) Animals were trained using a 1.2 mA (high) foot shock. Mean latencies ± s.e.m. at Test 2 of groups of rats systemically injected with 3 (n = 8), 30 (n = 7) or 120 (n = 8) mg/Kg of RU38486 or Veh (n = 8) immediately after Test 1. (D) Animals were trained using a 1.2 mA (high) foot shock. Mean latencies ± s.e.m. of groups of rats systemically injected twice, 5 hours apart, with 2.2 mg/Kg of cycloheximide (CXM) (n = 7) or Veh (n = 7) immediately after Test 1 and re-tested 48 hours later (Test 2). (***p < 0.001).
Rats trained with 0.9 mA and treated with RU38486 showed a significant reduction in IA retention compared to vehicle-injected controls (Figure 5B). A two-way ANOVA revealed a significant effect of treatment (F1,64 = 42.73, p < 0.0001), test (F3,64 = 4.39, p < 0.01) and treatment-test interaction (F3,64 = 4.98, p < 0.01). A Bonferroni post-hoc test revealed a significantly decreased latency in the 30 mg/Kg RU38486-injected animals compared to vehicle-injected controls at Test 2. The effect of RU38486 was contingent upon memory reactivation, as the same treatment in the absence of Test 1 did not affect memory retention (Figure 5B). Furthermore, the effect of RU38486 was persistent one week after Test 2 (Test 3, Figure 5B) and their memory retention remained significantly impaired compared to vehicle-injected controls after a reminder footshock administered one day later (Test 4, Figure 5B; Bonferroni post-hoc tests). Hence, systemic RU38486 treatment persistently disrupts the reconsolidation of an established fear memory of moderate intensity.
Conversely, rats that were trained with 1.2 mA shock intensity and 48 hours later received an RU38486 injection after reactivation showed normal memory retention (Figure 5C). We therefore tested whether an increased dosage of RU38486 could effectively decrease memory retention, but 120 mg/Kg RU38486 treatment also failed to significantly disrupt the reconsolidation of a 1.2 mA IA memory (Figure 5C). A two-way ANOVA revealed no effect of treatment, test, or treatment-test interaction. Since previous work suggested that a 2 day-old IA memory induced by a high intensity shock is disrupted by post-retrieval systemic inhibition of protein synthesis (30, 31), we tested the effect of a protein synthesis inhibitor on IA evoked by a 1.2 mA shock intensity. Rats were trained and forty-eight hours later they were tested (Test 1, Figure 5D). Immediately after Test 1 and five hours later (Figure 5A), they received a s.c. injection of either 2.2 mg/Kg cycloheximide or vehicle. This cycloheximide treatment blocks 70% of protein synthesis in the rat brain for at least 5 hours (29). As shown in Figure 5D, cycloheximide disrupted the retention of a memory induced by a 1.2 mA intensity shock. A two-way ANOVA revealed a significant effect of treatment (F1,24 = 6.45, p < 0.05), test (F1,24 = 5.45, p < 0.05) and treatment-test interaction (F1,24 = 19.18, p < 0.001). A Bonferroni post-hoc test revealed a significant reduction in latency in the cycloheximide-injected animals compared to controls at Test 2. Thus, a two-day old, high intensity fear memory is unaffected by RU38486 but can be disrupted by protein synthesis inhibition.
Time and number of interventions are critical factors that enable RU38486 to weaken a high intensity traumatic memory
As cycloheximide treatments revealed that a high intensity traumatic memory can be disrupted, in the next set of experiments, we investigated whether changing two parameters, that is, number of post-retrieval treatments and training-reactivation interval, enables RU38486 to weaken a high intensity traumatic memory. Clinical studies on PTSD suggest that an immediate intervention (e.g., debriefing) after trauma is less effective than a relatively delayed one (38-41), indicating that timing of treatment is critical for a successful outcome.
In the first set of experiments, we tested whether multiple treatments weaken an IA memory elicited by a high intensity shock. As depicted in Figure 6A, animals were trained with a 1.2 mA shock. Two days later, they underwent two subsequent testing trials separated by 24 hours (Test 1 and Test 2), each of which was followed by an injection of 30 mg/Kg RU38486. Finally, the animals were tested 48 hours later (Test 3).
