

Abstract
Lophocladines A (1) and B (2), two 2,7-naphthyridine alkaloids, were isolated from the marine red alga Lophocladia sp. collected in the Fijian Islands. Their structures were deduced on the basis of high-resolution mass spectra and one- and two-dimensional NMR spectroscopy. Lophocladine A (1) displayed affinity for NMDA receptors and was found to be a δ-opioid receptor antagonist, whereas lophocladine B (2) exhibited cytotoxicity to NCI-H460 human lung tumor and MDA-MB-435 breast cancer cell lines. Immunofluorescence studies indicated that the cytotoxicity of lophocladine B (2) was correlated with microtubule inhibition. This is the first reported occurrence of alkaloids based on a 2,7-naphthyridine skeleton from red algae.
Rhodophyta are known to be a rich source of diverse bioactive metabolites.1 High molecular weight red algal compounds of pharmacological interest include antiviral polysaccharides2 and the recently discovered potent HIV-inactivating protein griffithsin.3 Low molecular weight metabolites of red algae are dominated by halogenated terpenoids, acetogenins, bromophenols, bromoindoles as well as unusual amino acids. These metabolites possess a wide range of bioactivities, including cytotoxicity (e.g., halomon),4 antimalarial,5 anthelmintic (e.g., kainic acid),6 antimicrobial (e.g., allolaurinterol)7 and quorum sensing inhibition.8
The red algal genus Lophocladia, a member of the Rhodomelaceae (Ceramiales, Rhodophyta), is widespread throughout the tropics and subtropics.9 Six species of the genus are recognized worldwide; however, this genus has rarely been investigated for its chemical constituents. Previously reported metabolites include the carbohydrate mannoglyceric acid and several unusual amino acids such as γ-aminobutyric acid betaine, L-azetidine-2-carboxylic acid, L-baikiain, citrulline and 5-dimethylsulphonio-4-hydroxy-2-aminovalerate.10 The current examination of a Fijian sample of Lophocladia sp. was undertaken in order to identify the compounds responsible for the cytotoxicity of the crude extract, and led to the isolation of two new alkaloids with a 2,7-naphthyridine ring system11. Due to its pharmacological potential, this bicyclic ring system has been the subject of several synthetic studies.12 The only naturally occurring representative of this compound class is dipyridylmethylketone isolated from the roots and rhizomes of valerian (Valeriana officinalis).13 To date red algal alkaloids are rare and mostly based on the indole skeleton. Indeed, this is only the second report of a natural product with the copyrine skeleton and the first report of the occurrence of copyrine-based alkaloids in red algae.
Results and Discussion
The red alga Lophocladia sp. was collected by hand using SCUBA (6 m) near Savusavu on the coastline of Fiji's second largest island Vanua Levu in 1997. The alcohol-preserved tissue was extracted with CH2Cl2/MeOH (2:1) and the resulting cytotoxic crude organic extract (solid tumor selective at 15 μg/disk: Colon38Δmurine CFU-GM = 300 units)14 was vacuum chromatographed over silica gel. While the initial differential cytotoxicity could not be recovered from the resulting subfractions, their monitoring by 1H NMR spectra revealed one in particular that possessed interesting aromatic resonances. This fraction was further purified utilizing RP-SPE cartridges and RP-HPLC to yield lophocladine A (1) and B (2).
Compound 1 was obtained as a white solid and given the trivial name lophocladine A. The HRFABMS analysis of 1 gave an [M+H]+ peak at m/z 223.0840, consistent with the molecular formula C14H10N2O, indicating a structure with eleven degrees of unsaturation. The IR spectrum possessed absorptions for an amide carbonyl group (1677 cm-1) and an aromatic ring system (1624, 1591 cm-1). UV maxima observed at 224, 250 and 314 nm suggested a highly conjugated system. The 13C NMR data contained a total of 14 resonances for nine methine groups and five quarternary carbons, all downfield of 100 ppm. 1H-1H-COSY, TOCSY and homodecoupling experiments revealed the presence of an AA′BB′C and an ABX spin system. The five hydrogens of the former spin system at δ 7.44, 7.45 and 7.51 were readily assignable to a phenyl ring. Methine protons at δ 7.39, 8.71 and 9.40 delineated the ABX system of another aromatic ring. Furthermore, there was one additional methine proton at δ 7.39 and one exchangeable NH proton at δ 11.8. Thus, it was deduced that the basic skeleton of 1 consisted of two aromatic rings, one of them substituted with a cyclic amide (δC=O 160.9).
