

Abstract
Recent culture-independent studies have revealed that a healthy vaginal ecosystem harbors a surprisingly complex assemblage of microorganisms. However, the spatial distribution and composition of vaginal microbial populations have not been investigated using molecular methods. Here, we evaluated site-specific microbial composition within the vaginal ecosystem and examined the influence of sampling technique in detection of the vaginal microbiota. 16S rRNA gene clone libraries were prepared from samples obtained from different locations (cervix, fornix, outer vaginal canal) and by different methods (swabbing, scraping, lavaging) from the vaginal tracts of eight clinically healthy, asymptomatic women. The data reveal that the vaginal microbiota is not homogenous throughout the vaginal tract but differs significantly within an individual with regard to anatomical site and sampling method used. Thus, this study illuminates the complex structure of the vaginal ecosystem and calls for the consideration of microenvironments when sampling vaginal microbiota as a clinical predictor of vaginal health.
The vaginal microbiota is important for maintaining vaginal health and preventing infections of the reproductive tract (10, 25, 34). However, studies using 16S rRNA gene clone libraries for identifying vaginal microbes have revealed considerably more diversity in the vaginal microbial communities of healthy premenopausal women than previously realized (14, 17, 31, 33, 35, 36), thereby calling into question currently existing models for a healthy vaginal ecosystem and how it might be assessed. Although the vaginal microbiota in healthy individuals was traditionally thought to be dominated by Lactobacillus species, more recent studies have demonstrated that Lactobacillus is not the predominant bacterial genus within the vaginal tracts of a significant number of healthy women (1, 17, 31, 35, 36). Although 60 to 70% of the women had Lactobacillus-dominated vaginal microbiota, there were also individuals who lacked Lactobacillus altogether and instead had Gardnerella, Atopobium, Prevotella, Pseudomonas, or Streptococcus as the predominant bacteria in the vagina (1, 17, 31, 35, 36). The significant bacterial diversity observed among these individuals suggests that defining a healthy vaginal environment is more complex than originally thought.
Although the most common method of collecting vaginal samples for clinical analysis is to swab the middle or deep vaginal canal (6, 14, 31, 35), the extent to which this approach yields samples representative of the entire vaginal microbiota is unclear. Indeed, the size and anatomical complexity of the vaginal tract suggest the possibility that distinct microbial populations may reside at discrete sites (e.g., cervix, fornix, outer vaginal canal). However, within individuals, the presence or absence of vaginal microniches capable of supporting discrete microbial populations has not been evaluated. Differences in microbial population distributions within individuals may impede identification of the specific bacterium or group of bacteria responsible for vaginal health, which ultimately could impact clinical diagnostic evaluation and treatment of vaginal disease.
To evaluate the importance of considering both sampling site and sampling method for capturing representative vaginal microbial communities within individuals, we analyzed samples obtained from three different sites (cervix, fornix, outer vaginal canal) and by three different collection methods (swabbing, scraping, lavaging) from the vaginal tracts of eight clinically healthy (asymptomatic) women of reproductive age. We report here that the distribution of microbiota within the human vagina is not uniform but, instead, is distributed in a heterogeneous manner that varies according to anatomical location and sampling technique.
MATERIALS AND METHODS
Study participants and sample collection.
