

Abstract
The meta-vinylhalide fluoroalkyl ester nortropanes 1-4 were synthesized as ligands of the serotonin transporter (SERT) for use as positron emission tomography (PET) imaging agents. In vitro competition binding assays demonstrated that 1-4 have a high affinity for the SERT (Ki's = 0.3 - 0.4 nM) and are selective for the SERT over the dopamine and norepinephrine transporters (DAT and NET). MicroPET imaging in anesthetized cynomolgus monkeys with [18F]1-[18F]4 demonstrated that all four tracers behave similarly with peak uptake in the SERT-rich brain regions achieved after 45-55 min followed by a steady washout. An awake monkey study was performed with [18F]1 which demonstrated that the uptake of [18F]1 was not influenced by anesthesia. Chase studies with the SERT ligand 15 displaced [18F]1-[18F]4 but chase studies with the DAT ligand 16 did not displace [18F]1-[18F]4 thus indicating that the tracers were binding specifically to the SERT.
Introduction
The human serotonin transporter (SERT) is a 630 amino acid transmembrane protein located in the brain, peripheral organs, and blood platelets.3-6 Within the central nervous system (CNS), the SERT is localized on the presynaptic terminals of serotonergic neurons and functions to terminate neurotransmission by removing serotonin from the synapse. Serotonergic neurons originate primarily in the median and dorsal raphe nuclei of the brainstem and innervate discrete areas that include the hypothalamus, thalamus, striatum, and cerebral cortex.4, 7-10 Thus, the SERT can serve as a specific marker for serotonergic neuronal anatomy and integrity. Dysregulation of serotonin neurotransmission has been implicated in the pathophysiology of major depression and a reduction in SERT density has been observed postmortem in the tissues of depressed patients and suicide victims.7, 11-14 Therefore, the SERT is the target of the selective serotonin reuptake inhibitor (SSRI) class of antidepressants. The ability to image CNS SERT in vivo using positron emission tomography (PET)15-17 may provide insight into the pathophysiology of depression and suicide by enabling the SERT density of specific brain regions to be measured thereby indicating which regions of the brain have SERT density altered by the disease as well as allow for improved diagnostic techniques and monitoring of antidepressant therapy.18-22 Furthermore, the availability of SERT PET tracers may aid in the development of new SERT therapeutics by allowing for occupancy measurements of the therapeutic.23-28
This need for a SERT PET tracer has led to extensive research into the development of new tracers for this target17 with the majority of these new compounds belonging to the diarylsulfide24, 29-43 or nortropane44-46 classes. Many of the initial SERT PET tracers are 11C-labeled compounds and therefore are limited to use in the location where they are prepared due to the short half-life of 11C (t1/2 = 20.4 min). Tracers labeled with 18F are desirable because the longer half-life of 18F (t1/2 = 109.8 min) allows for longer radiosynthesis times and transport of 18F-labeled tracers to sites away from the production facility which thus enables PET imaging centers without onsite cyclotrons to utilize these tracers. Furthermore, 18F positrons have a lower maximum energy than 11C positrons (0.64 MeV vs. 0.97 MeV)17 and therefore deposit less energy into tissue, and also a shorter linear range47, 48 which allows for higher spatial resolution. These properties of 18F are fortuitous due to the valuable role that 19F plays in medicinal chemistry49-51 and numerous methods have now been developed to incorporate 18F or 19F into molecules.52-54
The goal of developing a viable SERT PET tracer is an important, but not easy, task as evidenced by the numerous compounds that have been, and continue to be, reported. Several criteria need to be met in order for a candidate molecule to become a useful tracer. The desirable properties for a candidate SERT PET tracer include (exceptions may exist): 1) high binding affinity55 (Ki = ~ 0.1 - 0.5 nM) for the SERT with high selectivity (>50:1) over the norepinephrine transporter (NET) and dopamine transporter (DAT); 2) moderate lipophilicity (logP = ~ 1 - 3)56-58 for good initial brain entry and low non-specific binding; 3) high uptake ratios vs. cerebellum59 (≥ 1.7) in SERT-rich brain regions such as thalamus, hypothalamus and raphe with uptake ratios ≥ 1.3 in low density regions such as anterior cingulate to enable delineation of specific SERT binding; 4) specific binding to brain SERT reaching peak equilibrium at ≤ 60 min followed by washout for both 11C- and 18F-tracers to allow quantification of SERT occupancy; 5) lack of radiolabeled metabolites generated in the brain; and 6) lack of lipophilic radiolabeled metabolites in peripheral organs that may enter brain and bind specifically or non-specifically to SERT-rich regions and cerebellum.
As part of an ongoing research project in our laboratories to develop SERT-selective tropane and nortropane PET and single-photon emission computed tomography (SPECT) imaging agents labeled with 11C or 18F (PET) or 123I (SPECT) for human diagnostic applications we have been exploring the effect of placing a vinyl halide60-66 or furyl substituent67 on the 3β-phenyl ring. We report here the synthesis and biological evaluation of 2β-carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane (1, FEmZIENT)68, 2β-carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane (2, FEmZBrENT)69, 2β-carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane (3, FPmZIENT)70, and 2β-carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane (4, FPmZBrENT)70 along with the radiosynthesis and microPET imaging of [18F]1-[18F]4 in anesthetized non-human primates and the PET imaging of [18F]1 in an awake non-human primate.
Chemistry
The synthesis of target compounds 1-4 is shown in Scheme 1. 2β-Carbomethoxy-3β-(3′-bromophenyl)tropane (5)63 was hydrolyzed in refluxing 1,4-dioxane/H2O71 to give the acid 6 which was converted to the acid chloride and then esterified to afford the 2-fluoroethyl ester 7 or the 3-fluoropropyl ester 8. N-demethylation72 afforded the nortropanes 9 and 10 which were coupled to (Z)-1,2-bis(trimethylstannyl)ethene73, 74 to give the vinyl-tin nortropanes 11 and 12 in varying cis:trans ratios with the major product being the cis isomer (~3:1). Halodestannylation then afforded the vinyl iodides 1 and 3 and the vinyl bromides 2 and 4 (after separation of the cis:trans isomers by semi-preparative HPLC).
Scheme 1.

Radiochemistry
The radiolabeling procedure is depicted in Scheme 2. N-Boc acid 13 or 1463 was dissolved in DMF, deprotonated with 0.1 M Bu4OH(aq), added to [18F]fluoroalkylbrosylate, and heated. The N-Boc group was cleaved under acidic conditions, the solution was neutralized, and the mixture was purified by semi-preparative HPLC. The desired HPLC fractions were combined and the product was isolated by solid phase extraction according to a previously reported procedure.75 We were able to obtain higher decay-corrected radiochemical yields with [18F]FEtOBs than with [18F]FPrOBs in our radiosyntheses (see Experimental Section). This is presumably because the carbon atom bonded to the brosylate leaving group bears a larger partial positive charge in [18F]FEtOBs than [18F]FPrOBs due to the electron-withdrawing properties of the fluorine atom which is two bonds away in [18F]FEtOBs but three bonds away in [18F]FPrOBs. The octanol/water partition coefficients56-58 of [18F]1-[18F]4 were measured according to a known procedure76, 77 and are shown in Table 1. These values are all in the range56 that will allow for diffusion of the tracer across the blood-brain-barrier.
Scheme 2.

Table 1.
Octanol/water partition coefficients.
| Compound | LogP7.4a | n = |
|---|---|---|
| [18F]1 | 1.69 ± 0.01 | 7 |
| [18F]2 | 1.40 ± 0.03 | 4 |
| [18F]3 | 1.64 ± 0.14 | 12 |
| [18F]4 | 1.87 ± 0.03 | 8 |
= Average value of n determinations ± the standard deviation.
In Vitro Competition Binding Assays
Vinylhalide nortropanes 1-4 and trans-1 were screened for binding to human monoamine transporters using in vitro competition binding assays with transfected SERT, DAT, or NET according to our previously reported procedure.61, 78 The binding affinities for each transporter were determined using [3H](R,S)-citalopram•HBr78, 79 ([3H]15) (SERT), [125I]RTI-5580 (DAT), or [3H]Nisoxetine81 (NET). The data in Table 2 indicate that 1-4 all have a high affinity for the SERT and are selective for the SERT over the DAT and NET.55 Trans-1 has a reduced affinity at all three transporters as expected based on previous reports.60, 74
Table 2.
Results of In Vitro Competition Binding Assays with Transfected Human Monoamine Transporters.
|
Ki (nM)a |
SERT selectivity |
||||
|---|---|---|---|---|---|
| Compd | hSERT | hDAT | hNET | DAT/SERT | NET/SERT |
| 1 | 0.43 ± 0.01b | 87.6 ± 10.2b | 109.9 ± 6.3c | ~204 | ~256 |
| 2 | 0.33 ± 0.10b | 35.5 ± 5.9b | 101.3 ± 2.5b | ~108 | ~307 |
| 3 | 0.26 ± 0.03b | 179.7 ± 17.8b | 67.5 ± 5.6b | ~691 | ~260 |
| 4 | 0.33 ± 0.03b | 118.7 ± 13.8b | 30.2 ± 0.4b | ~360 | ~92 |
| trans-1 | 15.2 ± 3.9b | 274.0 ± 9.3b | 1180 ± 426b | ~18 | ~78 |
= Average value of n determinations ± SEM (each determination performed in triplicate).
n = 2.
n = 3.
In Vivo Nonhuman Primate MicroPET Imaging
The in vivo regional brain uptake of [18F]1-[18F]4 was determined in anesthetized cynomolgus monkeys (a total of 4 different cynomolgus monkeys) using a Concorde microPET P4 according to our previously reported procedure.61 Baseline studies were initially performed to determine the extent of uptake of [18F]1-[18F]4 in the SERT-rich regions of the brain. The baseline time-activity curves (TACs) for [18F]1 are shown in Figure 1. Compound [18F]1 enters the brain rapidly and achieves peak uptake in the SERT-rich brain regions after ~ 45 minutes (Table S1) with the highest uptake observed in the midbrain followed by the putamen, thalamus, medulla, and caudate. This rank order of uptake is similar to that observed with previously reported 11C-labeled diarylsulfides30, 31, 33, 34, 37 and is consistent with the known distribution of SERT in the brain.34, 59, 82, 83 Lesser uptake is observed in the pons and the occipital and frontal cortices. The uptake in the midbrain, putamen, thalamus, medulla, and caudate remains nearly constant for ~ 20 minutes (~ 45-65 min post-injection) and then begins to steadily wash out. In the cerebellum, a region with low SERT density,59 peak uptake is achieved after ~ 27 minutes followed by a rapid washout of radioactivity down to uptake levels similar to that observed in the occipital and frontal cortices. Between 65-175 minutes post-injection the washout from the SERT-rich brain regions remains nearly parallel with the washout from the cerebellum indicating that a quasi-equilibrium (a condition where the ratio of radioactivity uptake in the region of interest to reference region stays relatively constant)37 has been established. The microPET images from this baseline study are shown in Figure 2.
Figure 1.

MicroPET baseline TACs obtained by injection of [18F]1 into an anesthetized cynomolgus monkey.
Figure 2.

MicroPET images (summed 0-235 min) obtained by injection of [18F]1 into an anesthetized cynomolgus monkey.
Metabolite analysis of [18F]1 (Figure 3) was performed with arterial plasma samples and determined by an HPLC method with radioactivity detection as previously described.61 The initial arterial plasma sample was taken at 1 minute post-injection of [18F]1 and consisted of 93 % of the total plasma radioactivity as unmetabolized [18F]1. The percent of total plasma radioactivity then decreased over time during the course of the study to 13 % unmetabolized [18F]1 after 180 minutes. The radioactive metabolite eluted immediately after the void volume during HPLC analysis and is believed to be [18F]fluoroethanol (or one of its metabolic oxidation products [18F]fluoroacetaldehyde or [18F]fluoroacetic acid) that would result from hydrolysis of the [18F]fluoroethyl ester.84 The percentage of protein-bound [18F]1 in plasma at each time point is shown in Table S15 and was in the range of 5.6 - 8.5 % during the course of the study.
Figure 3.

Metabolite analysis of [18F]1 in an anesthetized cynomolgus monkey.
It has been previously shown that anesthesia during PET imaging can interfere with radioligand binding.85-88 We therefore performed a baseline study in an awake rhesus monkey with [18F]1 using a Siemens/CTI High Resolution Research Tomograph (HRRT) in order to determine if there was a difference in the behavior of [18F]1 in an awake versus an anesthetized state. The TACs are shown in Figure 4 and the PET images are shown in Figure 5. High uptake is observed in the thalamus, putamen, midbrain, and caudate with peak uptake achieved after ~ 45 min (Table S2). The behavior of [18F]1 in an awake monkey is very similar to its behavior in an anesthetized monkey thus demonstrating that the imaging properties of [18F]1 are not affected by anesthesia. We were not able to follow the washout of [18F]1 in the awake study for the same amount of time as with the anesthetized study due to the difficulty of keeping an awake monkey motionless for an extended period of time.
Figure 4.