An ABX pattern in aromatic ring systems usually suggests a 1,3,4-substituted phenyl ring. However, the downfield proton and carbon values of H-6 and H-8 indicated the heteroaromatic nature of this ring system in lophocladine A (1). Subtraction of the proposed amide functionality from the molecular formula left nitrogen as the only available heteroatom; therefore, ring A was likely a substituted pyridine system. Analysis of the one bond coupling constants (1JCH) of the adjacent sites, CH-6 and CH-8 (see Table 1) confirmed this deduction and allowed the placement of N-7 between these two atoms.15 Thus, ring A was a 3,4-disubstituted pyridine. The observed coupling between the AB and X parts of the ABX spin system resulted from a p-coupling between H-5 and H-8 (5JHH = 0.83 Hz). This effect is known for 2-7-naphthyridines and revealed itself only in d5-pyridine (see Supporting Information).16 Methine protons H-5 and H-8 both showed HMBC correlations to quarternary carbons C-4a and C-8a, and thus completed the carbon framework of ring A (Figure 1).
Table 1.
NMR data for lophocladine A (1) and B (2) in d6-DMSO (δ in ppm, J in Hz).
| 1 | 2 | |||||
|---|---|---|---|---|---|---|
| position | δHa | δCb | HMBCc,d | 1JC,Hc | δHa | δCb |
| 1 | 160.9 qC | 157.5 qC | ||||
| N-2 | 11.80 (1H, brs) | |||||
| 3 | 7.39 (1H, d, J = 2.5) | 133.0 CH | 1, 4, 4a, 5, 8a, 1′ | 179.1 | 7.98 (1H, s) | 146.3 CH |
| 4 | 115.7 qC | 111.8 qC | ||||
| 4a | 141.9 qC | 138.2 qC | ||||
| 5 | 7.39 (1H, d, J = 5.6) | 117.3 CH | 1, 4, 4a, 6, 8, 8a | 165.1 | 7.51 (1H, d, J = 5.9) | 116.4 CH |
| 6 | 8.71 (1H, d, J = 5.6) | 151.2 CH | 4, 4a, 5, 8 | 180.2 | 8.57 (1H, d, J = 5.9) | 147.5 CH |
| N-7 | ||||||
| 8 | 9.40 (1H, s) | 150.4 CH | 1, 4a, 6, 8a | 180.2 | 9.59 (1H, s) | 149.2 CH |
| 8a | 120.6 qC | 120.1 qC | ||||
| 1′ | 134.8 qC | 136.5 qC | ||||
| 2′ | 7.51 (1H, m) | 128.8 CH | 4, 1′, 3′, 4′, 5′, 6′ | 160.7 | 7.49 (1H, m) | 128.7 CH |
| 3′ | 7.45 (1H, m) | 129.6 CH | 4, 2′, 4′, 5′, 6′ | 161.4 | 7.44 (1H, m) | 129.6 CH |
| 4′ | 7.44 (1H, m) | 127.7 CH | 2′,3′,5′,6′ | 158.7 | 7.42 (1H, m) | 127.1 CH |
| 5′ | 7.45 (1H, m) | 129.6 CH | 4, 2′, 3′, 4′, 6′ | 161.4 | 7.44 (1H, m) | 129.6 CH |
| 6′ | 7.51 (1H, m) | 128.8 CH | 4, 1′, 2′, 3′, 4′, 5′ | 160.7 | 7.49 (1H, m) | 128.7 CH |
Recorded at 300 MHz.
Recorded at 75 MHz, multiplicity determined by DEPT.
Obtained through 1D- and 2D-HMBC experiments (100 MHz) employing various delay times (42, 65 and 125 ms).
Protons showing long range correlation with indicated carbon.
Figure 1.

Partial structures of lophocladine A (1) including key COSY, HMBC and NOE correlations.
The phenyl ring (ring C) was confirmed by NOESY, HSQC-TOCSY, 1H-1H-COSY correlations between H-2′/H-3′, H-3′/H-4′, H-4′/H-5′, H-5′/H-6′, and HMBC correlations between H-4′/C-3′, H-4′/ C-5′, H-2′/C-1′ and H-6′/C-1′. The phenyl ring protons H-2′, H-3′, H-5′ and H-6′ showed long range couplings to C-4, indicating that ring C was attached to C-4. Further HMBC correlations observed between methine proton H-3 and C-4 and C-1′ placed CH-3 next to C-4. The 1JC,H for CH-3 (179.1 Hz) suggested that C-3 was in turn adjacent to the remaining nitrogen atom NH-2.15 Additional evidence was provided by COSY and NOESY correlations between H-3 and NH-2. Long range heteronuclear couplings between H-3 and C-1 demonstrated that NH-2 was adjacent to the carbonyl atom C-1, forming the required amide bond and consequently establishing the majority of ring B (Figure 1).