The Institutional Review Boards of the University of Illinois at Urbana-Champaign and the Carle Foundation Hospital approved this study. Eight healthy (asymptomatic), premenopausal women between the ages of 20 and 45 years were recruited for this study. Informed consent was obtained from all study participants. Individuals who were asymptomatic and showed no clinical signs of vaginal disease upon examination by a physician, including evidence of vaginal discharge, amine or fishy odor, and a vaginal pH of >4.5, were included in the study. Individuals were not eligible for the study if they were pregnant, lactating, menstruating, or had used antibiotics or spermicides within 2 months prior to specimen collection. Six vaginal samples were collected from each subject (Fig. 1). Swab samples were obtained from the cervix, fornix, and lower one-third outer region of the vaginal canal by using Copan sterile plastic Dacron transport swabs (HealthLink, Jacksonville, FL). This was followed by collection of an ectocervical lavage sample using a 15-ml sterile saline solution (Health Care Logistics, Circleville, OH). Two vaginal scrapings were obtained from the upper (proximal) one-third and lower (distal) one-third regions of the vaginal canal by using sterile Pap-Perfect plastic spatulas (Medscand, Trumbull, CT), which allowed for collection of both loose and more-adherent bacteria. All vaginal swab and scrape samples were placed in 1 ml sterile saline (Health Care Logistics, Circleville, OH) and frozen immediately upon collection and stored at −80°C for up to 3 years until used. To ensure that the microbial community profiles would be generated only from sites that had not been previously perturbed by swab or scrape procedures, a single sample was collected from each site, using the swab or scrape method. The one exception was the lower one-third region of the vaginal canal, which was swabbed first and then scraped. Vaginal smears were also obtained for each subject at the time of clinical examination by rolling a swab across and along the length of the vaginal wall and then onto a glass slide, which was stored at −80°C until Gram stained for evaluation according to the Nugent criteria (22), where a score of 0 to 3 corresponded to normal vaginal microbiota, a score of 4 to 6 was interpreted as disturbed vaginal microbiota corresponding to an intermediate state, and a score of 7 to 10 was classified as being consistent with bacterial vaginosis (BV). In addition, clinical diagnostic testing for sexually transmitted diseases was performed using the in-house laboratory services provided by the Carle Foundation Hospital, including evaluation for hepatitis B virus surface antigen, human immunodeficiency virus type 1 (HIV-1)/HIV-2 antibody, syphilis (rapid plasma reagin, qualitative test), Chlamydia, and Neisseria gonorrhoeae.
FIG. 1.
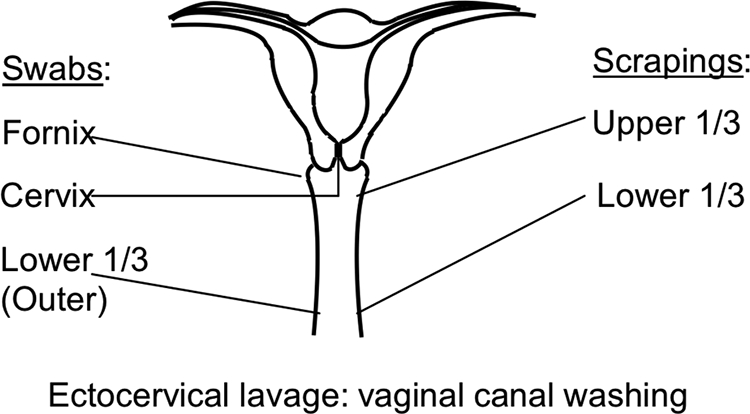
Schematic representation of vaginal sampling locations. Three swab samples were collected, from the fornix, cervix, and lower one-third (outer) region of the vaginal canal. Two vaginal scrapings were collected, from the upper one-third and the lower one-third of the vagina. An ectocervical lavage sample was obtained by washing the vaginal canal with sterile saline solution.
Genomic DNA extraction and PCR amplification.
Genomic DNA was isolated from 0.5-ml aliquots of each of the vaginal samples. To each sample, 125 μl of 0.5 M Na-EDTA (pH 8.0) containing 75 mg/ml lysozyme (Sigma-Aldrich, St. Louis, MO) was added, and the samples were incubated at 37°C for 30 min. To each sample, 70 μl of 10% sodium dodecyl sulfate and 5 μl of 10 mg/ml proteinase K (Fisher Scientific, Pittsburgh, PA) were added, and the resulting mixture was subjected to three successive freeze-thaw cycles, each consisting of incubation within a dry ice/ethanol bath for 5 min, followed by incubation at 37°C for 5 min. The samples were then incubated at 55°C for an additional 30 min to complete the proteinase K digestion of proteins in the disrupted cell suspension. Samples were centrifuged at 16,000 × g for 20 min following the addition of 70 μl of 5 M NaCl to the mixture and incubation on ice for 30 min. The samples were extracted sequentially with equal volumes of phenol and phenol-chloroform-isoamyl alcohol (Sigma-Aldrich, St. Louis, MO), followed by ethanol precipitation of the genomic DNA. Dried DNA pellets were resuspended in 100 μl of Tris-EDTA (10 mM Tris-Cl, 1 mM EDTA [pH 8.0]) and stored at −80°C until used. The quality and quantity of the extracted DNA were estimated by agarose gel analysis, using 5 μl of the extracted DNA.