Figure 6.
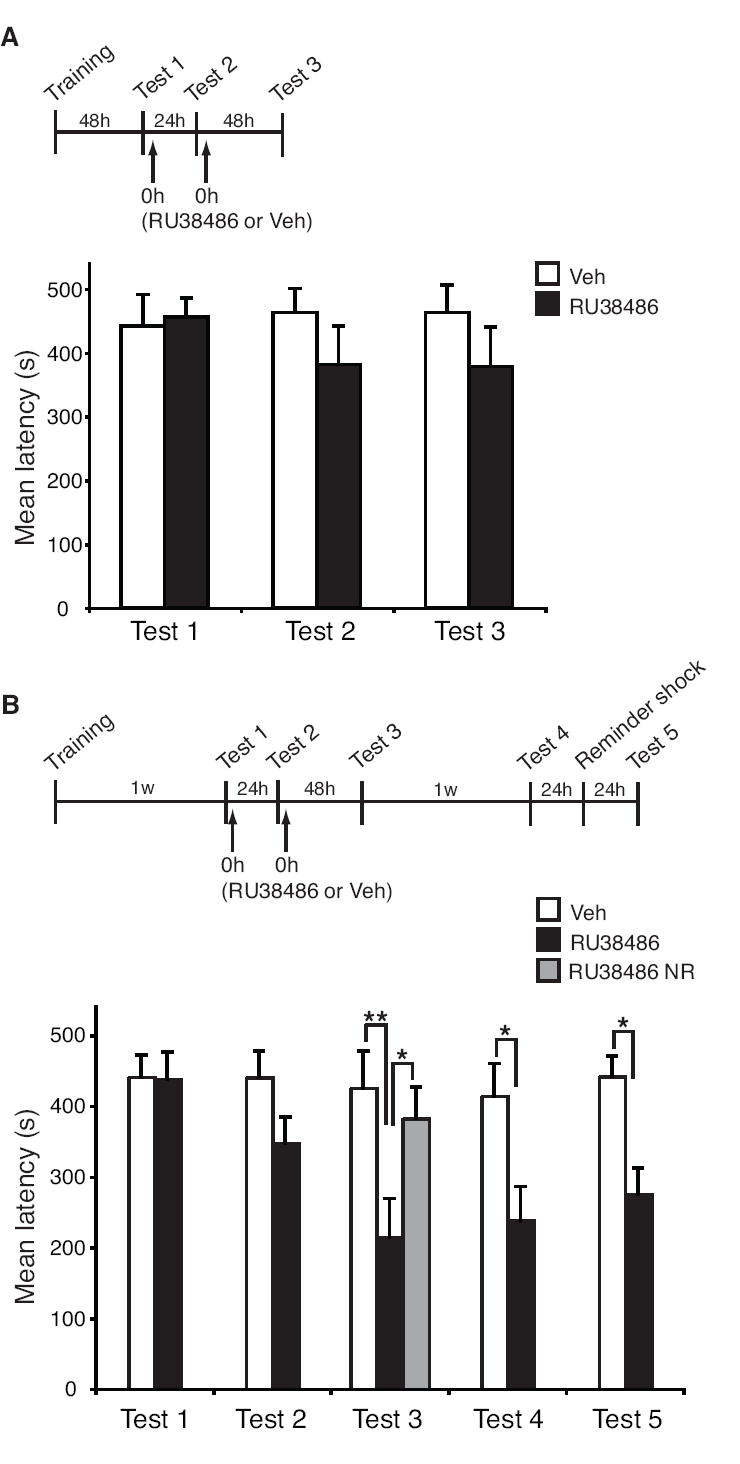
Time and number of interventions are critical parameters for RU38486 efficacy in disrupting a high intensity traumatic memory. (A) Mean latencies ± s.e.m. of groups of rats systemically injected (↑) with 30 mg/Kg of RU38486 (n = 12) or Veh (n = 9) immediately after Test 1 and Test 2, and then re-tested 48 hours later (Test 3). (B) Mean latencies ± s.e.m. of groups of rats systemically injected with 30 mg/Kg of RU38486 (n = 11) or Veh (n = 11) immediately after both Tests 1 and 2, and then re-tested 48 hours later (Test 3), 1 week later (Test 4) and after a reminder shock (Test 5). (*p < 0.05; **p < 0.01). RU38486 NR: rats that received RU38486 in the absence of reactivation (n = 6). (*p < 0.05).