The connection between ring A and B was accomplished by analysis of HMBC and selective 1D DPFGSE-NOE experiments.17 These correlations included long range CH couplings of H-5 and H-6 to C-4 and C-4a as well as couplings between H-8 and C-1 and C-8a, indicating that the ring closure occurred between C-4 and C-5 via C-4a and between C-1 and C-8 via C-8a, respectively. Through space interactions, observed between H-2′/6′ and H-5 confirmed this deduction. On the basis of the above data, the structure of 1 was determined as 4-phenyl[2,7]naphthyridin-1(2H)-one.18
Compound 2 was isolated by RP-HPLC from the same fraction containing lophocladine A (1) and was obtained as a yellowish amorphous solid. Accurate mass measurement of 2 revealed an [M+H]+ ion (m/z 222.1032) consistent with a molecular formula of C14H11N3. Analysis of the NMR data (Table 1) indicated that 2 shared many structural features with lophocladine A (1). The only significant differences between their NMR features were the absence of an exchangeable NH proton and the downfield shifts of H-3 and C-3 in the 1H and 13C NMR spectra of compound 2, respectively. Given the absence of oxygen in the molecular formula of 2, these differences could be accounted for by the presence of an amidine function instead of the lactam present in lophocladine A (1). Therefore, compound 2 was identified as 4-phenyl[2,7]naphthyridin-1-amine19 and was given the name lophocladine B (2).
The biological activity of compounds 1 and 2 was explored utilizing several cytotoxicity assays and through the PDSP/NIMH. The cytotoxic effects of lophocladine A (1) and B (2) were investigated using the NCI-H460 lung cancer, neuro-2a neuroblastoma and MDA-MB-435 breast cancer cell lines. Lophocladine A (1) was found to be inactive at 45 μM to the first two cell lines and 450 μM to the latter whereas lophocladine B (2) possessed moderate cytotoxicity to the MDA-MB-435 (IC50 = 3.1 μM) and NCI-H460 (IC50 = 64.6 μM) cell lines, but was inactive towards neuro-2a cells at 45 μM, the highest dose tested. However, NCI-H460 lung cancer cells treated with lophocladine B (2) also showed intriguing morphologic changes. At 13.5 μM (∼ IC50 the cell shape became flattened and developed short neuron-like projections. Changes in morphology appeared on day one and approximately 80% of the cells underwent the induction of morphologic changes by day six (for photomicrographs see Supporting Information). In a broad sense, changes in cell morphology have been correlated either with interference with signal transduction pathways or with breakdown of the cell skeleton.20 Thus, the effects of lophocladine B (2) on cellular microtubules and actin filaments were evaluated in A-10 cells. At 45 μM, 2 depolymerized 85 % of the microtubules while complete depolymerization of all cellular microtubules was detected at 113 μM which also caused breakdown of the nucleus into micronuclei (Figure 2). No loss of actin microfilaments was observed at these drug concentrations. Cell cycle analyses of MDA-MB-435 cells treated with lophocladine B (24 h, 20 μM) showed a significant reduction of cells in the G1 and S phases with an accumulation of cells in G2/M, indicating a G2/M cell cycle arrest (Figure 3). These biological effects of lophocladine B (2) are therefore qualitatively indistinguishable from those of dolastatin 10, symplostatin 1 or vinblastine. However, the potency of 2 is quite moderate when compared with that of the other tubulin depolymerizing agents.
Figure 2.
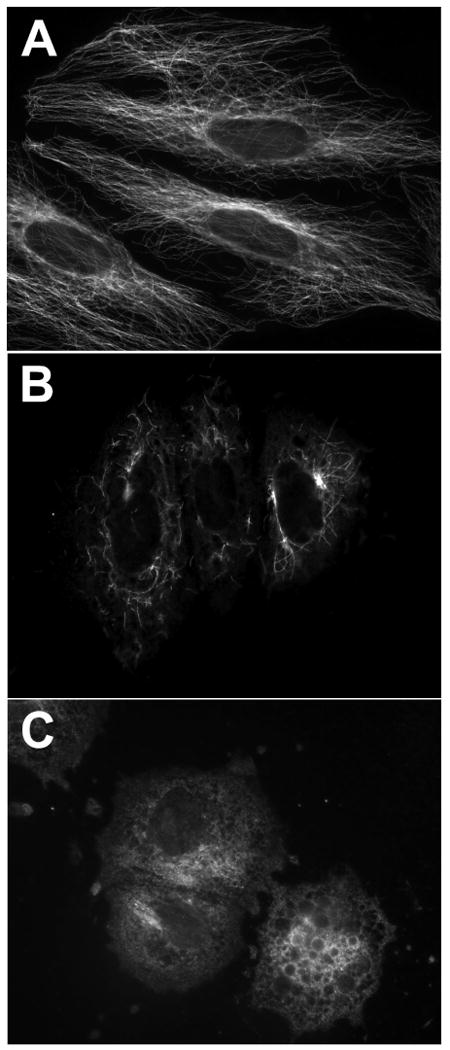
Effect of lophocladine B (2) on the microtubules of A-10 cells. Cellular microtubules were visualized in A-10 cells by indirect immunofluorescence techniques using a monoclonal β-tubulin antibody following a 24 h incubation with (A) vehicle control, (B) 45 μM lophocladine B (2) or (C) 113 μM lophocladine B (2).