PCR amplification of the nearly full-length 16S rRNA gene was performed using our optimized primer set, which we have previously shown to be superior for assessing vaginal samples, particularly for bacteria such as Gardnerella and Chlamydiales (13). This primer set was a combination of degenerate and species-specific 27f (numbering based on Escherichia coli 16S rRNA gene positions) forward primers (27f-YM, 27f-Bif, 27f-Bor, and 27f-Chl primers in a 4:1:1:1 ratio, respectively) (27f-YM, 5′-AGAGTTTGATYMTGGCTCAG-3′ [fourfold degenerate universal bacterial]; 27f-Chl, 5′-AGAATTTGATCTTGGTTCAG-3′ [Chlamydiales-specific]; 27f-Bor, 5′-AGAGTTTGATCCTGGCTTAG-3′ [Borrelia specific]; 27f-Bif, 5′-AGGGTTCGATTCTGGCTCAG-3′ [Bifidobacterium specific]) in conjunction with the 1492r bacterial reverse primer (1492r, 5′-TACCTTGTTACGACTT-3′). Primers were purchased from Integrated DNA Technologies (Coralville, IA). Briefly, 1 to 5 ng of total DNA was added to a PCR mixture containing 1× platinum Taq PCR buffer (Invitrogen, Carlsbad, CA), 2 mM MgCl2, 0.2 mM each deoxynucleoside triphosphate, 0.2 μM of the 27f-YM+3 primer mixture, 0.2 μM of the 1492r primer, and 0.25 U of platinum Taq in a total volume of 25 μl. PCR amplification was performed in the thermal cycler (DNA engine PTC-200; Bio-Rad, Hercules, CA) as follows: initial denaturation at 94°C for 4 min, followed by 24 cycles of 94°C for 1 min, 48°C for 30 s, and 72°C for 2 min. Upon completion of cycling, an additional 25 μl of a PCR mixture containing 1× platinum Taq buffer, 2 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 0.9 μM of the 27f-YM+3 primer mixture, 0.9 μM of the 1492r primer, and 0.25 U of platinum Taq was added to each PCR mixture. One additional cycle of amplification was performed as follows: 94°C for 1 min, 48°C for 30 s, and 72°C for 12 min. Amplification products were first purified using a PCR purification kit (Qiagen, Valencia, CA) and then further purified using 1% agarose gel for separation, followed by extraction using a QIAquick gel extraction kit (Qiagen, Valencia, CA).
16S rRNA gene clone library construction and sequencing.
Purified PCR products were cloned into the pCR2.1-TOPO vector and electroporated into electrocompetent Escherichia coli TOP10 cells by using the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA), according to the manufacturer's recommendations. For each sample, at least one 96-well plate containing a library with >90% of the colonies carrying cloned inserts, as determined by PCR analysis, was sequenced using the T7 promoter and M13 reverse primer sites at the high-throughput sequencing facility of the W. M. Keck Center for Comparative and Functional Genomics at University of Illinois at Urbana-Champaign (UIUC). For two subjects (no. 403 and 409), an additional, independent library for each of the six samples per subject was constructed and sequenced. The same extracted genomic DNA from each of the six samples from the two subjects was used as the starting material for the construction of the repeat libraries. All procedures were identical to those used for the original library construction.
Sequence processing and analysis.
Sequence processing was carried out using a series of Unix shell scripts. Forward and reverse reads from each clone were aligned using the BL2SEQ program in the stand-alone BLAST package from NCBI and then joined if the overlapping sequence was larger than 30 bp. For those pairs of reads that had less than 30 bp or no overlap and thus could not be aligned, each pair of reads was joined with five n's between the forward and reverse reads from each clone. These pseudojoined sequences were then subjected to a BLASTN search against all joined sequences obtained from the same clone library. If by aligning with a sequence from another clone it was revealed that the sequences still had overlap, then the two segments were joined appropriately. If the two matched segments had no overlap but each read could be matched to another clone sequence, additional n's were added to fill the gap between the segments based on the alignment with the matched sequence. In all cases, vector and primer sequences were removed from the joined clone sequences by using the BLASTN program with parameters similar to those of the VecScreen program from NCBI. Using this process, 4,555 sequences without gaps and 836 sequences with gaps were obtained.