HRRT baseline TACs obtained by injection of [18F]1 into an awake rhesus monkey.
Figure 5.

HRRT PET images (left, summed 60-120 min) obtained by injection of [18F]1 into an awake rhesus monkey. Composite MRI's of several rhesus monkeys (right), and overlaid images (center).
The TACs for the baseline studies with [18F]2-[18F]4 are shown in Figures 6-8, respectively. Compound [18F]2 reaches peak uptake after 55-65 minutes in the SERT-rich brain regions followed by a steady washout (Table S3). Peak uptake in the cerebellum is achieved after ~ 30 minutes followed by a rapid washout to a level of uptake slightly less than that observed in the occipital and frontal cortices. Peak uptake in the SERT-rich brain regions for [18F]3 was achieved after 45-55 minutes (Table S4) and for [18F]4 peak uptake was achieved after 35-45 minutes (Table S5) with both compounds showing a steady washout. The ratio of uptake in the SERT-rich brain regions vs. cerebellum uptake for compounds [18F]1-[18F]4 at 115 and 215 minutes post-injection is shown in Table 3 (the complete data is shown in Tables S6-S10 along with graphs of the uptake ratios vs. time in Figures S1-S5). The ratios for each tracer are very similar at these two time points thus indicating that these tracers all behave very similarly in vivo. The differences between the data in Table 3 are believed to be the result, at least partially, of individual differences between the monkeys studied rather than differences between the performance of each tracer. The uptake ratios for [18F]1 after 115 minutes in awake and anesthetized monkeys are also very similar which provides further evidence that isoflurane anesthesia does not influence the in vivo behavior of [18F]1.
Figure 6.

MicroPET baseline TACs obtained by injection of [18F]2 into an anesthetized cynomolgus monkey.
Figure 8.

MicroPET baseline TACs obtained by injection of [18F]4 into an anesthetized cynomolgus monkey.
Table 3.
Comparison of uptake of [18F]1-[18F]4 in SERT-rich brain regions to cerebellum uptake at 115 and 215 minutes post-injection.
| Brain | 115 min |
215 min |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Region | [18F]1a | [18F]1 | [18F]2 | [18F]3 | [18F]4 | [18F]1 | [18F]2 | [18F]3 | [18F]4 |
| Caudate | 1.6 | 1.4 | 1.5 | 1.2 | 1.4 | 1.5 | 1.6 | 1.2 | 1.7 |
| Putamen | 1.6 | 1.7 | 1.7 | 1.4 | 1.5 | 1.8 | 1.8 | 1.5 | 1.7 |
| Thalamus | 1.8 | 1.6 | 1.5 | 1.4 | 1.5 | 1.7 | 1.6 | 1.5 | 1.7 |
| Midbrain | 1.9 | 1.9 | 1.7 | 1.5 | 1.7 | 2.2 | 2.1 | 1.8 | 2.2 |
| Pons | 1.8 | 1.3 | 1.6 | 1.3 | 1.3 | 1.5 | 1.8 | 1.5 | 1.5 |
| Medulla | 1.3 | 1.5 | 1.5 | 1.5 | 1.5 | 1.7 | 1.7 | 1.8 | 1.8 |
| Occipital | N/A | 0.9 | 1.1 | 1.0 | 1.1 | 0.9 | 1.1 | 1.0 | 1.3 |
| Frontal | 1.2 | 1.0 | 1.0 | 0.8 | 1.0 | 1.1 | 1.1 | 1.0 | 1.0 |
= HRRT awake rhesus study.
In order to demonstrate that the observed uptake in the baseline studies with [18F]1-[18F]4 is the result of preferential binding to the SERT and not the DAT, chase studies were performed with the SERT ligand 15, and the DAT ligands (±)-methylphenidate•HCl89 (16) and RTI-113•HCl71, 90, 91 (17). Figures 9-12 show the TACs for the chase studies with 15 at 120 minutes post-injection of the tracer for compounds [18F]1-[18F]4, respectively. For all four chase studies the amount of radioactivity in the SERT-rich brain regions decreases to nearly the level of the cortices and cerebellum thus indicating that the observed uptake is the result of binding to the SERT. These results also indicate that [18F]1-[18F]4 can be used for occupancy studies of SERT-selective therapeutics.26 Graphs of the uptake ratio vs. time for these chase studies are shown in Figures S6-S9. Figure S10 shows the results of chasing [18F]1 with the DAT ligand 16 and Figure S11 shows the results of chasing [18F]1 with the DAT ligand 17. We chose to perform chase studies with both 16 and 17 because we wanted to chase [18F]1 with a tropane-based DAT ligand (17) and a non-tropane-based DAT ligand (16). In neither case did the amount of radioactivity uptake decrease after administration of the chase compound (other than normal washout) thus indicating that [18F]1 is not bound to the DAT. Chase studies using 16 were also performed with [18F]2-[18F]4 (Figures S12-S14, respectively) and, similarly to what was observed with [18F]1, the radioactivity was not displaced. We did not perform any chase studies with the NET ligand 1892, 93 because 18 also has an affinity for the SERT60 and we have previously demonstrated that 18 will displace SERT PET tracers.63, 64
Figure 9.

MicroPET TACs showing the results of injection of 15 (1.5 mg/kg) into an anesthetized cynomolgus monkey at 120 min post-injection of [18F]1.
Figure 12.

MicroPET TACs showing the results of injection of 15 (1.5 mg/kg) into an anesthetized cynomolgus monkey at 120 min post-injection of [18F]4.