Figure 3.

Cell cycle distribution of cells treated with lophocladine B (2). MDA-MB-435 cells were treated with (A) vehicle or (B) 20 μM lophocladine B (2) for 24 h and then stained with Krishan's Reagent and analyzed by flow cytometry.
Due to the structural resemblance of lophocladine A and B to biologically-active pyridopyridine compounds,21 metabolites 1 and 2 were screened in a panel of eleven cloned G-protein coupled receptors and ion channels at the Psychoactive Drug Screening Program of the National Institute of Mental Health (PDSP/NIMH). Lophocladine A (1) moderately inhibited [3H]-[D-Ala2,D-Leu5]-enkephalin binding to cloned δ-opioid receptors (50% inhibition at 10 μM) as well as radiolabeled MK801 binding to NMDA receptors (50% inhibition at 10 μM). Lophocladine B (2) showed no significant activity in the PDSP. The intrinsic activity of lophocladine A (1) at the δ-opiate receptor was further assessed using adenylyl cyclase assays. Lophocladine A (1) exhibited no significant inhibition of forskolin-stimulated adenylyl cyclase activity over a concentration range from 0.0001 to 100 μM in contrast to the reference [D-Pen2,D-Pen5]-enkephalin (DPDPE, 10 nM), indicating that compound 1 exhibits no agonist activity (see Supporting Information). Hence, lophocladine A (1) appears to be a micromolar antagonist of the δ-opioid receptor.
Experimental Section
General Experimental Procedures
UV and FT-IR spectra were obtained employing Hewlett Packard 8452A and Nicolet 510 instruments, respectively. All NMR spectra were recorded on Bruker Avance DRX300 and DPX400 spectrometers. Spectra were referenced to residual solvent signals with resonances at δH/C 2.50/39.5 (d6-DMSO), δH/C 3.31/49.0 (CD3OD), δH/C 8.74/150.35 (d5-pyridine) and δH 11.50 (CF3COOD). ESI and APCI mass spectra were obtained on a Thermo Finnigan LCQ Advantage mass spectrometer and FAB mass spectra were recorded on a Kratos MS50TC mass spectrometer. HPLC was carried out using a Waters system consisting of a Rheodyne 7725i injector, two 515 pumps, a pump control module and a 996 photodiode array detector. TLC grade (10-40 μm) Si gel was used for vacuum chromatography. All solvents were purchased as HPLC grade.
Algal Collection and Taxonomy
The marine red alga Lophocladia sp. was collected by SCUBA in February 1997 near Savusavu, Fiji Islands from a depth of 6 m and stored in 2-propanol at -20°C until extraction. With a degree of uncertainty, it was assigned to the genus Lophocladia (Rhodomelaceae, Rhodophyta). Lophocladia is comprised of six species recognized worldwide; with five being Pacific in their distribution. Lophocladia shares with Spirocladia the characteristic features of a polysiphonous axis that is characterized in part by descending rhizoids lying between the pericentral cells and by spirally twisted tetrasporangial stichidia. Spirocladia is principally differentiated from Lophocladia on the basis of the trichoblasts associated with spermatangial branches and this Fijian specimen was exclusively tetrasporophytic. Of the short-statured, heavily corticated species of both genera, this specimen most closely resembled L. kupukaia, sharing the feature a staggered pericentral cell arrangement. Nevertheless, this specimen differs from L. kupukaia in possessing longer tetrasporangial stichidia, raising the possibility of it being an undescribed species. A voucher specimen, D.L.B. 6474, has been deposited at the Herbario Marino Puertorriqueño (MSM), Department of Marine Sciences, University of Puerto Rico, Mayagüez.