A total of 5,391 sequences were submitted to the Seqmatch program in the Ribosomal Database Project II (RDP II) to identify related bacterial 16S rRNA gene sequences (7). A total of 5,340 clone sequences, which had related bacterial 16S rRNA gene sequences in the RDP II database with high Seqmatch scores (>0.8), were selected and used for further analysis. The mean Seqmatch score was 0.97. Analysis of the 51 clone sequences, which did not match and were thus not used for further analysis, revealed that nearly all of them were mixed clones and probably not unique or unknown phylotypes. These 5,340 clone sequences were again submitted to the Seqmatch program in RDP II to identify type strain sequences most closely related to each clone. Based on the Seqmatch results, the clone sequences could be assigned to 67 type strains (i.e., 67 phylotypes) in the RDP II. The 16S rRNA gene sequences of the 67 type strains from RDP II and the 5,340 clone sequences from our libraries were aligned using ClustalX (version 1.83) or Fast Aligner in the ARB package (20, 30). The PHYLIP package was used to construct a distance matrix from the aligned 67 type strains using DNADIST (Kimura 2 correction), and a phylogenetic tree was generated using FITCH (the Fitch-Margoliash method) (11; http://evolution.genetics.washington.edu/phylip.html). For bootstrap values, three methods (Fitch, parsimony, and maximum likelihood) with 100 resamplings were performed using FITCH, DNAPARS, DNAMLK, SEQBOOT, and CONSENSE in PHYLIP.
Statistical analysis.
We used Bray-Curtis distance analysis to determine the differences in microbial population profiles. The Bray-Curtis dissimilarity index (5, 12), which is equivalent to a doubly weighted form of the Sørensen-Dice dissimilarity index, was calculated according to the equation
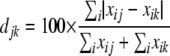 |
or
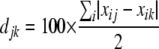 |
when 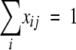 and
and 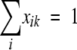 , where djk is the dissimilarity or distance between communities j and k, and xij and xik are the fractional or percentage populations of component i found in communities j and k. This distance was used to calculate the distance matrix for cluster analysis using the unweighted-pair group method using average linkages in the PHYLIP package.
, where djk is the dissimilarity or distance between communities j and k, and xij and xik are the fractional or percentage populations of component i found in communities j and k. This distance was used to calculate the distance matrix for cluster analysis using the unweighted-pair group method using average linkages in the PHYLIP package.
Phylotype diversity of the combined vaginal libraries within each subject was estimated by Simpson's reciprocal and Shannon-Wiener's diversity indices, using the EstimateS software (http://purl.oclc.org/estimates). The reciprocal of Simpson's diversity index (D), which provides information about the number of distinct phylotypes present in a sample, was calculated using the formula D−1 = (Σpi2)−1, where pi is the proportion of the ith species. The Shannon-Wiener diversity index (H), which provides information about the community composition both in terms of number and relative abundance of distinct phylotypes, was calculated using the formula H = −Σpi ln (pi), where pi is the proportion of the ith species.
To determine whether the observed differences in the microbiotas between libraries were statistically significant, the clone libraries of the six samples from each of the eight subjects were compared using the ∫-LIBSHUFF program (24). DNA distance matrices of the 5,340 clone sequences were calculated in ARB with the Jukes-Cantor correction (20) and subjected to ∫-LIBSHUFF analysis to compute the significance of the differences between libraries.
Bray-Curtis distance ratios.
To evaluate the significance of the differences found among the libraries from different sample types, we devised a distance ratio parameter, which is defined as the ratio of the “mean between-sample distance” to the “mean within-sample distance”:
 |
Bray-Curtis distance, as with other distance indices used to compare two samples, can be treated as the difference between two normally distributed variates X ∼ N(μX, σ2) and Y ∼ N(μY, σ2). The distribution of unsigned differences between these two variates, 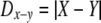 , can be treated as a folded normal distribution (18) with mean
, can be treated as a folded normal distribution (18) with mean
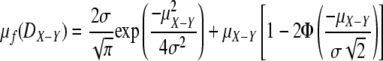 |
where 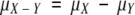 and
and 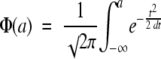 . Because
. Because 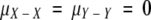 , the “mean within-sample distance” simplifies to
, the “mean within-sample distance” simplifies to 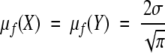 , and the unsigned distance ratio simplifies to
, and the unsigned distance ratio simplifies to
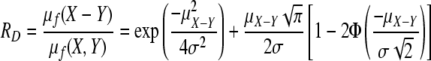 |
If 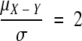 , X and Y have a 5% chance of being the same distribution, which corresponds to an RD value of ∼1.8. In the case that comparison is made between samples i and j with two repeats for each sample, the distance ratio for these two samples could be calculated as
, X and Y have a 5% chance of being the same distribution, which corresponds to an RD value of ∼1.8. In the case that comparison is made between samples i and j with two repeats for each sample, the distance ratio for these two samples could be calculated as
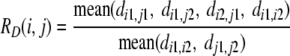 |
If the variation between two sample types is similar to the variation between repeat sampling events of the same sample, the distance ratio would be close to 1, and thus, the hypothesis that these two samples are different is not supported. On the other hand, for two samples with a distance ratio of >2, there is a <5% chance that two samples are from the same distribution. Single-factor analysis of variance was used to determine the P values for each within-sample versus between-sample comparison. To evaluate intersample differences within subjects 403 and 409, we repeated the library cloning process for each sample type. The Bray-Curtis dissimilarity indices based on 67 phylotypes (operational taxonomic units) were used as the distance for this calculation.