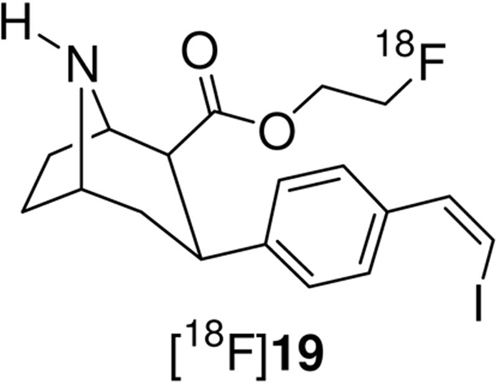
We have recently reported the microPET and HRRT PET imaging properties of [18F]19 in non-human primates (anesthetized and awake, respectively).64 Compound 19 is the para-substituted isomer of 1 and has a higher affinity at all three transporters than 1 (Ki = 0.08 nM SERT, 13 nM DAT, and 28 nM NET) but a lower SERT vs. DAT selectivity (~162). Comparison between [18F]19 and [18F]1 will reveal the effect of placing the vinyl iodide group in the para- or meta-position. The microPET baseline TACs for [18F]19 are reproduced in Figure S15 and the awake rhesus monkey HRRT baseline TACs are reproduced in Figure S17. Table 5 compares the times of peak uptake between [18F]19 and [18F]1 for the microPET and HRRT studies with each compound. In both the anesthetized and awake studies [18F]1 reaches peak uptake significantly faster than [18F]19 and [18F]1 also shows a greater washout (also see Tables S1, S2, S11, and S13). Thus, [18F]1 has superior imaging kinetics relative to [18F]19. Table 6 compares the ratio of uptake in SERT-rich brain regions to cerebellum uptake for [18F]1 and [18F]19 (also see Tables S6, S7, S12, and S14). Compound [18F]19 shows higher uptake ratios than [18F]1 in both anesthetized and awake states, and for both compounds the uptake ratios increase with time throughout the course of the studies (This is represented by Figures S1, S2, S16, and S18 where the uptake ratio is plotted vs. time). The differences in uptake ratio are a result of the differences in kinetics for each tracer. For compound [18F]1 (Figure 1) washout from the cerebellum begins after ~ 27 minutes and washout from the SERT-rich brain regions begins after ~65 minutes. This washout then remains fairly steady and the uptake ratios slowly increase until finally stabilizing after ~175 minutes (Figure S1). In contrast, [18F]19 reaches peak uptake in the cerebellum after 45 minutes and then begins to wash out while uptake in the SERT-rich brain regions continues to increase until 85-105 minutes followed by a very slow washout. This results in a continuously increasing uptake ratio with time (Figure S16). Similar differences between [18F]1 and [18F]19 are also observed in the awake monkey studies (Figures S2 and S18). Therefore, [18F]1 may be more ideally suited for measuring SERT density in humans due to its superior kinetics and the ability to achieve a quasi-equilibrium. Alternatively, as shown in Figure S19, chasing [18F]19 with 15 produces a more drastic displacement of [18F]19 than occurs with [18F]1 (Figure 9) which suggests that [18F]19 may be better suited to be used for occupancy determination studies of SSRI's.15, 25-27 Thus, compounds [18F]1 and [18F]19 each have their own unique imaging properties and are both promising candidates for use in human PET studies. The decision of which tracer to use, [18F]1 or [18F]19, would therefore have to be determined by the objective of the pending study.
Table 5.
Comparison of the times of peak uptake between [18F]1 and [18F]19 for anesthetized and awake monkey studies.a
| Time (min) |
||||
|---|---|---|---|---|
| Brain | Anesthetized |
Awake |
||
| Region | [18F]1 | [18F]19 | [18F]1 | [18F]19 |
| Caudate | 55 | 105 | 45 | 85 |
| Putamen | 45 | 95 | 45 | 85 |
| Thalamus | 45 | 85 | 45 | >75 |
| Midbrain | 45 | 95 | 45 | >75 |
| Pons | 45 | 115 | 85 | >75 |
| Medulla | 35 | 85 | 35 | 85 |
| Cerebellum | 28 | 45 | 45 | 35 |
Table 6.
Comparison of the uptake ratios between [18F]1 and [18F]19 for anesthetized and awake monkey studies at selected time points.a
| Anesthetized |
Awake |
|||||||
|---|---|---|---|---|---|---|---|---|
| Brain | 55 min |
115 min |
225 min |
95 min |
||||
| Region | [18F]1 | [18F]19 | [18F]1 | [18F]19 | [18F]1 | [18F]19 | [18F]1 | [18F]19 |
| Caudate | 1.1 | 1.1 | 1.4 | 1.7 | 1.6 | 2.5 | 1.5 | 2.0 |
| Putamen | 1.2 | 1.6 | 1.7 | 2.4 | 1.9 | 3.5 | 1.7 | 2.1 |
| Thalamus | 1.2 | 1.5 | 1.6 | 2.2 | 1.8 | 3.1 | 1.7 | 2.4 |
| Midbrain | 1.4 | 1.6 | 1.9 | 2.5 | 2.4 | 4.2 | 1.6 | 2.0 |
| Pons | 0.9 | 1.2 | 1.3 | 2.0 | 1.6 | 3.1 | 1.5 | 1.6 |
| Medulla | 1.1 | 1.5 | 1.5 | 2.1 | 1.9 | 3.0 | 1.1 | 1.5 |
Summary
The SERT ligands 1-4 were synthesized from m-bromophenyl tropane 5 and evaluated for binding to the SERT, DAT, and NET with in vitro competition binding assays using transfected cells. Compounds 1-4 all have a high and nearly equal affinity for the SERT and are selective for the SERT over the DAT and NET. Radiolabeling afforded tracers [18F]1-[18F]4 with higher decay-corrected radiochemical yields obtained with [18F]FEtOBs than with [18F]FPrOBs. Tracers [18F]1-[18F]4 were found to have lipophilicities in the range logP7.4 = 1.4 - 1.9. MicroPET imaging studies in anesthetized cynomolgus monkeys demonstrated that [18F]1-[18F]4 behave very similarly in vivo with peak uptake achieved after 45-55 min followed by a steady washout. The ratios of uptake in SERT-rich brain regions to cerebellum uptake for [18F]1-[18F]4 were also very similar. An HRRT study was performed with [18F]1 in an awake rhesus monkey to determine if anesthesia was influencing the behavior of [18F]1. The time to peak uptake and the uptake ratio of [18F]1 were similar between anesthetized and awake states indicating that anesthesia does not affect the imaging properties of [18F]1. Chase studies with the SERT ligand 15 displaced tracers [18F]1-[18F]4 but chase studies with the DAT ligand 16 did not displace tracers [18F]1-[18F]4 thus indicating that the observed uptake in the brain is a result of preferential binding to the SERT and not the DAT. Comparison between [18F]1 and the para-substituted isomer [18F]19 demonstrates that the position of the vinyl iodide group can have a significant affect on the tracer properties of the two compounds. Peak uptake in the putamen and caudate is achieved after 45 and 55 min, respectively, with [18F]1 but it takes 95 and 105 min, respectively, with [18F]19. Similar effects are observed in the midbrain and thalamus. Alternatively, the ratio of uptake in the SERT-rich brain regions to the cerebellum is higher with [18F]19 than with [18F]1. Both [18F]1 and [18F]19 are, therefore, promising candidates for advancement to human studies because [18F]1 offers faster kinetics than [18F]19 but [18F]19 offers higher uptake ratios than [18F]1 and it will be interesting to see if these differences are maintained in human subjects. Additionally, each compound can be radiolabeled as [123I]1 or [123I]19 for SPECT imaging which will further expand the possible uses of these two tracers. We have, therefore, chosen to further pursue [18F]1 out of this series of tracers reported above in combination with [18F]19 due to the higher radiochemical yields that can be obtained by O-alkylation with [18F]FEtOBs, the ability to use both [18F]1 and [18F]19 and to make direct comparisons between the two, and the possibility of radiolabeling each with 123I for SPECT imaging. Thus, we intend to evaluate both [18F]1 and [18F]19 in healthy normal human volunteers under baseline conditions as well as perform test-retest variability in dose-dependent blocking studies to determine their suitability for measuring occupancy in subjects with neuropsychiatric disorders.
Experimental Section
General
Solvents were purchased from VWR and had originated from EMD or Burdick & Jackson. Anhydrous solvents (100-mL septum-capped bottles) were purchased from Aldrich. TLC plates used were EMD glass-backed Silica Gel 60 F254, 20 × 20 cm, 250 μm. Preparatory TLC plates used were Analtech Uniplate Silica Gel GF 20 × 20 cm, 2000 μm. Silica gel used was EMD Silica Gel 60, 40-63 μm. Radial chromatography was performed with a Harrison Research Chromatotron. Semi-preparatory HPLC: Waters XTerra Prep RP18, 5 μm, 19 × 100 mm + guard cartridge (19 × 10 mm), 60:40:0.1 v/v/v MeOH/H2O/NEt3. Analytical HPLC: Waters NovaPak 3.9 × 150 mm, 75:25:0.1 v/v/v MeOH/H2O/NEt3. HRMS was performed by the Emory University Mass Spectrometry Center. NMR spectrometry was performed on Varian Inova and Unity spectrometers at the specified frequencies.
3β-(3′-Bromophenyl)tropane-2β-carboxylic acid (6)
2β-Carbomethoxy-3β-(3′-bromophenyl)tropane (5) (0.74 g, 2.19 mmol), 1,4-dioxane (15 mL) and H2O (15 mL) were stirred at reflux for 16 h. The solvent was removed azeotropically with EtOH to give a white solid that was dried under vacuum for 3 h. The solid was suspended in CHCl3 (5 mL), cooled to 0 °C, and the precipitate was isolated by filtration and dried under vacuum to afford 0.48 g (68%) of a white solid: 1H NMR (600 MHz, CD3OD) δ 7.51 (br s, 1 H), 7.36 (d, 1 H, J = 7.8 Hz), 7.31 (d, 1 H, J = 7.8 Hz), 7.20 (dd, 1 H, J = 7.8 Hz), 3.94 (m, 2 H), 3.36 (dt, 1 H, J = 6.0 Hz, J = 13.8 Hz), 2.81 (m, 1 H), 2.79 (s, 3 H), 2.69 (m, 1 H), 2.40 (m, 2 H), 2.17 (m, 2 H), 1.84 (m, 1 H); HRMS (APCI) [MH]+ Calcd for C15H19O2N79Br: 324.0594, Found: 324.0593; Calcd for C15H19O2N81Br: 326.0578, Found: 326.0574.
2β-Carbo(2-fluoroethoxy)-3β-(3′-bromophenyl)tropane (7)