Extraction and Isolation
Lophocladia sp. (31.8 g dry wt) was extracted repeatedly with CH2Cl2-MeOH (2:1, v/v) to produce 1.6 g of crude organic extract. A portion of the extract (1.3 g) was subjected to normal phase vacuum liquid chromatography (VLC), using stepwise gradient elution from hexanes containing increasing proportions of EtOAc followed by MeOH, to produce nine subfractions. 1H NMR profiling of these fractions indicated the 25% MeOH in EtOAc fraction to be of further interest due to a large number of aromatic and heteroaromatic resonances in its 1H NMR spectrum. This fraction was further fractionated over a reverse phase solid phase extraction (SPE) cartridge (Varian Mega Bond Elut C18, 2 g) using gradient elution from MeOH-H2O (80:20) to MeOH (100%) to give three more fractions. RP-HPLC separation of the first of these fractions (YMC ODS AQ-323, 250 × 10.0 mm, 5 μm; MeOH-H2O (65:35), 2 mL/min) yielded compounds 1 (20.0 mg) and 2 (10.9 mg). Rechromatography of the remaining two SPE fractions using the same RP-HPLC conditions provided additional quantities of compounds 1 (8.3 mg) and 2 (5.2 mg).
Lophocladine A, 4-phenyl[2,7]naphthyridin-1(2H)-one (1): thin colorless radiating needles from DMSO (28.3 mg); mp 211-213 °C; UV (MeOH) λmax 224 nm (ε 4.06), 250 nm (ε 4.03), 314 nm (ε 3.88); IR (KBr) νmax 2918, 2849, 1677, 1624, 1591 cm-1; 1H and 13C NMR data (d6-DMSO), see Table 1 (for 1H and 13C NMR data recorded in d5-pyridine and 1H NMR data recorded in CF3COOD, see Supporting Material); positive ESIMS m/z 223.3 [M+H]+; positive APCIMS m/z 223.3 [M+H]+ (100); negative APCIMS m/z 221.5 [M-H]- (100); positive HRFABMS (3-NBA) m/z 223.0840 [M+H]+ (calcd for C14H11N2O, 223.0871).
Lophocladine B, 4-phenyl[2,7]naphthyridin-1-amine (2): yellowish gum (16.1 mg); UV (MeOH) λmax 220 nm (ε 4.01), 254 nm (ε 4.02), 336 nm (ε 3.89); IR (KBr) νmax 3333, 3220, 1636, 1612, 1543, 1465, 1357, 1229 cm-1; 1H and 13C NMR data (d6-DMSO), see Table 1 (for 1H NMR data recorded in d4-MeOH and CF3COOD, see Supporting Material); positive APCIMS m/z 222.3 [M+H]+ (100); negative APCIMS m/z 220.3 [M-H]- (100); positive HRFABMS (3-NBA) m/z 222.1032 [M+H]+ (calcd for C14H12N3, 222.1031).
Cytotoxicity against NCI-H460 and Neuro-2a Cell Lines
Toxicity towards mouse neuro-2a neuroblastoma cells and NCI-H460 lung tumor cells was measured using the method of Alley et al.22 with cell viability being determined spectrophotometrically by MTT reduction as previously described.23 Cells cultured in RPMI-1640 medium containing L-glutamine (Sigma, St. Louis, MO) and supplemented with 1 mM sodium pyruvate, 50 μg/mL streptomycin, 50 units/mL penicillin, 0.2% sodium bicarbonate and 10% fetal bovine serum were seeded in 96-well plates at 6000 cells/well in 180 μL. After 24 h, pure compounds were dissolved in DMSO and diluted into medium without fetal bovine serum; 20 μL of this mixture containing 2 μg of the test compound (= 10 μg/mL final assay concentration) was added for screening, or in a dilution series for determining IC50 values. DMSO was less than 1% of the final concentration. After 48 h, the medium was removed and cell viability determined.
A-10 and MDA-MB-435 Cell Cultures
A-10 cells, a rat aortic smooth muscle cell line, were purchased from the American Type Culture Collection (Manassas, VA) and were grown in Basal Medium Eagle containing Earle's salts, 50 μg/mL gentamycin sulfate, and 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). MDA-MB-435 breast cancer cells were obtained from the Lombardi Cancer Center, (Georgetown University, Washington, DC) and were grown in IMEM (Richter's Medium; Biosource, Camarillo, CA), with 10% FBS and 25 μg/mL gentamycin sulfate.