RESULTS
Clinical evaluation and sample collection.
The eight premenopausal nonpregnant women were asymptomatic for vaginal infections, as determined by self-assessment and clinical examination by the physician (J. S. Weisbaum). The median age of the women was 26.5 years (range 20 to 45 years), with seven self-identifying as Caucasian and one as Asian-Indian. Six vaginal samples were obtained from each subject, providing a total of 48 samples, from which 16S rRNA gene clone libraries were constructed.
Overall structure and diversity of vaginal bacterial communities.
Approximately 500 clone sequences from the six samples combined were available for most subjects, while repeat library construction provided over 1,000 clone sequences each for subjects 403 and 409. The combined 5,340 16S rRNA gene clones from all eight subjects were grouped into sequences representing 67 phylotypes, identified from 67 type strains from the RDP II database.
Phylogenetic analysis yielded a tree comprising five phyla, namely Proteobacteria, Firmicutes, Fusobacteria, Bacteroidetes, and Actinobacteria, and 32 genera (Fig. 2). Members of the phylum Firmicutes (mainly the genus Lactobacillus) were the most frequently detected clones recovered from the subjects, accounting for 55% of all clones. Clones with sequences belonging to the phyla Proteobacteria and Actinobacteria (mainly the genera Pseudomonas and Gardnerella, respectively) also constituted significant portions of the libraries, with each accounting for 15% of all the clones, while those belonging to Fusobacteria and Bacteroidetes were far less prevalent.
FIG. 2.

Phylogenetic distribution of vaginal bacteria from eight healthy, asymptomatic subjects. Numbers of clones for each phylotype are indicated in parentheses. The tree was produced using DNADIST and FITCH in PHYLIP. Bootstrap values of >50 from 100 resamplings (* indicates bootstrap value of <50, and − represents no clustering in the method) are indicated at each node (Fitch/parsimony/maximum likelihood). The scale bar represents 0.02 substitutions per site. Thermotoga maritima was used as the outgroup. The right side of the figure displays relative abundances of the phylotypes by subject and by group. The numbers above the abundance columns for the subjects correspond to the subject identification number. G1 to G4 correspond to the four groups into which the subjects were divided.
Genus level diversity was variable depending on the phylum, although this may be a reflection of database bias (16). The phyla Firmicutes and Proteobacteria possessed phylotypes belonging to 14 and 12 genera, respectively, but the other three phyla contained phylotypes belonging to only one to three genera each. At the species level, the genus Pseudomonas showed the highest diversity, with 19 species, while Lactobacillus had a total of six species. Five type strain sequences (Lactobacillus iners, Lactobacillus crispatus, Lactobacillus gasseri, Gardnerella vaginalis, and Pseudomonas gessardii) closely matched more than 500 clones each, while 22 type strain sequences closely matched only one or two clones each.
Variation in microbial composition among subjects.
To examine the overall microbial profile of each individual, a dendrogram comparing the eight subjects was constructed based on the Bray-Curtis distances of the phylum-level composition of the combined clone libraries for each individual (Fig. 3). Subjects clustered into four groups with a cutoff distance of 20%: group 1 (subjects 401 and 402), group 2 (subjects 404, 405, 406, and 408), group 3 (subject 403), and group 4 (subject 409). The presence and relative abundance of the 67 phylotypes varied among the groups (Fig. 2 and 3). Group 1 showed the lowest diversity compared to those of the other groups, according to Shannon's and Simpson's diversity indices (Table 1), whereas group 4, which had members from all five phyla represented, exhibited the highest diversity. Group 1 was comprised predominantly of Lactobacillus species, constituting approximately 90% of the total clones sequenced from each individual. Lactobacillus and Pseudomonas constituted the two most abundant bacterial genera found in group 2, with Lactobacillus species accounting for 60 to 70% of the clones and Pseudomonas species 20 to 30%. The microbial compositions of groups 3 and 4 differed substantially from those of groups 1 and 2, with high proportions of sequences closely related to Gardnerella vaginalis. In group 3, 46% and 42% of the clones were closely related to G. vaginalis and Lactobacillus species, respectively. In group 4, G. vaginalis represented 27% of the clones, followed by clones related to Pseudomonas species (21%) and Leptotrichia amnionii (16%).