As shown in Figure 6A, this treatment protocol was unable to significantly affect the retention of a high intensity traumatic memory. Indeed, although a small trend was observed, a two-way ANOVA comparing vehicle- and RU38486-treated animals revealed no effect of treatment, test or treatment-test interaction.
In a second set of experiments, we investigated whether an IA memory of high intensity could be disrupted by RU38486 if the time elapsing between training and reactivation was extended. Rats were trained and one week later underwent two consecutive treatments with 30 mg/Kg RU38486 or vehicle following memory reactivation separated by 24 hours (Test 1 and Test 2, Figure 6B). A retention test, two days later (Test 3), revealed that the RU38486 treatments significantly disrupted the memory (Figure 6B). A two-way ANOVA (significant effect of treatment: F1,100 = 24.02, p < 0.0001 and test: F4,100 = 2.84, p < 0.05) followed by a Bonferroni post-hoc test revealed that, while after the first of the two treatments there was only a small trend, after the second treatment (Test 3) a significant reduction in latency was evident in the RU38486-injected rats compared to vehicle-injected controls. The effect was contingent upon memory reactivation, as RU38486 injections in the absence of both Test 1 and Test 2 had no effect on memory retention. Hence, two consecutive treatments a week after training disrupt the reconsolidation of a high intensity traumatic memory. The memory disruption was persistent when the animals were retested a week later (Test 4) and memory retention remained impaired at Test 5, 24 hours following a reminder footshock (Bonferroni post-hoc test, Figure 6B).
Together, these results establish that both timing and number of interventions are crucial parameters for designing treatment protocols that effectively disrupt the retention of traumatic memories using RU38486.
Discussion
This study provides evidence that systemic treatments with RU38486 persistently disrupt memory retention. Memory impairment did not revert after a reminder footshock, suggesting that the RU38486 targets memory reconsolidation and, therefore, ultimately, the storage of information. We propose that RU38486 in tandem with memory reactivation sessions represents a novel therapeutic approach for the treatment of psychiatric disorders linked to traumatic experiences and fear conditioning.
Dosage, persistent effect and target mechanism of RU38486
RU38486 is a safe, well-tolerated FDA approved medication used for termination of pregnancy (http://www.fda.gov/cder/drug/infopage/mifepristone/default.htm). In addition, it has been studied in clinical trials for treatment of psychotic depression (42-46), Alzheimer’s Disease (47-49), schizophrenia (50) and bipolar disorder (51). Our results used a preclinical model in rats to show that RU38486 in tandem with memory reactivation can be employed to disrupt the retention of traumatic memories, suggesting that disorders resulting from traumatic experiences, including but not restricted to ASD and PTSD, may benefit from such treatment. The dosage of 30 mg/Kg, that in our studies optimally and persistently disrupts traumatic memories, is within the range of the 1200 mg dosage found to be effective in treating psychotic major depression in human adults (43). The effect of RU38486 is significant and persistent whether it is administered before or after memory retrieval, suggesting that an extended time window of treatment around the retrieval trial exists. Importantly, administration before retrieval does not affect the recall of the memory itself, a condition necessary for the treatment to be effective and disrupt memory reconsolidation.
The mechanism targeted by RU38486 administered after retrieval appears to be the reconsolidation of the memory of the original experience. Determining whether the target mechanism is the reconsolidation of the memory and not other processes such as extinction, which is elicited by similar reactivation (exposure) protocols, is important for elucidating the temporal evolution of the amnesic effects. Retrieval is generally evoked by the exposure to the context or cues (conditioned stimulus, CS) in the absence of shock (unconditioned stimulus, US). This exposure can elicit both the reconsolidation of the original memory and extinction learning, by which the animal learns that the CS is no longer associated to the US, and therefore “safe”(52). Both disrupting reconsolidation and inducing extinction lead to the same end result, that is, a decrease in the expression of the conditioned response. However, whereas disrupting reconsolidation weakens the original memory, inducing extinction leaves the original memory intact but counteracts its expression by adding a new memory (52, 53). This represents a very different therapeutic outcome. Agonists of glucocorticoid receptors, including dexamethasone and corticosterone administered after CS exposures enhance extinction learning (54-56), while administration of the corticosterone synthesis inhibitor metyrapone disrupts the consolidation of extinction (57). Conversely, our study provides evidence that antagonists of glucocorticoid receptors likely target the reconsolidation of the memory of original experience. In fact, the hallmarks of extinction learning, namely spontaneous recovery and memory reinstatement following a footshock reminder (53), fail to take place in our paradigm. In line with this conclusion previous studies in rodents have revealed that RU38486 disrupts memory consolidation (58, 59) as well as extinction itself (60).