Sulforhodamine B (SRB) Assay
Inhibition of MDA-MB-435 cell proliferation and cytotoxicity were measured using the SRB assay.24 Cells were plated in 96-well plates and allowed to attach and grow for 24 h. The compounds or vehicle (EtOH) were added and incubated with the cells for 48 h. Following drug exposure, the cells were fixed with 10% trichloroacetic acid at 4 °C for 1 h and then the cell layer was stained with an SRB solution (0.4%) for 30 min. Excess stain was washed off with 1% HOAc and the SRB was solubilized with 10 mM TRIS base for 1 h on an orbital shaker. The absorbance of the SRB solution was measured at 560 nm. Dose-response curves were generated, and the IC50 values calculated from the linear portion of the log dose-response curves.
Indirect Immunofluorescence
Phenotypic changes in the structure of the cells were evaluated by indirect immunofluorescent techniques. A-10 cells were plated onto glass coverslips, treated with a range of compounds for 18-24 h. The cells were fixed with MeOH, blocked with 10% calf serum and then incubated with a β-tubulin antibody (T-4026; Sigma, St. Louis, MO). After a series of washes the secondary antibody, FITC conjugated sheep anti-mouse (F-3008; Sigma) was added for 1 h. After another series of washes the nuclei were stained with 0.1 μg/mL 4,6-diamidino-2-phenylindol (DAPI), and then washed and the coverslips mounted. The coverslips were examined with a Nikon ES800 fluorescence microscope, and pictures taken with a Photometrics Cool Snap FX3 camera.
Microfilament Disrupting Assay
Lophocladine B (2) was tested for microfilament-disrupting activity using rhodamine-phalloidin. A-10 cells were grown on glass coverslips in Basal Medium Eagle (BME) containing 10% fetal calf serum. The cells were incubated with the test compound for 24 h and then fixed with 3% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 2 min, and chemically reduced with sodium borohydride (1 mg/mL in PBS) three times for 5 min each. Following a 45 min incubation with 100 nM TRITC-phalloidin in phosphate buffered saline (to visualize the actin cytoskeleton), the coverslips were washed, stained with 4,6-diamidino-2-phenylindole (DAPI) to visualize DNA, mounted on microscope slides, and examined and photographed using a Nikon E800 Eclipse fluorescence microscope with a Photometrics Cool Snap FX3 camera. The images were colorized and overlayed using Metamorph® software.
Cell Cycle analysis
MDA-MB-435 cells were treated for 24 h with vehicle or sample. The cells were harvested and stained with Krishan's Reagent. This suspension was analyzed using a Becton Dickinson FACScan flow cytometer, and the data were plotted as propidium iodide intensity versus the number of events. Quality control samples were run with each assay to insure instrument linearity and resolution. Percentages of cells in each phase of the cell cycle were determined using ModFit LT™ 3.0 software (manual analysis mode).
Receptor Affinity Screen
Assays for the following receptors were performed by the PDSP/NIMH: (1) muscarinic receptors: M1, M5; (2) opiate receptors: μ, δ, κ; (3) GABA receptors: benzodiazepine (rat brain site), GABA A; (4) histaminergic receptors: H1, H2, H3, H4. Detailed on-line protocols are available for all assays at the PDSP/NIMH web site (http://pdsp.cwru.edu). For screening purposes, 10 μM of each compound was incubated with the appropriate receptor preparation and percent inhibition determined.
Adenylyl Cyclase Assay
Adenylyl cyclase assays were performed according to a method described previously.25 δ-opioid receptor-expressing CHO cell cultures were washed twice with serum-free F12 Ham's medium and then incubated for 8 h in 1 mL of the same medium containing 1.2 μCi [3H]adenine. The tritium-containing medium was then aspirated and replaced with serum-free F12 Ham's medium containing a phosphodiesterase inhibitor 50 μM Ro20-1724 and peptidase inhibitors (10 μM bestatin, 30 μM captopril, 50 μM L-leucyl-L-leucine). These cultures were incubated at 37 °C for 40 min in the presence of 50 μM forskolin and various concentrations of opioids. Incubations were terminated by the addition of 300 μL of Stop Solution (2% SDS, 1.3 mM cyclic AMP), followed by addition of 100 μL concentrated HClO4, and 750 μL H2O. [14C]Cyclic AMP (5000 cpm in 50 μL) was added to each plate to correct for recovery. After transferring the contents of culture dishes to 1.5 mL centrifuge tubes, 12 M KOH was added to neutralize the samples. The resulting precipitate was pelleted by centrifuging at 10,000 × g for 10 min. Cyclic AMP in the supernatants was isolated by sequential chromatography over Bio-Rad AG-50W-X4 cation exchange resin and neutral alumina. Concentrations of [3H]cyclic AMP and [14C]cyclic AMP in eluates were determined simultaneously using a Beckman LS 6000SC scintillation counter (3H channel 0–250, 14C channel 350–670). Counts were corrected for crossover and recovery.