FIG. 3.

Grouping of subjects by bacterial composition at the phylum level. The histogram (lower panel) shows the phylum level bacterial composition of each subject, and the dendrogram (upper panel) displays the clustering of the eight subjects. Four groups were identified using a cutoff distance value of 20%. Over 500 clone sequences each were analyzed for groups 1 and 2, while about 1,000 clone sequences each were analyzed for groups 3 and 4.
TABLE 1.
Bacterial diversity of vaginal microbiota by subjects
| Diversity index | Value for indicated subjects in group:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 1
|
2
|
3
|
4
|
|||||
| 401 | 402 | 408 | 404 | 405 | 406 | 403 | 409 | |
| Shannon's | 0.72 | 0.32 | 1.70 | 1.79 | 1.97 | 2.00 | 1.25 | 2.62 |
| Simpson's | 1.34 | 1.13 | 3.01 | 4.17 | 4.95 | 3.78 | 2.58 | 7.95 |
Nugent scoring of phylogenetic groups.
Nugent scoring of Gram-stained smears was performed to compare the microbial profiles determined by 16S rRNA gene clone library analysis to the vaginal bacteria detected microscopically in each subject. Examination of the microbiota by Gram staining revealed that the six subjects in groups 1 and 2 had normal vaginal microbiota (Nugent score = 0), while the subject in group 3 (no. 403) had intermediate or altered vaginal microbiota (Nugent score = 6) and the subject in group 4 (no. 409) had a Nugent score of 9, indicative of vaginal microbiota consistent with BV, despite self-reporting as asymptomatic and having no overt clinical symptoms.
Variation in microbial composition by sample type within subjects.
The influence of anatomical site and sampling method on the composition of bacterial communities within each individual is reflected by the heterogeneity of the population distribution patterns among samples within individuals (Fig. 4). Clustering based on the microbial composition of the vaginal samples within each individual revealed that grouping of samples varied from subject to subject, indicative of the heterogeneity of samples both between and within individuals. Lactobacillus species were the most abundant bacteria in all the sample types in subjects 401 and 402. In subjects 404, 405, 406, and 408, Lactobacillus and Pseudomonas species were dominant in all the samples, but the relative abundances of each varied significantly among the sample types within and between individuals. G. vaginalis was abundant in all the samples from subject 403, but again, the relative abundances varied among the sample types. Substantial variation in bacterial composition was observed among all of the samples in subject 409, with none of the samples showing similarity to each other in their bacterial compositions.
FIG. 4.

Clustering of vaginal samples within individuals based on microbial composition. The histograms (lower portion of each panel) show the phylum level bacterial composition of each subject, and the dendrograms (upper portion of each panel) display the clustering of the sample types within each of the eight subjects. CX, swab cervix library; FX, swab fornix library; OT, swab outer library; SL, scrape lower library; SU, scrape upper library; LA, lavage library.
Diversity of microbiota within and among different sample types.
Clustering of subjects varied considerably depending on the sample type (see Fig. S1 in the supplemental material). We performed pairwise comparisons among the 48 sample types from all eight subjects using ∫-LIBSHUFF analysis (24), which tests the differences among bacterial 16S rRNA gene libraries. The results revealed the bacterial content of some samples within the same subject to be significantly (P < 0.001) different from other samples, while a number of samples were found to be subsets of each other (see Table S1 in the supplemental material). Libraries from the lavage samples of most of the subjects were subsets of the other libraries from the same subject. Compared to the other samples, the lavage samples detected the least diversity in the vaginal microbiota and exhibited a complete absence of Proteobacteria sequences (e.g., Pseudomonas) (see Fig. S2 in the supplemental material). The lavage samples were also the only samples that had no unique sequences that were not also present in one of the other sample types, and removing the lavage library from the combined libraries of each subject did not affect the types of bacteria detected for that individual (see Fig. S3 in the supplemental material). As swabs are the most commonly collected vaginal samples in published studies, the bacterial diversity detected by the combined swab samples alone was also examined. In most subjects, fewer bacterial types were detected using the three swab samples alone than those seen in the clone libraries of all six samples combined for each individual (see Fig. S3 in the supplemental material). The scrape samples showed the greatest bacterial diversity. Indeed, the two scrape samples alone detected as much diversity as all six vaginal samples combined (see Fig. S3 in the supplemental material).