Several studies have reported that acute administration of glucocorticoids impairs memory retrieval, which may represent a different approach to weaken the strength of traumatic memories by either preventing reconsolidation or facilitating extinction (55, 61). In contrast, here, we report that glucocorticoid receptors antagonists do not affect retrieval per se but rather disrupt the reconsolidation process.
Although we cannot completely exclude that the memory impairment seen with RU38486 might be due to a failure in subsequent retrievals rather than an interference with the reconsolidation process, we favor the latter hypothesis for the following reasons: memory impairment is evident at testing times that are remote from the times of treatment, the administration of RU38486 before testing does not affect retrieval per se and footshock reminder exposure fails to reinstate the disrupted memory.
Selectivity of the treatment
The post-retrieval effect of RU38486 is selective for the reactivated memory. To our knowledge this is the first evidence showing such a memory-selective effect.
On a different level, selectivity of the treatments targeting memory reconsolidation has been also supported by the fact that, in all reconsolidation studies, including the present one, if the pharmacological treatment is applied in the absence of reactivation, no effect is observed on the memory retention (13, 17, 19, 37). Furthermore, only directly reactivated associations become labile, while indirectly reactivated associations do not, suggesting that memory reactivation produces content-limited vulnerability rather than global changes in a memory and its associations (62).
Thus, pharmacological intervention in combination with behavioral therapy that target memory reconsolidation will not likely cause a general disruption of other memories including those that are linked associatively to the reactivated memory.
Moreover, RU38486 treatments administered after memory reactivation result in a decrease in the retention of the memory rather than a complete amnesia, suggesting that this treatment will likely weaken the intensity of the memory and not result in a complete deletion of memory contents.
Intensity of the initial traumatic experience, age of the memory and number of treatments
Whereas a single administration of RU38486 affected 0.6 and 0.9 mA IA memories reactivated 2 days following training, it did not affect a 1.2 mA IA memory. However, since the latencies resulting from a 1.2 mA footshock mostly reached the maximum latency (540 sec), we cannot exclude that memory disruption occurred but was masked by a ceiling effect. Nevertheless, this memory was significantly weakened by inhibition of protein synthesis, suggesting that it was potentially disruptable. Importantly, when one week elapsed between training and reactivation, the IA memory could then be disrupted by RU38486. Thus, for high intensity traumatic experiences, a delayed intervention is likely to be more beneficial than an immediate one. Interestingly, clinical studies on PTSD have reported that, whereas early interventions, such as psychological debriefing, are critical for managing the stress response to trauma (63, 64), to be more efficacious, intervention should not occur too early (e.g. immediately after the trauma). Indeed, intervening too early, when the stress of the experience and trauma has not yet waned, might exacerbate the relapse to fear (38-41). Similarly, in rodents, recent fear is resistant to extinction (65). Thus, it is plausible that experiencing a highly traumatic event evokes a stronger and longer lasting stress response compared to experiencing a mild trauma. In this case, an immediate post-trauma re-exposure may additionally increase the stress response, which may become yet more resistant to disruption. Since stress and arousal are known to mediate memory consolidation (66, 67), and systemic administration of corticosterone or glucocorticoid receptor agonists shortly after training can facilitate memory retention (68-70), an immediate treatment with RU38486 may not be sufficient to counteract a strong trauma. However, an intervention that occurs at later times, when the stress response is decreased, might be more efficacious. Additionally, it is possible that a high intensity stress response may recruit different mechanisms that are more resistant to disruption than those activated by a milder stress.