3β-(3′-Bromophenyl)tropane-2β-carboxylic acid (6) (0.43 g, 1.33 mmol) was suspended in anhydrous CH2Cl2 (25 mL) under Ar followed by addition of anhydrous NEt3 (0.30 mL, 2.15 mmol, 1.6 equiv.) and cooling to -4 °C. Oxalyl chloride (1.0 mL, 2 M CH2Cl2, 2.0 mmol, 1.5 equiv.) was diluted with anhydrous CH2Cl2 (5 mL) and added dropwise through the condenser to the reaction flask over a period of 5 min. The reaction mixture was stirred under Ar at -4 °C for 20 min, warmed to rt, and the solvent was removed to give a brown solid that was dried under vacuum for 20 min. The solid was dissolved in anhydrous CH2Cl2 (25 mL) and cooled to -4 °C under Ar. FEtOH (0.80 mL, 13.62 mmol 10.2 equiv.) and anhydrous NEt3 (0.30 mL, 2.15 mmol, 1.6 equiv) were dissolved in anhydrous CH2Cl2 (10 mL) and added dropwise through the condenser to the acid chloride solution over a period of 3 min. The reaction mixture was stirred under Ar at -4 °C for 5 min, warmed to rt, stirred at rt for 80 min, and then the solvent was removed to give a brown solid that was dried under vacuum for 20 min. The solid was dissolved in CH2Cl2, poured onto dry silica (43 mm h × 43 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), v/v/v hexane/EtOAc/NEt3 75:20:5 (300 mL), 50:45:5 (100 mL) to afford 0.31 g (63 %) of a colorless syrup: TLC Rf = 0.38 (50:45:5 v/v/v hexane/EtOAc/NEt3); 1H NMR (600 MHz, CDCl3) δ 7.39 (s, 1 H), 7.29 (d, 1 H, J = 7.8 Hz), 7.21 (d, 1 H, J = 7.8 Hz), 7.14 (dd, 1 H, J = 7.8 Hz), 4.45 (dddd, 2 H, 2JHF = 46.8 Hz, 3JHH = 5.8 Hz, 3JHH = 2.6 Hz), 4.29 (dddd, 1 H, 3JHF = 29.4 Hz,2JHH = 13.2 Hz, 3JHH = 5.2 Hz, 3JHH = 2.6 Hz), 4.08 (dddd, 1 H, 3JHF = 28.2 Hz, 2JHH = 12.6 Hz, 3JHH = 5.9 Hz, 3JHH = 2.4 Hz), 3.63 (m, 1 H), 3.36 (m, 1 H), 2.98 (m, 2 H), 2.53 (td, 1 H, J = 3.0 Hz, J = 12.3 Hz), 2.22 (s, 3 H), 2.20 (m, 1 H), 2.11 (m, 1 H), 1.70 (m, 2 H), 1.60 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 171.29, 145.79, 130.73, 129.69, 129.16, 126.10, 122.38, 81.74 (d, 1JCF = 169.2 Hz), 65.47, 62.91 (d, 2JCF = 20.6 Hz), 62.33, 52.65, 42.08, 34.04, 33.71, 25.95, 25.45; HRMS (APCI) [MH]+ calcd for C17H22O2N79Br: 370.0813, found: 370.0812; calcd for C17H22O2N81Br: 372.0797, found: 372.0793.
2β-Carbo(3-fluoropropoxy)-3β-(3′-bromophenyl)tropane (8)
3β-(3′-Bromophenyl)tropane-2β-carboxylic acid (6) (0.11 g, 0.34 mmol) was suspended in anhydrous CH2Cl2 (8 mL) under Ar followed by addition of anhydrous NEt3 (0.08 mL, 0.57 mmol, 1.7 equiv.) and cooling to -4 °C. Oxalyl chloride (0.27 mL, 2 M CH2Cl2, 0.54 mmol, 1.6 equiv.) was diluted with anhydrous CH2Cl2 (2 mL) and added dropwise to the reaction flask over a period of 90 s. The reaction mixture was stirred under Ar at -4 °C for 20 min, warmed to rt, and the solvent was removed to give a brown solid that was dried under vacuum for 20 min. The solid was dissolved in anhydrous CH2Cl2 (10 mL) and cooled to -4 °C under Ar. FPrOH (0.32 g, 4.10 mmol 12.1 equiv.) and anhydrous NEt3 (0.08 mL, 0.57 mmol, 1.7 equiv.) were dissolved in anhydrous CH2Cl2 (5 mL) and added dropwise to the acid chloride solution. The reaction mixture was stirred under Ar at -4 °C for 5 min, warmed to rt, and the solvent was removed to give a brown solid that was dried under vacuum for 10 min. The solid was dissolved in CH2Cl2, poured onto dry silica (39 mm h × 43 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), v/v/v hexane/EtOAc/NEt3 75:20:5 (300 mL), 50:45:5 (100 mL) to afford 88 mg (67%) of a colorless syrup: TLC Rf = 0.41 (50:45:5 v/v/v hexane/EtOAc/NEt3); 1H NMR (600 MHz, CDCl3) δ 7.36 (s, 1 H), 7.29 (d, 1 H, J = 7.8 Hz), 7.19 (d, 1 H, J = 7.8 Hz), 7.14 (dd, 1 H, J = 7.8 Hz), 4.37 (2 m, 2 H, 2JHF = 47.1 Hz), 4.14 (m, 1 H), 3.98 (m, 1 H), 3.58 (m, 1 H), 3.35 (m, 1 H), 2.98 (dt, 1 H, J = 5.7 Hz, J = 12.0 Hz), 2.91 (m, 1 H), 2.50 (td, 1 H, J = 2.4 Hz, J = 12.6 Hz), 2.21 (s, 3 H), 2.18 (m, 1 H), 2.11 (m, 1 H), 1.85 (2 m, 2 H, 3JHF = 24.6 Hz), 1.70 (m, 2 H), 1.60 (m, 1 H); 13C NMR (150 MHz, CDCl3) δ 171.62, 146.01, 130.57, 129.72, 129.09, 125.98, 122.38, 80.89 (d, 1JCF = 165.0 Hz), 65.55, 62.29, 59.96 (d, 3JCF = 6.2 Hz), 52.80, 42.10, 34.11, 33.61, 29.94 (d, 2JCF = 20.6 Hz), 26.00, 25.43; HRMS (APCI) [MH]+ Calcd for C18H24O2N79BrF: 384.0969, Found: 384.0971; Calcd for C18H24O2N81BrF: 386.0954, Found: 386.0953.
2β-Carbo(2-fluoroethoxy)-3β-(3′-bromophenyl)nortropane (9)
2β-Carbo(2-fluoroethoxy)-3β-(3′-bromophenyl)tropane (7) (0.26 g, 7.02 × 10-4 mol), 2,2,2-trichloroethyl chloroformate (1.0 mL, 7.27 mmol, 10.4 equiv.), Na2CO3(s) (36 mg, 0.34 mmol, 0.5 equiv.), and toluene (15 mL) were stirred at reflux under Ar for 4 h, cooled, poured onto dry silica (43 mm h × 43 mm i.d.), and eluted under vacuum with CH2Cl2 (75 mL) and then 75:20:5 v/v/v/ hexane/EtOAc/NEt3. The solvent was removed to give a colorless residue that was dried under vacuum (0.37 g). To the residue was added Zn dust (0.46 g), AcOH (10 mL), and H2O (0.3 mL), and the mixture was stirred at rt for 21 h. The reaction mixture was filtered, the filtrate was diluted with CH2Cl2 (50 mL) and H2O (50 mL), and cooled to 0 °C. The aqueous phase was basified to pH 11 with conc. NH4OH(aq), the layers were separated, and the aqueous layer was extracted with CH2Cl2 (20 mL× 2). The combined CH2Cl2 layers were dried over MgSO4 and the solvent was removed to give a colorless oil. The oil was dissolved in CH2Cl2, poured onto dry silica (35 mm h × 43 mm i.d.), and eluted under vacuum: CH2Cl2 (75 mL), then v/v/v hexane/EtOAc/NEt3 75:20:5 (50 mL), 50:45:5 (50 mL), 20:75:5 (500 mL). The solvent was removed to give 0.18 g (72%) of a light yellow oil: TLC Rf = 0.10 (50:45:5 v/v/v hexane/EtOAc/NEt3); 1H NMR (600 MHz, CDCl3) δ 7.35 (s, 1 H), 7.33 (m, 1 H), 7.15 (m, 2 H), 4.36 (dddd, 1 H, 2JHF = 46.8 Hz, 2JHH = 10.8 Hz, 3JHH = 2.4 Hz, 3JHH = 5.4 Hz), 4.23 (dddd, 1 H, 2JHF = 48.0 Hz, 2JHH = 10.8 Hz, 3JHH = 2.4 Hz, 3JHH = 6.6 Hz), 4.12 (dddd, 1 H, 3JHF = 26.4 Hz, 2JHH = 13.2 Hz, 3JHH = 2.4 Hz, 3JHH = 6.6 Hz), 4.01 (dddd, 1 H, 3JHF = 30.0 Hz, 2JHH = 13.2 Hz, 3JHH = 2.4 Hz, 3JHH = 5.4 Hz), 3.75 (m, 2 H), 3.25 (dt, 1 H, J = 6.0 Hz, J = 12.6 Hz), 2.82 (m, 1 H), 2.37 (td, 1 H, J = 3.0 Hz, J = 13.2 Hz), 2.12 (m, 1 H), 2.01 (m, 1 H), 1.76 (m, 1 H), 1.67 (m, 3 H); 13C NMR (150 MHz, CDCl3) δ 172.98, 144.85, 130.64, 130.00, 129.82, 126.21, 122.61, 81.25 (d, 1JCF = 169.2 Hz), 62.99 (d, 2JCF = 20.7 Hz), 56.54, 53.76, 51.08, 35.44, 33.60, 29.33, 27.89; HRMS (APCI) [MH]+ Calcd for C16H20O2N79BrF: 356.0656, Found: 356.0656; Calcd for C16H20O2N81BrF: 358.0635, Found: 358.0636.
2β-Carbo(3-fluoropropoxy)-3β-(3′-bromophenyl)nortropane (10)
2β-Carbo(3-fluoropropoxy)-3β-(3′-bromophenyl)tropane (8) (78 mg, 2.03 × 10-4 mol), 2,2,2-trichloroethyl chloroformate (0.30 mL, 2.18 mmol, 10.7 equiv.), Na2CO3(s) (11 mg, 0.10 mmol, 0.5 equiv.), and toluene (5 mL) were stirred at reflux under Ar for 4 h, cooled, poured onto dry silica (25 mm h × 33 mm i.d.), and eluted under vacuum with CH2Cl2 (25 mL) and then 75:20:5 v/v/v/ hexane/EtOAc/NEt3 (100 mL). The solvent was removed to give a colorless residue that was dried under vacuum (0.11 g). To the residue was added Zn dust (0.13 g), AcOH (4 mL), and H2O (0.1 mL), and the mixture was stirred at rt for 21 h. The reaction mixture was filtered, the filtrate was diluted with CH2Cl2 (25 mL) and H2O (25 mL), and cooled to 0 °C. The aqueous phase was basified to pH 10 with conc. NH4OH(aq), the layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined CH2Cl2 layers were dried over MgSO4 and the solvent was removed to give a colorless oil. The oil was dissolved in CH2Cl2, poured onto dry silica (26 mm h × 33 mm i.d.), and eluted under vacuum: CH2Cl2 (75 mL), then v/v/v hexane/EtOAc/NEt3 75:20:5 (50 mL), 50:45:5 (75 mL), 20:75:5 (100 mL). The solvent was removed to give 44 mg (59%) of a light yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 2 H), 7.15 (m, 2 H), 4.17 (ddd, 2 H, 2JHF = 47.4 Hz, JHH = 5.7 Hz), 4.01 (m, 1 H), 3.91 (m, 1 H), 3.73 (m, 2 H), 3.24 (dt, 1 H, J = 5.9 Hz, J = 13.2 Hz), 2.73 (d, 1 H, J = 5.4 Hz), 2.36 (td, 1 H, J = 2.4 Hz, J = 13.2 Hz), 2.13 (m, 1 H), 2.02 (m, 1 H), 1.69 (m, 6 H); 13C NMR (150 MHz, CDCl3) δ 173.30, 144.95, 130.74, 130.04, 129.83, 126.21, 122.61, 80.46 (d, 1JCF = 165.2 Hz), 60.11 (d, 3JCF = 5.9 Hz), 56.45, 53.67, 51.00, 35.62, 33.56, 29.68 (d, 2JCF = 20.7 Hz), 29.22, 27.79; HRMS (APCI) [MH]+ Calcd for C17H22O2N79BrF: 370.0813, Found: 370.0814; Calcd for C17H22O2N81BrF: 372.0792, Found: 372.0793.
(Z)-1,2-Bis(trimethylstannyl)ethene
Purified acetylene (passed successively through a -78 °C cold trap, conc. H2SO4(aq), NaOH(s), CaCl2(s), and then Drierite) was bubbled through a solution of hexamethylditin (2.52 g, 7.69 mmol), Pd(PPh3)4 (0.89 g, 0.77 mmol), and 1,4-dioxane (20 mL, purged with Ar for 45 min prior to use) at 65 °C for 4 h. The solution was cooled to rt, stirred at rt for 20 min, and filtered. The filtrate was poured onto silica gel (14 cm high × 4 cm i.d.) that had been pretreated with 10% NEt3/hexane (100 mL) and then 1% NEt3/hexane (100 mL). The product was eluted under vacuum with 1% NEt3/hexane (200 mL) and the solvent was removed to give a dark orange oil that was briefly dried under vacuum (2.48 g, 91%). TLC Rf = 0.66 (1% NEt3/Hexane); 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 2 H), 0.17 (t, 18 H, 2JSnH = 26.6 Hz); 13C NMR (150 MHz, CDCl3) δ 155.17, - 8.10.
(Z)-1,2-Bis(trimethylstannyl)ethene was found to be stable (by 1H NMR spectroscopy) for at least 1 year when stored at -15 °C under Ar and protected from moisture and light (the vial was flushed with Ar, capped, wrapped with Parafilm, completely wrapped in Al foil, and then stored in a resealable plastic bag in a freezer).
2β-Carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (11)