Distance relationships of microbial profiles from different sample types.
Bray-Curtis distance analysis, which addresses the differences in population profiles based on specified classification criteria (i.e., binning by closely related representatives) rather than strictly considering phylogenetic sequence similarity, was performed for all sample types within each subject (see Table S2 in the supplemental material). When the distances were calculated at three different levels (67 phylotypes, 32 genera, and five phyla), the intersample variation could be identified clearly from the distance values in each case. However, consistent patterns in the sample-related variation of the bacterial populations were not observed among the eight subjects. In some subjects, only one sample (e.g., the lower scrape sample for subject 401) appeared different from all the other samples within that individual. In other subjects, the six samples clearly fell into separate groups, such as those of subject 405, where the lower scrape and upper scrape samples were distinguishable from the swab and lavage samples. And yet, all of the samples for subject 409 appeared to be quite heterogeneous with respect to each other.
Evaluation of potential cloning bias.
To test the possibility that the observed heterogeneity of the distribution patterns for the microbial populations might be due to cloning bias, we repeated the construction and sequencing of the libraries for two subjects. Subjects 403 and 409, whose samples showed the most microbial diversity and differed substantially from those of the other subjects in the composition of their vaginal bacteria, were selected for the construction of repeat libraries to confirm the presence of the highly diverse microbiota observed in these individuals. No differences were found in the bacterial compositions of the original and repeat clone libraries by using ∫-LIBSHUFF analysis (data not shown). Bray-Curtis dissimilarity distances for the repeat libraries were also used to calculate distance ratios of between-sample versus within-sample distances, which is equivalent to the statistical distance, as detailed in Materials and Methods. This allowed us to determine whether the distance between two different samples within the same subject was greater than the distance between repeat libraries of the same sample from that individual (Fig. 5). The ratios for the sample types within each of these subjects (Fig. 5A), most of which were >1, confirmed that the variation found among sampling sites or sampling techniques was not the result of variation that would be expected from repeat library cloning of the same sample. The distance ratios obtained correlated well with the Bray-Curtis distances from the combined libraries of the repeats (Fig. 5B). Using this distance ratio index, after considering the sampling variation for the repeat libraries from subject 403, the cervix and fornix swab samples were not different from each other (distance ratio of ∼1). Likewise, the cervix swab and lavage samples were not different from each other (distance ratio of ∼1). However, the fornix swab and outer swab samples were different from each other (distance ratio of 4.6). For subject 409, most of the samples were different from each other (distance ratios of 1.8 to 5.9).
FIG. 5.

Distance ratios of between-sample versus within-sample distances for repeat libraries from subjects 403 and 409. (A) Bray-Curtis dissimilarity distances for the repeat libraries for the two most diverse subjects (no. 403 and 409) used to calculate distance ratios. The P values for single factor analysis of variance comparing between-sample distances versus within-sample distances are denoted as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001. (B) Linear correlation of Bray-Curtis distances with the calculated distance ratios.
DISCUSSION
Normal as well as abnormal microbiotas play a critical role in the progression and outcome of complex microbial diseases (e.g., see references 19, 28, 32, and 36). In order to better understand the disease process, to gain clearer understanding of the influence of the microbiota that are present, and to better evaluate methods for diagnosing and treating these complex diseases, it is important to understand the impact that variability in collection protocols and sampling techniques may have on diagnosis and data interpretation.
Here, we report considerable variation between the microbiotas among samples collected from three vaginal sites (cervix, fornix, outer vaginal canal) and by three different sampling methods (lavaging, swabbing, scraping). Previous culture-based studies have reported differences in site-specific vaginal microbial profiles (2, 3, 21), but the scope of these studies was limited due to cultivation biases. This is the first study examining niche variation in the vaginal tract by using molecular methods, which allowed for greater resolution in the detection of site-specific differences. In addition, this is the first study to address potential sampling bias introduced by use of swabs only, which may not adequately collect adherent bacteria. Finally, results for culture-independent analysis of the influence of sampling methods on the detection of vaginal microbes have also not been previously reported.