In agreement with our results, temporal dynamics of memory reconsolidation in mice are dependent on the strength of the memory, such that weaker memories are more easily disrupted after reactivation than stronger ones (71). Similarly, overtraining in rat auditory fear conditioning renders memories transiently resistant to protein synthesis inhibition (Wang and Nader, personal communication).
In apparent contrast, previous studies using fear conditioning types of tasks in different species have shown that the passage of time (on the order of several weeks) influences the stability of a memory, and that older memories become less susceptible to disruption following their reactivation (30, 71-76). This process likely reflects a lingering of the consolidation phase (13, 17). Similarly, it is possible that over time older memories become resistant to the effect of RU38486 and further studies are need to address this point. To unravel the effect of time on the stabilization of IA memory in relationship to the effect of RU38486, co-investigating the contribution of the intensity of the original training and type and number of reactivations and treatments will be necessary. Indeed, highly traumatic experiences may result in memories that are initially resistant to disruption (stress effect), but subsequently become labile, and then finally consolidate over time to a resistant state once again (consolidation effect).
Hence, all these parameters should be kept in mind when designing therapeutic protocols: an immediate intervention may be inefficient for a highly traumatic experience, whereas a very late intervention may not be efficacious because memory has become more consolidated and therefore resistant to post-reactivation intervention.
In conclusion, our findings demonstrate that RU38486 administered systemically in the context of memory reactivation is effective at disrupting reconsolidation of fear-related memories. We have also shown that it is effective when given at the dosage range within that used in clinical applications of RU38486 for other purposes and that this disruption occurs after only one or two exposures. Finally, the effect of RU38486 is selective for the reactivated memory. Our preclinical findings therefore provide the parameters for very promising clinical trials using RU38486 for the treatment of the re-experiencing symptoms of PTSD.
Supplementary Material
Acknowledgments
The authors thank Dillon Chen, Virginia Gao, and Gabriella Pollonini for helpful discussions and comments on both the manuscript and experimental design; Reginald Miller and the Center for Comparative Medicine and Surgery facility at Mount Sinai School of Medicine for technical support. This work was supported by National Institute of Mental Health grants R01 MH074736 and R01 MH065635 and the NARSAD Independent Investigator Award (to CMA).
Footnotes
The authors reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association Press; 2000. [Google Scholar]
- 2.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 3.Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. Jama. 2000;283:1837–1844. doi: 10.1001/jama.283.14.1837. [DOI] [PubMed] [Google Scholar]
- 4.Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, Pitts CD. Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry. 2001;62:860–868. doi: 10.4088/jcp.v62n1105. [DOI] [PubMed] [Google Scholar]
- 5.Fairbank JA, Nicholson RA. Theoretical and empirical issues in the treatment of post-traumatic stress disorder in Vietnam veterans. J Clin Psychol. 1987;43:44–55. doi: 10.1002/1097-4679(198701)43:1<44::aid-jclp2270430107>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 6.Keane TM, Scott WO, Chavoya GA, Lamparski DM, Fairbank JA. Social support in Vietnam veterans with posttraumatic stress disorder: a comparative analysis. J Consult Clin Psychol. 1985;53:95–102. doi: 10.1037//0022-006x.53.1.95. [DOI] [PubMed] [Google Scholar]
- 7.Bouton ME, Mineka S, Barlow DH. A modern learning theory perspective on the etiology of panic disorder. Psychol Rev. 2001;108:4–32. doi: 10.1037/0033-295x.108.1.4. [DOI] [PubMed] [Google Scholar]
- 8.Grillon C, Morgan CA, Southwick SM, Davis M, Charney DS. Baseline startle amplitude and prepulse inhibition in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res. 1996;64:169–178. doi: 10.1016/s0165-1781(96)02942-3. [DOI] [PubMed] [Google Scholar]