2β-Carbo(2-fluoroethoxy)-3β-(3'-bromophenyl)nortropane (9) (0.23 g, 6.46 × 10-4 mol), (Z)-1,2-bis(trimethylstannyl)ethene (0.72 g, 2.04 mmol, 3.2 equiv), Pd(PPh3)4 (80 mg, 6.92 × 10-5 mol, 0.1 equiv), and toluene (25 mL, purged with Ar for 2 h) were stirred at reflux under Ar for 18 h. The reaction mixture was cooled to rt, stirred at rt for 1.5 h, poured onto dry silica (40 mm h × 43 mm i.d.), and eluted under vacuum: CH2Cl2 (100 mL), hexane (50 mL), v/v/v hexane/EtOAc/NEt3 75:20:5 (100 mL), 50:45:5 (100 mL), 20:75:5 (300 mL). The solvent was removed to give an orange oil that was radially chromatographed (4 mm silica, v/v/v hexane/EtOAc/NEt3 90:8:2 (300 mL), 75:20:5 (1 L)) to afford 0.13 g of a colorless syrup that was ~73:27 cis/trans by integration of the 1H NMR vinyl resonances. The product was again radially chromatographed (2 mm silica, v/v/v hexane/EtOAc/NEt3 95:4:1 (1 L), 90:8:2 (800 mL), 75:20:5 (200 mL), 50:45:5 (200 mL)) to afford 64 mg (21%) of a colorless syrup that was ~93:7 cis/trans, 29 mg (10%) of a colorless syrup that was ~76:24 cis/trans, and 29 mg (10%) of a colorless syrup that was ~47:53 cis/trans. TLC Rf = 0.28 (20:75:5 v/v/v hexane/EtOAc/NEt3); 1H NMR (600 MHz, CDCl3) δ 7.54 (td, 1 H, 3JHH = 13.8 Hz, 3JSnH = 75.0 Hz), 7.23 (dd, 1 H, J = 7.8 Hz), 7.11 (m, 3 H), 6.18 (td, 1 H, 3JHH = 13.8 Hz, 2JSnH = 32.0 Hz), 4.33 (dddd, 1 H, 2JHF = 46.9 Hz, 2JHH = 10.8 Hz, 3JHH = 5.4 Hz, 3JHH = 2.4 Hz), 4.20 (dddd, 1 H, 2JHF = 47.7 Hz, 2JHH = 10.8 Hz, 3JHH = 6.9 Hz, 3JHH = 2.4 Hz), 4.10 (partially resolved dddd, 1 H, 3JHH = 2.4 Hz), 3.95 (dddd, 1 H, 3JHF = 30.1 Hz, 2JHH = 13.0 Hz, 3JHH = 5.4 Hz, 3JHH = 2.4 Hz), 3.74 (m, 2 H), 3.27 (dt, 1 H, J = 5.4 Hz, J = 13.2 Hz), 2.83 (m, 1 H), 2.43 (td, 1 H, J = 3.0 Hz, J = 12.9 Hz), 2.13 (m, 1 H), 2.02, (m, 1 H), 1.77 (m, 1 H), 1.68 (m, 2 H), 1.61 (br s, 1 H), 0.08 (t, 9 H, 2JSnH = 27.3 Hz); 13C NMR (100 MHz, CDCl3) δ 173.18, 147.41, 142.29, 141.13, 133.71, 128.30, 126.86, 126.47, 125.57, 81.22 (d, 1JCF = 169.1 Hz), 62.85 (d, 2 JCF = 20.5 Hz), 56.53, 53.84, 51.24, 35.75, 33.84, 29.37, 27.88, -7.89; HRMS (EeSI) [M+H]+ Calcd for C21H31O2NF120 Sn: 468.1355, Found: 468.1354.
2β-Carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (12)
2β-Carbo(3-fluoropropoxy)-3β-(3′-bromophenyl)nortropane (10) (0.15 g, 4.05 × 10-4 mol), (Z)-1,2-bis(trimethylstannyl)ethene (0.54 g, 1.53 mmol, 3.8 equiv), Pd(PPh3)4 (73 mg, 6.32 × 10-5 mol, 0.16 equiv), and toluene (15 mL) were stirred at reflux under Ar for 16 h. The reaction mixture was cooled to rt, poured onto dry silica (40 mm h × 43 mm i.d.) that had been pretreated with 10% NEt3/hexane (50 mL), and eluted under vacuum: CH2Cl2 (50 mL), hexane/EtOAc/NEt3 v/v/v 75:20:5 (50 mL), 50:45:5 (50 mL), 20:75:5 (400 mL). The solvent was removed to give a brown oil (0.17 g) that was ~78:22 cis/trans (+ impurities) by integration of the 1H NMR vinyl resonances. Purification by radial chromatography (2 mm silica, v/v/v hexane/EtOAc/NEt3 95:4:1 (100 mL), 90:8:2 (700 mL), 85:12:3 (100 mL), 80:16:4 (100 mL), 75:20:5 (300 mL)) afforded 52 mg (27%) of a faint yellow syrup that was ~91:9 cis/trans and 41 mg (21%) of a faint yellow syrup that was ~68:32 cis/trans. TLC Rf = 0.24 (silica, 20:75:5 v/v/v hexane/EtOAc/NEt3); 1H NMR (600 MHz, CDCl3) δ 7.52 (td, 1 H, 3JHH = 13.8 Hz, 3JSnH = 73.4 Hz), 7.22 (dd, 1 H, J = 7.5 Hz), 7.10 (s, 1 H), 7.08 (m, 2 H), 6.17 (td, 1 H, 3JHH = 13.8 Hz, 2JSnH = 32.1 Hz), 4.17 and 4.09 (dm, 1 H + 1 H, 2JHF = 46.8 Hz), 3.92 (m, 2 H), 3.73 (m, 1 H), 3.70 (m, 1 H), 3.25 (dt, 1 H, J = 5.9 Hz, J = 13.2 Hz), 2.73 (m, 1 H), 2.62 (br s, 1 H, NH - conc. dependent), 2.41 (td, 1 H, J = 13.2 Hz, J = 2.6 Hz), 2.11 (m, 1 H), 2.02 (m, 1 H), 1.76-1.58 (m, 5 H), 0.08 (t, 9 H, 2JSnH = 27.0 Hz); 13C NMR (150 MHz, CDCl3) δ 173.53, 147.32, 142.42, 141.09, 133.71, 128.35, 126.86, 126.50, 125.69, 80.53 (d, 1JCF = 165.5 Hz), 59.97 (d, 3JCF = 6.2 Hz), 56.52, 53.79, 51.20, 35.97, 33.90, 29.68 (d, 2JCF =19.9 Hz), 29.33, 27.86, -7.84; HRMS (ESI) [M+H]+ Calcd for C22H33O2NF120Sn: 482.1512, found: 482.1513.
2β-Carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane (1)
2β-Carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (11) (~96:4 cis/trans, 35 mg, 7.51 × 10-5 mol) was dissolved in CHCl3 (5 mL) and cooled to -2 °C under Ar. ICl (0.11 mL, 1.0 M CH2Cl2, 0.11 mmol, 1.5 equiv) was added dropwise, the reaction mixture was stirred at -2 °C under Ar for 15 min, and quenched by addition of Na2S2O3•5 H2O (0.307 g, 1.24 mmol, in 5 mL H2O). The mixture was diluted with CHCl3 (5 mL) and H2O (5 mL), the layers were separated, and the aqueous phase extracted with CHCl3 (5 mL × 2). The combined CHCl3 layers were dried over MgSO4, the solvent was removed, and the residue was purified by preparative TLC (20:75:5 v/v/v hexane/EtOAc/NEt3 × 2) to give 23 mg (71%) of a colorless residue that was ~95:5 cis/trans by integration of the 1H NMR vinyl resonances. Separation of the isomers by semi-preparative HPLC (cistR = 19.7 min, 8.8 ml/min) afforded 16 mg (50 %) of 1 as a white residue: 1H NMR (600 MHz, CDCl3) δ 7.47 and 7.46 (overlapping resonances, 2 H), 7.30 (m, 2 H), 7.20 (d, 1 H, J = 7.8 Hz), 6.56 (d, 1 H, J = 8.4 Hz), 4.31 (dddd, 1 H, 2JHF = 47.1 Hz, 2JHH = 10.6 Hz, 3JHH = 5.2 Hz, 3JHH = 2.4 Hz), 4.20 (partially resolved ddd, 0.5 H, 2JHH = 10.6 Hz, 3JHH = 7.1 Hz, 3JHH = 2.4 Hz), 4.11 (overlapping resonances, 0.5 H + 0.5 H, 3JHH = 2.4 Hz), 4.06 (ddd, 0.5 H, 2JHH = 12.9 Hz, 3JHH = 7.1 Hz, 3JHH = 2.4 Hz), 3.97 (dddd, 1 H, 3JHF = 30.3 Hz, 2JHH = 12.9 Hz, 3JHH = 5.2 Hz, 3JHH = 2.4 Hz), 3.77 (m, 2 H), 3.32 (dt, 1 H, J = 6.0 Hz, J = 12.6 Hz), 2.88 (m, 1 H), 2.44 (td, 1 H, J = 13.2 Hz, J = 3.0 Hz), 2.14 (m, 1 H), 2.03 (m, 1 H), 1.80 (m, 1 H), 1.71 (overlapping m + br s, 2 H + 1 H); 13C NMR (150 MHz, CDCl3) δ 172.90, 141.59, 138.68, 136.93, 128.37, 127.55, 127.45, 126.93, 81.26 (d, 1JCF = 170.1 Hz), 79.79, 63.26 (d, 2JCF = 18.7 Hz), 56.27, 53.91, 50.76, 35.28, 32.84, 28.71, 27.31; HRMS (ESI) [MH]+ calcd for C18H22O2NF127I: 430.0674, found: 430.0660.
trans-1
1H NMR (600 MHz, CDCl3) δ 7.40 (d, 1 H, J = 15.0 Hz), 7.24 (dd, 1 H, J = 7.8 Hz), 7.14 (s, 1 H), 7.13 (m, 2 H), 6.82 (d, 1 H, J = 15.0 Hz), 4.31 (dddd, 1 H, 2JHF = 47.2 Hz, 2JHH = 10.7 Hz, 3JHH = 5.5 Hz, 3JHH = 2.4 Hz), 4.19 (partially resolved ddd, 0.5 H, 2JHH = 10.7 Hz, 3JHH = 6.7 Hz, 3JHH = 2.4 Hz), 4.11 (overlapping resonances, 0.5 H + 0.5 H, 3JHH = 2.4 Hz), 4.06 (ddd, 0.5 H, 2JHH = 12.9 Hz, 3JHH = 6.7 Hz, 3JHH = 2.4 Hz), 3.95 (dddd, 1 H, 3JHF = 30.1 Hz, 2JHH = 12.9 Hz, 3JHH = 5.5 Hz, 3JHH = 2.4 Hz), 3.77 (m, 2 H), 3.27 (dt, 1 H, J = 12.6 Hz, J = 6.0 Hz), 2.83 (m, 1 H), 2.62 (br s, 1 H, NH), 2.41 (td, 1 H, J = 12.9 Hz, J = 3.0 Hz), 2.15 (m, 1 H), 2.04 (m, 1 H), 1.78 (m, 1 H), 1.68 (m, 2 H); semi-preparative HPLC (tR = 26.9 min, 8.8 mL/min).
2β-Carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane (2)
2β-Carbo(2-fluoroethoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (11) (~80:20 cis/trans, 38 mg, 8.15 × 10-5 mol), was dissolved in CH2Cl2 (4 mL) and cooled to 0 °C under Ar. Br2 (35 mg, 0.22 mmol) was dissolved in CH2Cl2 (1 mL) and added dropwise until a faint yellow color remained. The reaction mixture was stirred at 0 °C under Ar for 6 min and then quenched by addition of Na2S2O3•5 H2O (0.57 g in 4 mL H2O). The reaction mixture was diluted by addition of CH2Cl2 (10 mL) and H2O (10 mL), the layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 2). The combined CH2Cl2 layers were dried over MgSO4 and the solvent was removed to give a colorless residue that was purified by preparative TLC (silica, 20:75:5 v/v/v hexane/EtOAc/NEt3) to afford 21 mg (67%) of a colorless residue that was ~91:9 cis/trans by integration of the 1H NMR vinyl resonances. The isomers were separated by semi-preparative HPLC (9 mL/min, cistR = 15.9 min, transtR = 20.4 min) to afford 16 mg (51%) of 2 as a colorless residue: 1H NMR (600 MHz, CDCl3) δ 7.53 (d, 1 H, J = 7.8 Hz), 7.49 (s, 1 H), 7.30 (dd, 1 H, J = 7.8 Hz), 7.18 (d, 1 H, J = 7.2 Hz), 7.04 (d, 1 H, J = 8.4 Hz), 6.42 (d, 1 H, J = 7.8 Hz), 4.30 (dddd, 1 H, 2JHF = 47.1 Hz, 2JHH= 10.7 Hz,3JHH= 2.4 Hz, 3JHH= 5.3 Hz), 4.15 (unresolved dddd, 1 H, 2JHF = 47.1 Hz, 2JHH = 2.4 Hz), 4.09 (unresolved dddd, 1 H, 2JHH = 2.4 Hz), 3.96 (dddd, 1 H, 3JHF = 30.6 Hz, 2JHH= 12.9 Hz, 3JHH= 2.4 Hz, 3JHH= 5.3 Hz), 3.82 (br s, 1 H), 3.31 (dt, 1 H, J = 6.0 Hz, J = 13.2 Hz), 2.87 (m, 1 H), 2.45 (unresolved td, 1 H, J = 12.6 Hz), 2.19 (m, 1 H), 2.07 (m, 1 H), 1.80 (m, 1 H), 1.72 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 173.13, 142.03, 135.13, 132.51, 128.40, 128.10, 127.56, 127.47, 106.71, 81.27 (d, 1JCF = 169.2 Hz), 63.08 (d, 2JCF = 18.6 Hz), 56.42, 53.89, 51.00, 35.47, 33.31, 29.07, 27.64; Analytical HPLC (tR = 4.4 min, 1 mL/min); HRMS (ESI) [MH]+ Calcd for C18H22O2NF79 Br: 382.0813 , Found: 382.0815; Calcd for C18H22O2NF81Br: 384.0792 , Found: 384.0796.
2β-Carbo(3-fluoropropoxy)-3β-(3′((Z)-2-iodoethenyl)phenyl)nortropane (3)
2β-Carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (12) (~ 95:5 cis/trans, 84 mg, 1.75 × 10-4 mol) was dissolved in CHCl3 (10 mL) and cooled to -7 °C under Ar(g). ICl (0.27 mL, 1.0 M CH2Cl2, 0.27 mmol, 1.5 equiv.) was added dropwise, the reaction mixture was warmed to rt, stirred at rt for 10 min, and quenched by addition of Na2S2O3•5 H2O (0.683 g, 2.75 mmol, in 10 mL H2O). The reaction mixture was diluted with CHCl3 (25 mL) and H2O (25 mL), the layers were separated, and the aqueous layer was extracted with CHCl3 (10 mL × 2). The combined CHCl3 layers were dried over MgSO4 and the solvent removed to give a yellow residue (85 mg) that was purified on Waters silica Sep-Pak Classics (2 in series): loaded with CH2Cl2 (1 mL), eluted with CH2Cl2 (1 mL), then hexane/EtOAc/NEt3 v/v/v 90:8:2 (2 mL), 75:20:5 (5 mL), 50:45:5 (10 mL), 20:75:5 (25 mL). The desired fractions were combined and the solvent was removed to give 43 mg (55%) of a yellow oil that was ~86:14 cis/trans by integration of the 1H NMR vinyl resonances. The isomers were separated by semi-preparative HPLC (8.8 mL/min; cistR = 25.2 min; transtR = 34.3 min) to afford 26 mg (34%) of 3 as an opaque residue: 1H NMR (600 MHz, CDCl3) δ 7.47 and 7.46 (overlapping resonances, 2 H, J = 8.4 Hz), 7.29 (m, 2 H), 7.18 (d, 1 H, J = 7.8 Hz), 6.56 (d, 1 H, J = 8.4 Hz), 4.14 (m, 1 H), 4.06 (m, 1 H), 3.98 (p, 1 H, J = 6.0 Hz), 3.87 (m, 1 H), 3.76 (m, 1 H), 3.73 (m, 1 H), 3.31 (dt, 1 H, J = 13.2 Hz, J = 5.4 Hz), 2.78 (d, 1 H, J = 5.4 Hz), 2.53 (br s, 1 H, NH), 2.43 (td, 1 H, J = 2.4 Hz, J = 12.9 Hz), 2.14 (m, 1 H), 2.03 (m, 1 H), 1.78 (m, 1 H), 1.68 (m, 3 H), 1.58 (m, 1 H); 13C NMR (150 MHz, CDCl3) δ 173.52, 142.47, 138.64, 136.79, 128.32, 127.63, 126.78, 80.55 (d, 1JCF = 165.0 Hz), 79.56, 59.97 (d, 3JCF = 4.5 Hz), 56.47, 53.75, 51.19, 35.73, 33.58, 29.70 (d, 2JCF = 20.6 Hz), 29.31, 27.83; HRMS (ESI) [MH]+ Calcd for C19H24O2NF127I: 444.0830, found: 444.0825;