Supplementary Material
1D NMR spectra of lophocladin A (1) and B (2) recorded in various solvents (d6-DMSO, d5-pyridine, d4-MeOH and CF3COOD), key homodecoupling and 1D DPFGSE-NOE experiments for compound 1, comparison of NMR spectroscopic data of 1 and 2 with those published for closely related compounds, a graph of the adenylyl cyclase assay and photomicrographs of the morphological changes of NCI-H460 lung cancer cells induced by lophocladine B (2). This material is available free of charge via the Internet at http://pubs.acs.org.

Acknowledgments
The authors gratefully acknowledge the government of Fiji for permission to make these algal collections, the NMR facility of the Department of Chemistry at Oregon State University and the OSU mass spectrometry facility (J. Morre). We also would like to thank the NIMH Psychoactive Drug Screening Program for biological screening. Financial support for this work at OSU and SIO came from the National Institutes of Health (GM63554 and CA100851), the National Institute of Environmental Health Sciences (P30 ES00210) and at SFBR from the William Randolph Hearst Foundation. H.G. acknowledges fellowship support from the German Research Foundation (GR 2673/1-1).
References and Notes
- 1.Blunt JW, Copp BR, Munro MHG, Northcote PT, Prinsep MR. Nat Prod Rep. 2005;22:15–61. doi: 10.1039/b415080p. and earlier reviews in the series. [DOI] [PubMed] [Google Scholar]
- 2.(a) Che CT. Drug Dev Res. 1991;23:201–218. [Google Scholar]; (b) Bourgougnon N, Lahaye M, Chermann JC, Kornprobst JM. Bioorg Med Chem Lett. 1993;3:1141–1146. [Google Scholar]; (c) Haslin C, Lahaye M, Pellegrini M, Chermann JC. Planta Med. 2001;67:301–305. doi: 10.1055/s-2001-14330. [DOI] [PubMed] [Google Scholar]; (d) Mazumder S, Ghosal PK, Pujol CA, Carlucci MJ, Damonte B, Ray B. Int J Biol Macromol. 2002;31:87–95. doi: 10.1016/s0141-8130(02)00070-3. [DOI] [PubMed] [Google Scholar]
- 3.Mori T, O'Keefe BR, Sowder RC, II, Bringans S, Gardella R, Berg S, Cochran P, Turpin JA, Buckheit RW, Jr, McMahon JB, Boyd MR. J Biol Chem. 2005;280:9345–9353. doi: 10.1074/jbc.M411122200. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fuller RW, Cardellina JH, II, Kato Y, Brinen LS, Clardy J, Snader KM, Boyd MR. J Med Chem. 1992;35:3007–3011. doi: 10.1021/jm00094a012. [DOI] [PubMed] [Google Scholar]; (b) Yotsu-Yamashita M, Yasumoto T, Yamada S, Bajarias FFA, Formeloza MA, Romero ML, Fukuyo Y. Chem Res Toxicol. 2004;17:1265–1271. doi: 10.1021/tx0498556. [DOI] [PubMed] [Google Scholar]
- 5.(a) Etahiri S, Bultel-Poncé V, Caux C, Guyot M. J Nat Prod. 2001;64:1024–1027. doi: 10.1021/np0002684. [DOI] [PubMed] [Google Scholar]; (b) Topcu G, Aydogmus Z, Imre S, Gören AC, Pezzuto JM, Clement JA, Kingston DGI. J Nat Prod. 2003;66:1505–1508. doi: 10.1021/np030176p. [DOI] [PubMed] [Google Scholar]
- 6.Daigo K. Yakugaku Zasshi. 1959;79:350–353. [Google Scholar]
- 7.Vairappan CS, Kawamoto T, Miwa H, Suzuki M. Planta Med. 2004;70:1087–1090. doi: 10.1055/s-2004-832653. [DOI] [PubMed] [Google Scholar]
- 8.Manefield M, de Nys R, Kumar N, Read R, Givskov M, Steinberg P, Kjelleberg S. Microbiol. 1999;145:283–291. doi: 10.1099/13500872-145-2-283. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ballantine DL, Gerwick WH, Velez SM, Alexander E, Guevara P. Hydrobiologica. 1987;151/152:463–469. [Google Scholar]; (b) Millar AJK. Hydrobiologica. 2000;398/399:65–74. [Google Scholar]; (c) Nam KW, Maggs CA, McIvor L, Stanhope MJ. J Phycol. 2000;36:759–772. doi: 10.1046/j.1529-8817.2000.00013.x. [DOI] [PubMed] [Google Scholar]; (d) Coppejans E, Millar AJK. Bot Marina. 2000;43:315–346. [Google Scholar]
- 10.(a) Impellizzeri G, Mangiafico S, Oriente G, Piattelli M, Sciuto S. Phytochemistry. 1975;14:1549–1557. [Google Scholar]; (b) Sciuto S, Chillemi R, Patti A, Piattelli M. J Nat Prod. 1988;51:1017–1020. doi: 10.1021/np50059a042. [DOI] [PubMed] [Google Scholar]; (c) Sciuto S, Chillemi R, Morrone R, Patti A, Piattelli M. Biochem Syst Ecol. 1989;17:5–10. [Google Scholar]
- 11.Synonyms: 2,7-diazanaphthalene, copyrine Allen CFH. Chem Rev. 1950;47:275–305. doi: 10.1021/cr60147a004.