- 9.Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29:1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 10.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 11.Miller MM, McEwen BS. Establishing an agenda for translational research on PTSD. Ann N Y Acad Sci. 2006;1071:294–312. doi: 10.1196/annals.1364.023. [DOI] [PubMed] [Google Scholar]
- 12.Siegmund A, Wotjak CT. Toward an animal model of posttraumatic stress disorder. Ann N Y Acad Sci. 2006;1071:324–334. doi: 10.1196/annals.1364.025. [DOI] [PubMed] [Google Scholar]
- 13.Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Dudai Y. Reconsolidation: the advantage of being refocused. Curr Opin Neurobiol. 2006;16:174–178. doi: 10.1016/j.conb.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- 16.Yehuda R, Flory JD, Southwick S, Charney DS. Developing an agenda for translational studies of resilience and vulnerability following trauma exposure. Ann N Y Acad Sci. 2006;1071:379–396. doi: 10.1196/annals.1364.028. [DOI] [PubMed] [Google Scholar]
- 17.Dudai Y, Eisenberg M. Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron. 2004;44:93–100. doi: 10.1016/j.neuron.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 19.Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- 20.Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5(Suppl 2):61–72. doi: 10.1111/j.1601-183X.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- 21.Blundell J, Kouser M, Powell CM. Systemic inhibition of mammalian target of rapamycin inhibits fear memory reconsolidation. Neurobiol Learn Mem. 2008;90:28–35. doi: 10.1016/j.nlm.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben Mamou C, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9:1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- 23.Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129:267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Przybyslawski J, Roullet P, Sara SJ. Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci. 1999;19:6623–6628. doi: 10.1523/JNEUROSCI.19-15-06623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin XC, Lu YF, Yang XF, Ma L, Li BM. Glucocorticoid receptors in the basolateral nucleus of amygdala are required for postreactivation reconsolidation of auditory fear memory. Eur J Neurosci. 2007;25:3702–3712. doi: 10.1111/j.1460-9568.2007.05621.x. [DOI] [PubMed] [Google Scholar]
- 26.Tronel S, Alberini CM. Persistent disruption of a traumatic memory by postretrieval inactivation of glucocorticoid receptors in the amygdala. Biol Psychiatry. 2007;62:33–39. doi: 10.1016/j.biopsych.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pugh CR, Fleshner M, Rudy JW. Type II glucocorticoid receptor antagonists impair contextual but not auditory-cue fear conditioning in juvenile rats. Neurobiol Learn Mem. 1997;67:75–79. doi: 10.1006/nlme.1996.3741. [DOI] [PubMed] [Google Scholar]
- 28.Flint RW, Jr, Marino CL. Cycloheximide impairs reconsolidation of a contextually reactivated memory in a conditioned taste aversion paradigm. Behav Neurosci. 2007;121:433–438. doi: 10.1037/0735-7044.121.2.433. [DOI] [PubMed] [Google Scholar]
- 29.Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J Neurosci. 2006;26:3010–3020. doi: 10.1523/JNEUROSCI.4818-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milekic MH, Alberini CM. Temporally graded requirement for protein synthesis following memory reactivation. Neuron. 2002;36:521–525. doi: 10.1016/s0896-6273(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 31.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- 32.Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duvarci S, Nader K. Characterization of fear memory reconsolidation. J Neurosci. 2004;24:9269–9275. doi: 10.1523/JNEUROSCI.2971-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc Natl Acad Sci U S A. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anokhin KV, Tiunova AA, Rose SP. Reminder effects - reconsolidation or retrieval deficit? Pharmacological dissection with protein synthesis inhibitors following reminder for a passive-avoidance task in young chicks. Eur J Neurosci. 2002;15:1759–1765. doi: 10.1046/j.1460-9568.2002.02023.x. [DOI] [PubMed] [Google Scholar]