2β-Carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane (4)
2β-Carbo(3-fluoropropoxy)-3β-(3′-((Z)-2-trimethylstannylethenyl)phenyl)nortropane (12) (~ 91:9 cis/trans, 52 mg, 1.08 × 10-4 mol) was dissolved in CHCl3 (5 mL) and cooled to 0 °C under Ar. Br2 (58 mg, 3.63 × 10-4 mol, 3.4 equiv.) was dissolved in CHCl3 (1 mL) and added dropwise until a faint yellow color persisted (not all of the solution was added). The reaction mixture was stirred at 0 °C under Ar for 15 min and then quenched by addition of Na2S2O3•5 H2O (98 mg, 3.95 × 10-4 mol, dissolved in 10 mL H2O). The mixture was diluted with CHCl3 (10 mL) and H2O (10 mL), the layers were separated, and the aqueous layer was extracted with CHCl3 (10 mL × 2). The combined CHCl3 layers were dried over MgSO4 and the solvent was removed to give a colorless syrup that became an opaque residue when dried under vacuum. The residue was dissolved in CH2Cl2, poured onto dry silica (26 mm h × 33 mm i.d.), and eluted under vacuum: CH2Cl2 (25 mL), hexanes/EtOAc/NEt3 v/v/v 75:20:5 (25 mL), 50:45:5 (50 mL), 20:75:5 (250 mL). The isolated product was further purified by preparative TLC (20:75:5 v/v/v hexanes/EtOAc/NEt3) to give a faint yellow syrup (26 mg) that was ~93:7 cis/trans by integration of the 1H NMR vinyl resonances. The isomers were separated by semi-preparative HPLC (cistR = 20.3 min, 9.0 mL/min) to afford 21 mg (49%) of a colorless residue: 1H NMR (600 MHz, CDCl3) δ 7.53 (d, 1 H, J = 7.2 Hz), 7.49 (s, 1 H), 7.30 (dd, 1 H, J = 7.2 Hz), 7.15 (d, 1 H, J = 7.2 Hz), 7.04 (d, 1 H, J = 8.4 Hz), 6.44 (d, 1 H, J = 8.4 Hz), 4.11 (m, 1 H), 4.03 (m, 1 H), 3.97 (quintet, 1 H, J = 5.9 Hz), 3.87 (m, 2 H), 3.82 (m, 1 H), 3.32 (dt, 1 H, J = 6.0 Hz, J = 12.6 Hz), 2.80 (overlapping resonances: br s + m, 2 H), 2.46 (td, 1 H, J = 2.4 Hz, J = 13.2 Hz), 2.25 (m, 1 H), 2.14 (m, 1 H), 1.82 (m, 1 H), 1.74 (m, 2 H), 1.65 (m, 1 H), 1.54 (m, 1 H); 13C NMR (150 MHz, CDCl3) δ 173.55, 141.61, 135.24, 132.32, 128.51, 128.11, 127.69, 127.51, 106.90, 80.41 (d, 1JCF = 166.0 Hz), 60.38 (d, 3JCF = 4.2 Hz), 56.31, 53.86, 50.64, 35.63, 33.00, 29.57 (d, 2JCF = 18.7 Hz), 28.63, 27.34; HRMS (ESI) [M+H]+ Calcd for C19H24O2FN79Br: 396.0969, found: 396.0974.
[18F]Fluoroethylbrosylate
H18F was produced with a Siemens 11-MeV RDS 112 cyclotron by employing the 18O(p,n)18F reaction in H218O. The H18F(aq) was transferred to a chemical processing control unit (CPCU), collected on a trap/release cartridge, released with K2CO3(aq) (0.9 mg in 0.6 mL H2O), and added to a CH3CN solution of Kryptofix 222 (5 mg in 1 mL). The solution was placed in a 110 °C oil bath, the solvent was evaporated under a N2(g) flow, and CH3CN (3 mL) was added and evaporated in order to azeotropically dry the Kryptofix 222/ K18F. 1,2-Dibrosylethane (4 mg in 1 mL CH3CN) was added, the reaction mixture was heated at 90 °C for 10 min, and the [18F]fluoroethylbrosylate was trapped on a Waters silica Sep-Pak Classic (WAT051900) (previously prepped with 10 mL EtOEt). The [18F]fluoroethylbrosylate was eluted with EtOEt, the EtOEt solution was transferred to a hot cell under N2(g) pressure and collected in a V-tube to give [18F]fluoroethylbrosylate in 74 % radiochemical yield (decay corrected from transfer of H18F(aq) to the CPCU). The V-tube was placed in an 80 °C oil bath and the EtOEt was evaporated with an Ar(g) flow. The solution of radiolabeling precursor was then added to this V-tube (see below).
[18F]Fluoropropylbrosylate
Prepared in an analogous manner as [18F]fluoroethylbrosylate using 1,3-dibrosylpropane.
2β-Carbo(2-[18F]fluoroethoxy)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane ([18F]1)
N-(t-butoxycarbonyl)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane-2β-carboxylic acid (13) (~ 1.1 mg, ~98:2 cis/trans) was dissolved in DMF (0.3 mL), deprotonated by addition of 0.1 M Bu4NOH(aq) (16 μL, 0.7 equiv), and added to [18F]fluoroethylbrosylate. The solution was heated at 90 °C for 10 min, 6 M HCl(aq) (~ 0.16 mL, ~ 422 equiv) was added, the solution was heated at 90 °C for 10 min, cooled to 0 °C, and neutralized by addition of 6 M NH4OH(aq) (~ 0.16 mL, ~ 422 equiv). The solution was diluted with HPLC solvent and purified by semi-preparative HPLC (tR (range) = 15-19 min, 9.2 mL/min). The desired fractions were combined, diluted 1:2 v/v with H2O and loaded onto a Waters C18 Sep-Pak. The Sep-Pak was washed with 0.9 % NaCl(aq) (40 mL) and then EtOH (0.5 mL). The product was eluted from the Sep-Pak with EtOH (1.5 mL) and collected in a sealed sterile vial containing 0.9 % NaCl(aq) (3.5 mL). This solution was passed successively through a 1 μm filter and then a 0.2 μm filter (Acrodisc PTFE) under Ar-pressure and collected in a sealed sterile dose vial containing 0.9 % NaCl(aq) (10 mL). The total synthesis time was ~ 80 min from the delivery of [18F]fluoroethylbrosylate to the hot cell with a 6.3 ± 1.8 % (n = 4) radiochemical yield (decay corrected). The product was then analyzed by analytical HPLC (tR = 4.9 min, 1 mL/min) to determine the radiochemical purity (97 ± 2 %, n = 4).
2β-Carbo(2-[18F]fluoroethoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane ([18F]2)
N-(t-butoxycarbonyl)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane-2β-carboxylic acid (14) (~0.6 mg, ~95:5 cis/trans) was dissolved in DMF (0.3 mL), deprotonated by addition of 0.1 M Bu4NOH(aq) (10 μL, 0.7 equiv), and added to [18F]fluoroethylbrosylate. The solution was heated at 90 °C for 10 min, 6 M HCl(aq) (~ 0.11 mL, ~ 480 equiv) was added, the solution was heated at 90 °C for 10 min, cooled to 0 °C, and neutralized by addition of 6 M NH4OH(aq) (~ 0.11 mL, ~ 480 equiv). The solution was diluted with HPLC solvent and purified by semi-preparative HPLC (tR (range) = 10-16 min, 9.2 mL/min). The desired fractions were combined, diluted 1:2 v/v with H2O and loaded onto a Waters C18 Sep-Pak. The Sep-Pak was washed with 0.9 % NaCl(aq) (40 mL) and then EtOH (0.5 mL). The product was eluted from the Sep-Pak with EtOH (1.5 mL) and collected in a sealed sterile vial containing 0.9 % NaCl(aq) (3.5 mL). This solution was passed successively through a 1 μm filter and then a 0.2 μm filter (Acrodisc PTFE) under Ar-pressure and collected in a sealed sterile dose vial containing 0.9 % NaCl(aq) (10 mL). The total synthesis time was ~75 min from the delivery of [18F]fluoroethylbrosylate to the hot cell with a 4.2 ± 2.7 % (n = 3) radiochemical yield (decay corrected). The product was then analyzed by analytical HPLC (tR = 4.4 min, 1 mL/min) to determine the radiochemical purity (95 ± 2 %, n = 3).
2β-Carbo(3-[18F]fluoropropoxy)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane ([18F]3)
N-(t-butoxycarbonyl)-3β-(3′-((Z)-2-iodoethenyl)phenyl)nortropane-2β-carboxylic acid (13) (~0.6 mg) was dissolved in DMF (0.3 mL), deprotonated by addition of 0.1 M Bu4NOH(aq) (11 μL, 0.9 equiv), and added to [18F]fluoropropylbrosylate. The solution was heated at 105 °C for 10 min, 6 M HCl(aq) (~0.12 mL, ~ 580 equiv) was added, the solution was heated at 105 °C for 10 min, cooled to 0 °C, and neutralized by addition of 6 M NH4OH(aq) (~0.12 mL, ~ 580 equiv). The solution was diluted with HPLC solvent and purified by semi-preparative HPLC (tR (range) = 21-25 min, 9.2 mL/min). The desired fractions were combined, diluted 1:2 v/v with H2O and loaded onto a Waters C18 Sep-Pak. The Sep-Pak was washed with 0.9 % NaCl(aq) (40 mL) and then EtOH (0.5 mL). The product was eluted from the Sep-Pak with EtOH (1.5 mL) and collected in a sealed sterile vial containing 0.9 % NaCl(aq) (3.5 mL). This solution was passed successively through a 1 μm filter and then a 0.2 μm filter (Acrodisc PTFE) under Ar-pressure and collected in a sealed sterile dose vial containing 0.9 % NaCl(aq) (10 mL). The total synthesis time was ~80 min from the delivery of [18F]fluoropropylbrosylate to the hot cell with a 1.9 ± 0.8 % (n = 3) radiochemical yield (decay corrected). The product was then analyzed by analytical HPLC (tR = 6.3 min, 1 mL/min) to determine the radiochemical purity (94 ± 4 %, n = 3).
2β-Carbo(3-[18F]fluoropropoxy)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane ([18F]4)
N-(t-butoxycarbonyl)-3β-(3′-((Z)-2-bromoethenyl)phenyl)nortropane-2β-carboxylic acid (14) (~0.6 mg) was dissolved in DMF (0.3 mL), deprotonated by addition of 0.1 M Bu4NOH (aq) (12 μL, 0.9 equiv), and added to [18 F]fluoropropylbrosylate. The solution was heated at 90 °C for 10 min, 6 M HCl(aq) (~0.15 mL, ~ 655 equiv) was added, the solution was heated at 90 °C for 10 min, cooled to 0 °C, and neutralized by addition of 6 M NH4OH(aq) (~0.15 mL, ~ 655 equiv). The solution was diluted with HPLC solvent and purified by semi-preparative HPLC (tR (range) = 15-20 min, 9.2 mL/min). The desired fractions were combined, diluted 1:2 v/v with H2O and loaded onto a Waters C18 Sep-Pak. The Sep-Pak was washed with 0.9 % NaCl(aq) (40 mL) and then EtOH (0.5 mL). The product was eluted from the Sep-Pak with EtOH (1.5 mL) and collected in a sealed sterile vial containing 0.9 % NaCl(aq) (3.5 mL). This solution was passed successively through a 1 μm filter and then a 0.2 m filter (Acrodisc PTFE) under Ar-pressure and collected in a sealed sterile dose vial containing 0.9 % NaCl(aq) (10 mL). The total synthesis time was ~77 min from the delivery of [18F]fluoropropylbrosylate to the hot cell with a 1.7 ± 0.3 % (n = 3) radiochemical yield (decay corrected). The product was then analyzed by analytical HPLC (tR = 6.0 min, 1 mL/min) to determine the radiochemical purity (93 ± 7 %, n = 3).
Supplementary Material
Figure 7.