- 12.(a) Baldwin JJ, Mensler K, Ponticello GS. J Org Chem. 1978;43:4878–4880. [Google Scholar]; (b) Sakamoto T, Kondo Y, Yamanaka H. Chem Pharm Bull. 1985;33:626–633. doi: 10.1248/cpb.33.5332. [DOI] [PubMed] [Google Scholar]; (c) Ikeura Y, Tanaka T, Kiyota Y, Morimoto S, Ogino M, Ishimaru T, Kamo I, Doi T, Natsugari H. Chem Pharm Bull. 1997;45:1642–1652. doi: 10.1248/cpb.45.1642. [DOI] [PubMed] [Google Scholar]; (d) Barbu E, Cuiban F. Heterocycl Comm. 2000;6:259–264. [Google Scholar]; (e) Barbu E, Wolff JJ, Bolocan I, Cuiban F. Heterocycl Comm. 2000;6:25–28. [Google Scholar]; (f) Paudler WW, Cornrich SJ. J Heterocycl Chem. 1970;7:419–421. [Google Scholar]
- 13.Janot MM, Guilhem J, Contz O, Venera G, Cionga E. Ann Pharm Fr. 1979;37:413–420. [PubMed] [Google Scholar]
- 14.Valeriote F, Grieshaber CK, Media J, Pietraszkewicz H, Hoffmann J, Pan M, McLaughlin S. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- 15.Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds: Tables of Spectral Data. 3rd engl. Springer-Verlag; Berlin: 2000. pp. 102–111. [Google Scholar]
- 16.Barbu E, Mihaiescu D, Cuiban F. Molecules. 2000;5:956–960. [Google Scholar]
- 17.Stott K, Keeler J, Van QN, Shaka AJ. J Magn Reson. 1997;125:302–324. [Google Scholar]
- 18.See Supporting Information. The NMR spectroscopic data of 1 were in good agreement with those published for closely related compounds (see ref. 12a-d).
- 19.See Supporting Information. The NMR data of 2 were in good agreement with those published for closely related synthetic derivatives: see ref. 12f and 16.
- 20.For examples of agents inducing cell morphology and their modes of action see Itayasu T, Shimizu T, Iizumi T, Oshio S, Umeda T, Takeda K. Anticancer Res. 1998;18:113–118.Erkel G, Gehrt A, Anke T, Sterner O. Z Naturforsch. 2002;57c:759–767. doi: 10.1515/znc-2002-7-834.West CML, Price P. Anti-Cancer Drugs. 2004;15:179–187. doi: 10.1097/00001813-200403000-00001.
- 21.Litvinov VP, Roman SV, Dyachenko VD. Russ Chem Rev. 2001;70:299–320. [Google Scholar]
- 22.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 23.Manger RL, Leja LS, Lee SY, Hungerford JM, Hokama Y, Dickey RW, Granade HR, Lewis R, Yasumoto T, Wekell MM. J AOAC Int. 1995;78:521–527. [PubMed] [Google Scholar]
- 24.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 25.Soderstrom K, Choi H, Berman FW, Aldrich JV, Murray TF. Europ J Pharmacol. 1997;338:191–197. doi: 10.1016/s0014-2999(97)81948-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1D NMR spectra of lophocladin A (1) and B (2) recorded in various solvents (d6-DMSO, d5-pyridine, d4-MeOH and CF3COOD), key homodecoupling and 1D DPFGSE-NOE experiments for compound 1, comparison of NMR spectroscopic data of 1 and 2 with those published for closely related compounds, a graph of the adenylyl cyclase assay and photomicrographs of the morphological changes of NCI-H460 lung cancer cells induced by lophocladine B (2). This material is available free of charge via the Internet at http://pubs.acs.org.