- 36.Milekic MH, Pollonini G, Alberini CM. Temporal requirement of C/EBPbeta in the amygdala following reactivation but not acquisition of inhibitory avoidance. Learn Mem. 2007;14:504–511. doi: 10.1101/lm.598307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nat Rev Neurosci. 2000;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- 38.Bisson JI, Jenkins PL, Alexander J, Bannister C. Randomised controlled trial of psychological debriefing for victims of acute burn trauma. Br J Psychiatry. 1997;171:78–81. doi: 10.1192/bjp.171.1.78. [DOI] [PubMed] [Google Scholar]
- 39.Gray MJ, Litz BT. Behavioral interventions for recent trauma: empirically informed practice guidelines. Behav Modif. 2005;29:189–215. doi: 10.1177/0145445504270884. [DOI] [PubMed] [Google Scholar]
- 40.McNally RJ, Bryant RA, Ehlers A. Does early psychological intervention promote recovery from posttraumatic stress? Psychol Sci Publ Inter. 2003;4:45–79. doi: 10.1111/1529-1006.01421. [DOI] [PubMed] [Google Scholar]
- 41.Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann N Y Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 42.Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol. 2001;21:516–521. doi: 10.1097/00004714-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52:386–392. doi: 10.1016/s0006-3223(02)01432-4. [DOI] [PubMed] [Google Scholar]
- 44.DeBattista C, Belanoff J. The use of mifepristone in the treatment of neuropsychiatric disorders. Trends Endocrinol Metab. 2006;17:117–121. doi: 10.1016/j.tem.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology. 2006;31:628–636. doi: 10.1038/sj.npp.1300884. [DOI] [PubMed] [Google Scholar]
- 46.Simpson GM, El Sheshai A, Loza N, Kingsbury SJ, Fayek M, Rady A, et al. An 8-week open-label trial of a 6-day course of mifepristone for the treatment of psychotic depression. J Clin Psychiatry. 2005;66:598–602. doi: 10.4088/jcp.v66n0509. [DOI] [PubMed] [Google Scholar]
- 47.Belanoff JK, Jurik J, Schatzberg LD, DeBattista C, Schatzberg AF. Slowing the progression of cognitive decline in Alzheimer’s disease using mifepristone. J Mol Neurosci. 2002;19:201–206. doi: 10.1007/s12031-002-0033-3. [DOI] [PubMed] [Google Scholar]
- 48.DeBattista C, Belanoff J. C-1073 (mifepristone) in the adjunctive treatment of Alzheimer’s disease. Curr Alzheimer Res. 2005;2:125–129. doi: 10.2174/1567205053585954. [DOI] [PubMed] [Google Scholar]
- 49.Pomara N, Doraiswamy PM, Tun H, Ferris S. Mifepristone (RU 486) for Alzheimer’s disease. Neurology. 2002;58:1436. doi: 10.1212/wnl.58.9.1436. [DOI] [PubMed] [Google Scholar]
- 50.Gallagher P, Watson S, Smith MS, Ferrier IN, Young AH. Effects of adjunctive mifepristone (RU-486) administration on neurocognitive function and symptoms in schizophrenia. Biol Psychiatry. 2005;57:155–161. doi: 10.1016/j.biopsych.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 51.Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology. 2004;29:1538–1545. doi: 10.1038/sj.npp.1300471. [DOI] [PubMed] [Google Scholar]
- 52.Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60:352–360. doi: 10.1016/j.biopsych.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 54.Abrari K, Rashidy-Pour A, Semnanian S, Fathollahi Y. Administration of corticosterone after memory reactivation disrupts subsequent retrieval of a contextual conditioned fear memory: dependence upon training intensity. Neurobiol Learn Mem. 2008;89:178–184. doi: 10.1016/j.nlm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 55.Cai WH, Blundell J, Han J, Greene RW, Powell CM. Postreactivation glucocorticoids impair recall of established fear memory. J Neurosci. 2006;26:9560–9566. doi: 10.1523/JNEUROSCI.2397-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang YL, Chao PK, Lu KT. Systemic and intra-amygdala administration of glucocorticoid agonist and antagonist modulate extinction of conditioned fear. Neuropsychopharmacology. 2006;31:912–924. doi: 10.1038/sj.npp.1300899. [DOI] [PubMed] [Google Scholar]
- 57.Barrett D, Gonzalez-Lima F. Behavioral effects of metyrapone on Pavlovian extinction. Neurosci Lett. 2004;371:91–96. doi: 10.1016/j.neulet.2004.08.046. [DOI] [PubMed] [Google Scholar]