MicroPET baseline TACs obtained by injection of [18F]3 into an anesthetized cynomolgus monkey.
Figure 10.

MicroPET TACs showing the results of injection of 15 (1.5 mg/kg) into an anesthetized cynomolgus monkey at 120 min post-injection of [18F]2.
Figure 11.

MicroPET TACs showing the results of injection of 15 (1.5 mg/kg) into an anesthetized cynomolgus monkey at 120 min post-injection of [18F]3.
Acknowledgement
This research was sponsored by the NIMH (1-R21-MH-66622-01). We acknowledge the use of shared instrumentation provided by grants from the NIH and the NSF.
Abbreviations
- PET
positron emission tomography
- SUV
- SPECT
single-photon emission computed tomography
- TAC
time-activity curve
- HRRT
high resolution research tomograph
- EOB
end-of-bombardment
- CNS
central nervous system
- SSRI
selective serotonin reuptake inhibitor
- SERT
serotonin transporter
- DAT
dopamine transporter
- NET
norepinephrine transporter
Footnotes
Supporting Information Available: Additional microPET data and elemental analysis data of 1-4. This material is available free of charge via the Internet at http://www.pubs.acs.org
References
- 1.Thie JA. Understanding the Standardized Uptake Value, Its Methods, and Implications for Usage. J. Nucl. Med. 2004;45:1431–1434. [PubMed] [Google Scholar]
- 2.Bentourkia M, Zaidi H. Tracer Kinetic Modeling in PET. PET Clin. 2007;2:267–277. doi: 10.1016/j.cpet.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Rudnick G. Active Transport of 5-Hydroxytryptamine by Plasma Membrane Vesicles Isolated from Human Blood Platelets. J. Biol. Chem. 1977;252:2170–2174. [PubMed] [Google Scholar]
- 4.Blakely RD, De Felice LJ, Hartzell HC. Molecular Physiology of Norepinephrine and Serotonin Transporters. J. Exp. Biol. 1994;196:263–281. doi: 10.1242/jeb.196.1.263. [DOI] [PubMed] [Google Scholar]
- 5.Torres GE, Gainetdinov RR, Caron MG. Plasma Membrane Monoamine Transporters: Structure, Regulation and Function. Nature Reviews Neuroscience. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 6.Linder AE, W. N, Diaz JL, Szasz T, Burnett R, Watts SW. Serotonin (5-HT) in Veins: Not All in Vain. J. Pharmacol. Exp. Ther. 2007;323:415–421. doi: 10.1124/jpet.107.122630. [DOI] [PubMed] [Google Scholar]
- 7.Owens MJ, Nemeroff CB. Role of Serotonin in the Pathophysiology of Depression: Focus on the Serotonin Transporter. Clin. Chem. 1994;40:288–295. [PubMed] [Google Scholar]
- 8.Fujita M, Shimada S, Maeno H, Nishimura T, Tohyama M. Cellular Localization of Serotonin Transporter mRNA in the Rat Brain. Neurosci. Lett. 1993;162:59–62. doi: 10.1016/0304-3940(93)90559-4. [DOI] [PubMed] [Google Scholar]
- 9.Austin MC, Bradley CC, Mann JJ, Blakely RD. Expression of Serotonin Transporter Messenger RNA in the Human Brain. J. Neurochem. 1994;62:2362–2367. doi: 10.1046/j.1471-4159.1994.62062362.x. [DOI] [PubMed] [Google Scholar]
- 10.Stockmeier CA, Shapiro LA, Haycock JW, Thompson PA, Lowy MT. Quantitative Subregional Distribution of Serotonin-1A Receptors and Serotonin Transporters in the Human Dorsal Raphe. Brain Res. 1996;727:1–12. doi: 10.1016/0006-8993(96)00239-9. [DOI] [PubMed] [Google Scholar]
- 11.Lucki I. The Spectrum of Behaviors Influenced by Serotonin. Biol. Psychiatry. 1998;44:151–162. doi: 10.1016/s0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- 12.Owens MJ, Nemeroff CB. The Serotonin Transporter and Depression. Depression and Anxiety. 1998;8(Suppl 1):5–12. [PubMed] [Google Scholar]
- 13.Mann JJ. Role of the Serotonergic System in the Pathogenesis of Major Depression and Suicidal Behavior. Neuropsychopharm. 1999;21:99S–105S. doi: 10.1016/S0893-133X(99)00040-8. [DOI] [PubMed] [Google Scholar]
- 14.Mann JJ, Brent DA, Arango V. The Neurobiology and Genetics of Suicide and Attempted Suicide: A Focus on the Serotonergic System. Neuropsychopharm. 2001;24:467–477. doi: 10.1016/S0893-133X(00)00228-1. [DOI] [PubMed] [Google Scholar]
- 15.Laakso A, Hietala J. PET Studies of Brain Monoamine Transporters. Curr. Pharm. Des. 2000;6:1611–1623. doi: 10.2174/1381612003398799. [DOI] [PubMed] [Google Scholar]
- 16.Laruelle M, Slifstein M, Huang Y. Positron Emission Tomography: Imaging and Quantification of Neurotransporter Availability. Methods. 2002;27:287–299. doi: 10.1016/s1046-2023(02)00085-3. [DOI] [PubMed] [Google Scholar]
- 17.Ametamey SM, Honer M, Schubiger PA. Molecular Imaging with PET. Chem. Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 18.Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, Frazer A. Effects of Chronic Antidepressant Treatments on Serotonin Transporter Function, Density, and mRNA Level. J. Neurosci. 1999;19:10494–10501. doi: 10.1523/JNEUROSCI.19-23-10494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin Clearance in vivo is Altered to a Greater Extent by Antidepressant-Induced Downregulation of the Serotonin Transporter than by Acute Blockade of this Transporter. J. Neurosci. 2002;22:6766–6772. doi: 10.1523/JNEUROSCI.22-15-06766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kugaya A, Sanacora G, Staley JK, Malison RT, Bozkurt A, Khan S, Anand A, van Dyck CH, Baldwin RM, Seibyl JP, Charney D, Innis RB. Brain Serotonin Transporter Availability Predicts Treatment Response to Selective Serotonin Reuptake Inhibitors. Biol. Psychiatry. 2004;56:497–502. doi: 10.1016/j.biopsych.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Mirza NR, Nielsen EØ, Troelsen KB. Serotonin Transporter Density and Anxiolytic-like Effects of Antidepressants in Mice. Prog. Neuro-Psychopharm. Biol. Psych. 2007;31:858–866. doi: 10.1016/j.pnpbp.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Meyer JH. Imaging the Serotonin Transporter During Major Depressive Disorder and Antidepressant Treatment. J. Psychiatry Neurosci. 2007;32:86–102. [PMC free article] [PubMed] [Google Scholar]
- 23.Aboagye EO, Price PM, Jones T. In Vivo Pharmacokinetics and Pharmacodynamics in Drug Development Using Positron-Emission Tomography. Drug Discovery Today. 2001;6:293–302. doi: 10.1016/s1359-6446(01)01684-1. [DOI] [PubMed] [Google Scholar]
- 24.Guilloteau D, Chalon S. PET and SPECT Exploration of Central Monoaminergic Transporters for the Development of New Drugs and Treatments in Brain Disorders. Curr. Pharm. Design. 2005;11:3237–3245. doi: 10.2174/138161205774424744. [DOI] [PubMed] [Google Scholar]
- 25.Lee C-M, Farde L. Using Positron Emission Tomography to Facilitate CNS Drug Development. Trends Pharmacol. Sci. 2006;27:310–316. doi: 10.1016/j.tips.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Takano A, Suzuki K, Kosaka J, Ota M, Nozaki S, Ikoma Y, Tanada S, Suhara T. A Dose-Finding Study of Duloxetine Based on Serotonin Transporter Occupancy. Psychopharmacology. 2006;185:395–399. doi: 10.1007/s00213-005-0304-0. [DOI] [PubMed] [Google Scholar]
- 27.Talbot PS, Laruelle M. The Role of In Vivo Molecular Imaging with PET and SPECT in the Elucidation of Psychiatric Drug Action and New Drug Development. Eur. Neuropsychopharmacol. 2002;12:503–511. doi: 10.1016/s0924-977x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 28.Hargreaves RJ. The Role of Molecular Imaging in Drug Discovery and Development. Clin. Pharmacol. Ther. 2008;83:349–353. doi: 10.1038/sj.clpt.6100467. [DOI] [PubMed] [Google Scholar]
- 29.Acton PD, Kung M-P, Mu M, Plössl K, Hou C, Siciliano M, Oya S, Kung HF. Single-Photon Emission Tomography Imaging of Serotonin Transporters in the Non-Human Primate Brain with the Selective Radioligand [123I]IDAM. Eur. J. Nucl. Med. 1999;26:854–861. doi: 10.1007/s002590050459. [DOI] [PubMed] [Google Scholar]
- 30.Wilson AA, Ginovart N, Schmidt M, Meyer JH, Threlkeld PG, Houle S. Novel Radiotracers for Imaging the Serotonin Transporter by Positron Emission Tomography: Synthesis, Radiosynthesis, and in Vitro and ex Vivo Evaluation of 11C-Labeled 2-(Phenylthio)araalkylamines. J. Med. Chem. 2000;43:3103–3110. doi: 10.1021/jm000079i. [DOI] [PubMed] [Google Scholar]
- 31.Ginovart N, Wilson AA, Meyer JH, Hussey D, Houle S. Positron Emission Tomography Quantification of [11C]-DASB Binding to the Human Serotonin Transporter: Modeling Strategies. J. Cereb. Blood Flow Metab. 2001;21:1342–1353. doi: 10.1097/00004647-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Emond P, Vercouillie J, Innis R, Chalon S, Mavel S, Frangin Y, Halldin C, Besnard J-C, Guilloteau D. Substituted Diphenyl Sulfides as Selective Serotonin Transporter Ligands: Synthesis and In Vitro Evaluation. J. Med. Chem. 2002;45:1253–1258. doi: 10.1021/jm010939a. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y, Hwang D-R, Narendran R, Sudo Y, Chatterjeee R, Bae S-A, Mawlawi O, Kegeles LS, Wilson AA, Kung HF, Laruelle M. Comparative Evaluation in Nonhuman Primates of Five PET Radiotracers for Imaging the Serotonin Transporters: [11C]McN5652, [11C]ADAM, [11C]DASB, [11C]DAPA, and [11C]AFM. J. Cereb. Blood Flow Metab. 2002;22:1377–1398. doi: 10.1097/01.WCB.0000040948.67415.05. [DOI] [PubMed] [Google Scholar]
- 34.Szabo Z, McCann UD, Wilson AA, Scheffel U, Owonikoko T, Mathews WB, Ravert HT, Hilton J, Dannals RF, Ricaurte GA. Comparison of (+)-11C-McN5652 and 11C-DASB as Serotonin Transporter Radioligands Under Various Experimental Conditions. J. Nucl. Med. 2002;43:678–692. [PMC free article] [PubMed] [Google Scholar]
- 35.Wellsow J, Kovar K-A, Machulla H-J. Molecular Modeling of Potential New and Selective PET Radiotracers for the Serotonin Transporter. J. Pharm. Pharmaceut. Sci. 2002;5:245–257. [PubMed] [Google Scholar]
- 36.Huang Y, Bae S, Zhu Z, Guo N, Roth BL, Laruelle M. Fluorinated Diaryl Sulfides as Serotonin Transporter Ligands: Synthesis, Structure-Activity Relationship Study, and in Vivo Evaluation of Fluorine-18-Labeled Compounds as PET Imaging Agents. J. Med. Chem. 2005;48:2559–2570. doi: 10.1021/jm0400808. [DOI] [PubMed] [Google Scholar]
- 37.Jarkas N, Votaw JR, Voll RJ, Williams L, Camp VM, Owens MJ, Purselle DC, Bremner JD, Kilts CD, Nemeroff CB, Goodman MM. Carbon-11 HOMADAM: A Novel PET Radiotracer for Imaging Serotonin Transporters. Nucl. Med. Biol. 2005;32:211–224. doi: 10.1016/j.nucmedbio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Fang P, Shiue GG, Shimazu T, Greenberg JH, Shiue C-Y. Synthesis and Evaluation of N,N-Dimethyl-2-(2-amino-5-[18F]fluorophenylthio)benzylamine (5-[18F]-ADAM) as a Serotonin Transporter Imaging Agent. Appl. Radiat. Isot. 2004;61:1247–1254. doi: 10.1016/j.apradiso.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Garg S, Thopate SR, Minton RC, Black KW, Lynch AJH, Garg PK. 3-Amino-4-(2-((4-[18F]fluorobenzyl)methylamino)methylphenylsulfanyl)benzonitrile, an F-18 Fluorobenzyl Analogue of DASB: Synthesis, in Vitro Binding, and in Vivo Biodistribution Studies. Bioconj. Chem. 2007;18:1612–1618. doi: 10.1021/bc070112g. [DOI] [PubMed] [Google Scholar]
- 40.Parhi AK, Wang JL, Oya S, Choi S-R, Kung M-P, Kung HF. 2-(2'-((Dimethylamino)methyl)-4'-(fluoroalkoxy)-phenylthio)benzenamine Derivatives as Serotonin Transporter Imaging Agents. J. Med. Chem. 2007;50:6673–6684. doi: 10.1021/jm070685e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarkas N, Voll RJ, Williams L, Votaw JR, Owens M, Goodman MM. Synthesis and In Vivo Evaluation of Halogenated N,N-Dimethyl-2-(2'-amino-4'-hydroxymethylphenylthio)benzylamine Derivatives as PET Serotonin Transporter Ligands. J. Med. Chem. 2008;51:271–281. doi: 10.1021/jm0707929. [DOI] [PubMed] [Google Scholar]
- 42.Mavel S, Vercouillie J, Garreau L, Raguza T, Ravna AW, Chalon S, Guilloteau D, Emond P. Docking Study, Synthesis, and In Vitro Evaluation of Fluoro-MADAM Derivatives as SERT Ligands for PET Imaging. Bioorg. Med. Chem. 2008;16:9050–9055. doi: 10.1016/j.bmc.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Wang JL, Parhi AK, Oya S, Lieberman B, Kung M-P, Kung HF. 2-(2'- ((Dimethylamino)methyl)-4'-(3-[18F]fluoropropoxy)-phenylthio)benzenamine for Positron Emission Tomography Imaging of Serotonin Transporters. Nucl. Med. Biol. 2008;35:447–458. doi: 10.1016/j.nucmedbio.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bois F, Baldwin RM, Kula NS, Baldessarini RJ, Innis RB, Tamagnan G. Synthesis and Monoamine Transporter Affinity of 3'-Analogs of 2-β-Carbomethoxy-3-β-(4'-iodophenyl)tropane (β-CIT) Bioorg. & Med. Chem. Lett. 2004;14:2117–2120. doi: 10.1016/j.bmcl.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 45.Peng X, Zhang A, Kula NS, Baldessarini RJ, Neumeyer JL. Synthesis and Amine Transporter Affinities of Novel Phenyltropane Derivatives as Potential Positron Emission Tomography (PET) Imaging Agents. Bioorg. & Med. Chem. Lett. 2004;14:5635–5639. doi: 10.1016/j.bmcl.2004.08.049. [DOI] [PubMed] [Google Scholar]