- 58.Donley MP, Schulkin J, Rosen JB. Glucocorticoid receptor antagonism in the basolateral amygdala and ventral hippocampus interferes with long-term memory of contextual fear. Behav Brain Res. 2005;164:197–205. doi: 10.1016/j.bbr.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 59.Roozendaal B, Okuda S, de Quervain DJ, McGaugh JL. Glucocorticoids interact with emotion-induced noradrenergic activation in influencing different memory functions. Neuroscience. 2006;138:901–910. doi: 10.1016/j.neuroscience.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 60.Yang YL, Chao PK, Ro LS, Wo YY, Lu KT. Glutamate NMDA receptors within the amygdala participate in the modulatory effect of glucocorticoids on extinction of conditioned fear in rats. Neuropsychopharmacology. 2007;32:1042–1051. doi: 10.1038/sj.npp.1301215. [DOI] [PubMed] [Google Scholar]
- 61.de Quervain DJ, Margraf J. Glucocorticoids for the treatment of post-traumatic stress disorder and phobias: A novel therapeutic approach. Eur J Pharmacol. 2008;583:365–371. doi: 10.1016/j.ejphar.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 62.Debiec J, Doyere V, Nader K, Ledoux JE. Directly reactivated, but not indirectly reactivated, memories undergo reconsolidation in the amygdala. Proc Natl Acad Sci U S A. 2006;103:3428–3433. doi: 10.1073/pnas.0507168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campfield KM, Hills AM. Effect of timing of critical incident stress debriefing (CISD) on posttraumatic symptoms. J Trauma Stress. 2001;14:327–340. doi: 10.1023/A:1011117018705. [DOI] [PubMed] [Google Scholar]
- 64.Everly GS, Jr, Mitchell JT. The debriefing “controversy” and crisis intervention: a review of lexical and substantive issues. Int J Emerg Ment Health. 2000;2:211–225. [PubMed] [Google Scholar]
- 65.Maren S, Chang CH. Recent fear is resistant to extinction. Proc Natl Acad Sci U S A. 2006;103:18020–18025. doi: 10.1073/pnas.0608398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roozendaal B. Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem. 2002;78:578–595. doi: 10.1006/nlme.2002.4080. [DOI] [PubMed] [Google Scholar]
- 67.Sandi C, Rose SP. Corticosteroid receptor antagonists are amnestic for passive avoidance learning in day-old chicks. Eur J Neurosci. 1994;6:1292–1297. doi: 10.1111/j.1460-9568.1994.tb00319.x. [DOI] [PubMed] [Google Scholar]
- 68.Cordero MI, Merino JJ, Sandi C. Correlational relationship between shock intensity and corticosterone secretion on the establishment and subsequent expression of contextual fear conditioning. Behav Neurosci. 1998;112:885–891. doi: 10.1037//0735-7044.112.4.885. [DOI] [PubMed] [Google Scholar]
- 69.Roozendaal B, McGaugh JL. Amygdaloid nuclei lesions differentially affect glucocorticoid-induced memory enhancement in an inhibitory avoidance task. Neurobiol Learn Mem. 1996;65:1–8. doi: 10.1006/nlme.1996.0001. [DOI] [PubMed] [Google Scholar]
- 70.Sandi C, Loscertales M, Guaza C. Experience-dependent facilitating effect of corticosterone on spatial memory formation in the water maze. Eur J Neurosci. 1997;9:637–642. doi: 10.1111/j.1460-9568.1997.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 71.Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boccia MM, Blake MG, Acosta GB, Baratti CM. Post-retrieval effects of icv infusions of hemicholinium in mice are dependent on the age of the original memory. Learn Mem. 2006;13:376–381. doi: 10.1101/lm.150306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eisenberg M, Dudai Y. Reconsolidation of fresh, remote, and extinguished fear memory in Medaka: old fears don’t die. Eur J Neurosci. 2004;20:3397–3403. doi: 10.1111/j.1460-9568.2004.03818.x. [DOI] [PubMed] [Google Scholar]
- 74.Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, Silva AJ. Stability of recent and remote contextual fear memory. Learn Mem. 2006;13:451–457. doi: 10.1101/lm.183406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kemenes G, Kemenes I, Michel M, Papp A, Muller U. Phase-dependent molecular requirements for memory reconsolidation: differential roles for protein synthesis and protein kinase A activity. J Neurosci. 2006;26:6298–6302. doi: 10.1523/JNEUROSCI.0890-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Litvin OO, Anokhin KV. Mechanisms of memory reorganization during retrieval of acquired behavioral experience in chicks: the effects of protein synthesis inhibition in the brain. Neurosci Behav Physiol. 2000;30:671–678. doi: 10.1023/a:1026698700139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.