- 46.Tamagnan G, Alagille D, Fu X, Kula NS, Baldessarini RJ, Innis RB, Baldwin RM. Synthesis and Monoamine Transporter Affinity of New 2β-Carbomethoxy-3β-[4-(Substituted Thiophenyl)]Phenyltropanes: Discovery of a Selective SERT Antagonist with Picomolar Potency. Bioorg. & Med. Chem. Lett. 2005;15:1131–1133. doi: 10.1016/j.bmcl.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 47.Levin CS, Hoffman EJ. Calculation of Positron Range and Its Effect on the Fundamental Limit of Positron Emission Tomography System Spatial Resolution. Phys. Med. Biol. 1999;44:781–799. doi: 10.1088/0031-9155/44/3/019. [DOI] [PubMed] [Google Scholar]
- 48.Wernick MN, Aarsvold JN. Emission Tomography: The Fundamentals of PET and SPECT. Elsevier Academic Press; San Diego, CA, USA: 2004. London, UK. [Google Scholar]
- 49.Böhm H-J, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. Fluorine in Medicinal Chemistry. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 50.Müller K, Faeh C, Diederich F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 51.Hagmann WK. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 52.Lasne M-C, Perrio C, Rouden J, Barré L, Roeda D, Dollé F, Crouzel C. Chemistry of β+-Emitting Compounds Based on Fluorine-18. Top. Curr. Chem. 2002;222:201–258. (Ch.6) [Google Scholar]
- 53.Cai L, Lu S, Pike VW. Chemistry with [18F]Fluoride Ion. Eur. J. Org. Chem. 2008:2853–2873. [Google Scholar]
- 54.Kirk KL. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Proc. Res. Dev. 2008;12:305–321. [Google Scholar]
- 55.Elfving B, Madsen J, Knudsen GM. Neuroimaging of the Serotonin Reuptake Site Requires High-Affinity Ligands. Synapse. 2007;61:882–888. doi: 10.1002/syn.20443. [DOI] [PubMed] [Google Scholar]
- 56.Dischino DD, Welch MJ, Kilbourn MR, Raichle ME. Relationship Between Lipophilicity and Brain Extraction of C-11-Labeled Radiopharmaceuticals. J. Nucl. Med. 1983;24:1030–1038. [PubMed] [Google Scholar]
- 57.Elfving B, Bjørnholm B, Ebert B, Knudsen GM. Binding Characteristics of Selective Serotonin Reuptake Inhibitors With Relation to Emission Tomography Studies. Synapse. 2001;41:203–211. doi: 10.1002/syn.1076. [DOI] [PubMed] [Google Scholar]
- 58.Waterhouse RN. Determination of Lipophilicity and Its Use as a Predictor of Blood-Brain Barrier Penetration of Molecular Imaging Agents. Mol. Imag. Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 59.Kish SJ, Furukawa Y, Chang L-J, Tong J, Ginovart N, Wilson A, Houle S, Meyer JH. Regional Distribution of Serotonin Transporter Protein in Postmortem Human Brain. Is the Cerebellum a SERT-Free Brain Region? Nuc. Med. Biol. 2005;32:123–128. doi: 10.1016/j.nucmedbio.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 60.Goodman MM, Chen P, Plisson C, Martarello L, Galt J, Votaw JR, Kilts CD, Malveaux G, Camp VM, Shi B, Ely TD, Howell L, McConathy J, Nemeroff CB. Synthesis and Characterization of Iodine-123 Labeled 2β-Carbomethoxy-3β-(4'-((Z)-2-iodoethenyl)phenyl)nortropane. A Ligand for in vivo Imaging of Serotonin Transporters by Single-Photon-Emission Tomography. J. Med. Chem. 2003;46:925–935. doi: 10.1021/jm0100180. [DOI] [PubMed] [Google Scholar]
- 61.Plisson C, McConathy J, Martarello L, Malveaux EJ, Camp VM, Williams L, Votaw JR, Goodman MM. Synthesis, Radiosynthesis, and Biological Evaluation of Carbon-11 and Iodine-123 Labeled 2β-Carbomethoxy-3β-[4'-((Z)-2-haloethenyl)phenyl]tropanes: Candidate Radioligands for in Vivo Imaging of the Serotonin Transporter. J. Med. Chem. 2004;47:1122–1135. doi: 10.1021/jm030384e. [DOI] [PubMed] [Google Scholar]
- 62.Plisson C, Jarkas N, McConathy J, Voll RJ, Votaw J, Williams L, Howell LL, Kilts CD, Goodman MM. Evaluation of Carbon-11-Labeled 2β-Carbomethoxy-3β-[4'-((Z)-2-iodoethenyl)phenyl]nortropane as a Potential Radioligand for Imaging the Serotonin Transporter by PET. J. Med. Chem. 2006;49:942–946. doi: 10.1021/jm050799v. [DOI] [PubMed] [Google Scholar]
- 63.Stehouwer JS, Jarkas N, Zeng F, Voll RJ, Williams L, Owens MJ, Votaw JR, Goodman MM. Synthesis, Radiosynthesis, and Biological Evaluation of Carbon-11 Labeled 2β-Carbomethoxy-3β-(3′-((Z)-2-haloethenyl)phenyl)nortropanes: Candidate Radioligands for In Vivo Imaging of the Serotonin Transporter with Positron Emission Tomography. J. Med. Chem. 2006;49:6760–6767. doi: 10.1021/jm060641q. [DOI] [PubMed] [Google Scholar]
- 64.Plisson C, Stehouwer JS, Voll RJ, Howell LL, Votaw JR, Owens MJ, Goodman MM. Synthesis and In Vivo Evaluation of Fluorine-18 and Iodine-123 Labeled 2β-Carbo(2-fluoroethoxy)-3β-(4'-((Z)-2-iodoethenyl)phenyl)nortropane as a Candidate Serotonin Transporter Imaging Agent. J. Med. Chem. 2007;50:4553–4560. doi: 10.1021/jm061303s. [DOI] [PubMed] [Google Scholar]
- 65.Seibyl J, Goodman M, Koren A, Stehouwer J, Jennings D, Staley J, Tamagnan G. Preclinical and Clinical Characterization of 123-I mZIENT, a Marker of Serotonin Transporter Density. J. Nucl. Med. 2007;48(Supplement 2):113P. Abstract 381. [Google Scholar]
- 66.Tamagnan G, Stehouwer J, Staley JK, Megyola C, Koren A, Goodman M, Seibyl J. In Vivo Characterization in Subhuman Primates of mZIENT: A New Serotonin Transporter, Effects of Selective SERT Displacement. NeuroImage. 2006;31:T131. [Google Scholar]
- 67.Stehouwer JS, Plisson C, Jarkas N, Zeng F, Voll RJ, Williams L, Martarello L, Votaw JR, Tamagnan G, Goodman MM. Synthesis, Radiosynthesis, and Biological Evaluation of Carbon-11 and Fluorine-18 (N-Fluoroalkyl) Labeled 2β-Carbomethoxy-3β-(4'-(3-furyl)phenyl)-tropanes and -nortropanes: Candidate Radioligands for in Vivo Imaging of the Serotonin Transporter with Positron Emission Tomography. J. Med. Chem. 2005;48:7080–7083. doi: 10.1021/jm0504095. [DOI] [PubMed] [Google Scholar]
- 68.Stehouwer J, Jarkas N, Zeng F, Voll R, Williams L, Votaw J, Goodman M. MicroPET Imaging of the Brain Serotonin Transporter with [18F]βFEmZIENT. J. Nucl. Med. 2006;47(Supplement 1):27P. Abstract No. 77. [Google Scholar]
- 69.Goodman M, Stehouwer J, Jarkas N, Voll R, Williams L, Votaw J. In Vivo Characterization in Non-Human Primates of βFEmZIENT and βFEmZBrENT: New Serotonin Transporter Imaging Agents: Effects of Selective Monoamine Transporter Displacement Using MicroPET. Neuroimage. 2006;31(Supplement 2):T27. [Google Scholar]
- 70.Stehouwer J, Jarkas N, Zeng F, Voll R, Williams L, Votaw J, Goodman M. MicroPET Imaging of the CNS Serotonin Transporter with [18]FPmZIENT and [18]FPmZBrENT. The Southeastern Regional Meeting of the American Chemical Society; Augusta, GA. November 1-4, 2006; 2006. Abstract #525. [Google Scholar]
- 71.Carroll FI, Kotian P, Dehghani A, Gray JL, Kuzemko MA, Parham KA, Abraham P, Lewin AH, Boja JW, Kuhar MJ. Cocaine and 3β-(4'-Substituted phenyl)tropane-2β-carboxylic Acid Ester and Amide Analogues. New High-Affinity and Selective Compounds for the Dopamine Transporter. J. Med. Chem. 1995;38:379–388. doi: 10.1021/jm00002a020. [DOI] [PubMed] [Google Scholar]
- 72.Blough BE, Abraham P, Lewin AH, Kuhar MJ, Boja JW, Carroll FI. Synthesis and Transporter Binding Properties of 3β-(4'-Alkyl-, 4'-alkenyl-, and 4'-alkynylphenyl)nortropane-2β-carboxylic Acid Methyl Esters: Serotonin Transporter Selective Analogs. J. Med. Chem. 1996;39:4027–4035. doi: 10.1021/jm960409s. [DOI] [PubMed] [Google Scholar]
- 73.Mitchell TN, Amamria A, Killing H, Rutschow D. Palladium Catalysis in Organotin Chemistry: Addition of Hexaalkylditins to Alkynes. J. Organomet. Chem. 1986;304:257–265. [Google Scholar]
- 74.Blough BE, Keverline KI, Nie Z, Navarro H, Kuhar MJ, Carroll FI. Synthesis and Transporter Binding Properties of 3β-[4'-(Phenylalkyl, -phenylalkenyl, and -phenyalkynl)phenyl]tropane-2β-carboxylic Acid Methyl Esters: Evidence of a Remote Phenyl Binding Domain on the Dopamine Transporter. J. Med. Chem. 2002;45:4029–4037. doi: 10.1021/jm020098n. [DOI] [PubMed] [Google Scholar]
- 75.Lemaire C, Plenevaux A, Aerts J, Del Fiore G, Brihaye C, Le Bars D, Comar D, Luxen A. Solid Phase Extraction - An Alternative to the Use of Rotary Evaporators for Solvent Removal in the Rapid Formulation of PET Radiopharmaceuticals. J. Labelled Compd. Radiopharm. 1999;42:63–75. [Google Scholar]
- 76.Wilson AA, Houle S. Radiosynthesis of Carbon-11 Labelled N-Methyl-2-(arylthio)benzylamines: Potential Radiotracers for the Serotonin Reuptake Receptor. J. Labelled Compd. Radiopharm. 1999;42:1277–1288. [Google Scholar]
- 77.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An Admonition When Measuring the Lipophilicity of Radiotracers Using Counting Techniques. Appl. Radiat. Isot. 2001;54:203–208. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]
- 78.Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter Receptor and Transporter Binding Profile of Antidepressants and Their Metabolites. J. Pharmacol. Exp. Ther. 1997;283:1305–1322. [PubMed] [Google Scholar]
- 79.Hiemke C, Härtter S. Pharmacokinetics of Selective Serotonin Reuptake Inhibitors. Pharmacol. Therap. 2000;85:11–28. doi: 10.1016/s0163-7258(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 80.Boja JW, Patel A, Carroll FI, Rahman MA, Philip A, Lewin AH, Kopajtic TA, Kuhar MJ. [125I]RTI-55: a potent ligand for dopamine transporters. Eur. J. Pharmacol. 1991;194:133–134. doi: 10.1016/0014-2999(91)90137-f. [DOI] [PubMed] [Google Scholar]
- 81.Cheetham SC, Viggers JA, Butler SA, Prow MR, Heal DJ. [3H]Nisoxetine - A Radioligand for Noradrenaline Reuptake Sites: Correlation With Inhibition of [3H]Noradrenaline Uptake and Effects of DSP-4 Lesioning and Antidepressant Treatments. Neuropharmacology. 1996;35:63–70. doi: 10.1016/0028-3908(95)00134-4. [DOI] [PubMed] [Google Scholar]
- 82.Hrdina PD, Foy B, Hepner A, Summers RJ. Antidepressant Binding Sites in Brain: Autoradiographic Comparison of [3H]Paroxetine and [3H]Imipramine Localization and Relationship to Serotonin Transporter. J. Pharmacol. Exp. Ther. 1990;252:410–418. [PubMed] [Google Scholar]
- 83.Cortés R, Soriano E, Pazos A, Probst A, Palacios JM. Autoradiography of Antidepressant Binding Sites in the Human Brain: Localization Using [3H]Imipramine and [3H]Paroxetine. Neuroscience. 1988;27:473–496. doi: 10.1016/0306-4522(88)90282-5. [DOI] [PubMed] [Google Scholar]
- 84.Potter PM, Wadkins RM. Carboxylesterases - Detoxifying Enzymes and Targets for Drug Therapy. Curr. Med. Chem. 2006;13:1045–1054. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- 85.Elfving B, Bjørnholm B, Knudsen GM. Interference of Anaesthetics With Radioligand Binding in Neuroreceptor Studies. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:912–915. doi: 10.1007/s00259-003-1171-8. [DOI] [PubMed] [Google Scholar]
- 86.Votaw J, Byas-Smith M, Hua J, Voll R, Martarello L, Levey AI, Bowman FD, Goodman M. Interaction of Isoflurane With the Dopamine Transporter. Anesthesiology. 2003;98:404–411. doi: 10.1097/00000542-200302000-00021. [DOI] [PubMed] [Google Scholar]
- 87.McCormick PN, Ginovart N, Vasdev N, Seeman P, Kapur S, Wilson AA. Isoflurane Increases Both the Specific Binding Ratio and Sensitivity to Amphetamine Challenge of [11C]-(+)-PHNO. Neuroimage. 2006;31:T33. Abstracts. [Google Scholar]
- 88.Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N. Ketamine Alters the Availability of Striatal Dopamine Transporter as Measured by [11C]β-CFT and [11C]β-CIT-FE in the Monkey Brain. Synapse. 2001;42:273–280. doi: 10.1002/syn.10012. [DOI] [PubMed] [Google Scholar]
- 89.Katz JL, Izenwasser S, Terry P. Relationships Among Dopamine Transporter Affinities and Cocaine-Like Discriminative-Stimulus Effects. Psychopharmacology. 2000;148:90–98. doi: 10.1007/s002130050029. [DOI] [PubMed] [Google Scholar]
- 90.Cook CD, Carroll FI, Beardsley PM. RTI 113, a 3-Phenyltropane Analog, Produces Long-Lasting Cocaine-Like Discriminative Stimulus Effects in Rats and Squirrel Monkeys. Eur. J. Pharmacol. 2002;442:93–98. doi: 10.1016/s0014-2999(02)01501-7. [DOI] [PubMed] [Google Scholar]
- 91.Wilcox KM, Lindsey KP, Votaw JR, Goodman MM, Martarello L, Carroll FI, Howell LL. Self-Administration of Cocaine and the Cocaine Analog RTI-113: Relationship to Dopamine Transporter Occupancy Determined by PET Neuroimaging in Rhesus Monkeys. Synapse. 2002;43:78–85. doi: 10.1002/syn.10018. [DOI] [PubMed] [Google Scholar]
- 92.Wong EHF, Sonders MS, Amara SG, Tinholt PM, Piercey MFP, Hoffmann WP, Hyslop DK, Franklin S, Porsolt RD, Bonsignori A, Carfagna N, McArthur RA. Reboxetine: A Pharmacologically Potent, Selective, and Specific Norepinephrine Reuptake Inhibitor. Biol. Psychiatry. 2000;47:818–829. doi: 10.1016/s0006-3223(99)00291-7. [DOI] [PubMed] [Google Scholar]
- 93.Patient Selection and Antidepressant Therapy With Reboxetine, A New Selective Norepinephrine Reuptake Inhibitor. J. Clin. Psychiatry. 1998;59(Supplement 14) [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.