The lavage sample detected the least diversity in the vaginal microbiota and may not be representative of the true diversity of the vaginal bacteria. Though the lavage sample was the most dilute of the six samples collected, dilution would not be expected to alter the overall microbial composition of the samples, merely the total quantity of microbes within that sample. An interesting observation was the complete lack of Proteobacteria sequences from the lavage samples of all the subjects, even though Proteobacteria constituted the second largest representation in the overall vaginal clone library. Little or no Fusobacteria, Actinobacteria, or Bacteroidetes were found in the three swab or lavage clone libraries for six of the eight subjects (groups 1 and 2), even though Fusobacteria, Actinobacteria, and, to a lesser extent, Bacteroidetes were present in the scrape samples, which presumably would facilitate the collection of more adherent organisms. Prior studies have shown that the vaginal microbiota includes organisms that form loosely and tightly tissue-adherent biofilms (9, 29), possibly accounting for the differences in the bacteria detected among the three sampling methods utilized. G. vaginalis, a member of the phylum Actinobacteria, forms adherent biofilms (29), and this may explain the greater prevalence of Actinobacteria in the scrape samples of some subjects. These results suggest that a single sample from an individual might not be sufficient to reflect the complexity of the vaginal microbiota within a subject, and these results provide a framework for microbial ecologists and population biologists who seek to identify and characterize factors that underlie the establishment and maintenance of barriers that separate and define microenvironments.
In this study, we were particularly concerned about causing perturbations in the microbial community composition at sites that were swabbed or scraped and, thus, collected a single sample from each site by any particular method. Nonetheless, we collected both a swab sample and a scrape sample from the lower one-third region of the vaginal canal of each individual. For six of the eight subjects, the microbial community compositions of the swab and scrape samples were considerably different. It is not currently clear whether these differences were strictly due to differences in the sampling methods or due to perturbations at the sampling site during collection, but this work underscores the importance of considering the potential effects of sample collection methodology on microbial community structure.
The composition of the vaginal microbiota varied substantially among the subjects in this study, in agreement with observations from other culture-independent studies of the vaginal microbiota (14, 17, 31, 33, 35, 36). Individuals from group 1 and group 2 all had Nugent scores of 0, consistent with what is generally considered a “healthy” vaginal microbiota (22). The vaginal microbiotas of group 1 subjects exhibited the least microbial diversity, and all the sample types were dominated by Lactobacillus spp. and, thus, most closely resembled what is conventionally considered to be a “healthy” microbial composition (1, 8, 10). Lactobacillus and Pseudomonas species dominated all the samples from group 2, but the relative abundances of each varied widely among the sample types within and between individuals. Given the abundance of Pseudomonas in most of the subjects in this study and in those from a recent study by Hyman et al. (17), Pseudomonas spp. may be previously unrecognized common members of the healthy vaginal ecosystem.
Gardnerella vaginalis was present in all the samples from subject 403 (group 3) and in most samples from subject 409 (group 4), but the relative abundances varied among the sample types. The presence of G. vaginalis sequences in the clone libraries of these two subjects is consistent with the Gram staining results for these individuals. While all of the subjects in this study were asymptomatic and did not exhibit any clinical signs of BV, the Nugent scores and the greater microbial diversities observed for subjects 403 and 409 suggest that subject 403 may have been transitioning into or out of disease and subject 409 may have had asymptomatic BV at the time of specimen collection. Alternatively, it is possible that our understanding of BV is incomplete, and a high Nugent score does not necessarily reflect a diseased state in all cases. Moreover, subject 409, who exhibited the greatest heterogeneity in bacterial composition among all of the sample types, reported having been infected with chlamydia in the 6 months prior to the study, indicating that the recently perturbed microbiota may have not yet recovered from the infection. However, this subject tested negative for chlamydia at the time of sample collection, and Chlamydia 16S rRNA gene sequences were not detected in the clone libraries from this individual. Interestingly, subjects 403 and 409 were also the only two subjects reporting new sexual partners within the 6 months prior to sample collection. History of multiple sexual partners is a known risk factor for BV (4, 26, 27), but given the small sample size, it is not possible to draw any conclusions regarding sexual history and vaginal microbiota in these study participants.
These results are highly significant for medical microbiologists who seek to correlate microbial community composition to the pathophysiology of vaginal diseases, and especially for clinical practitioners interested in predictive measures for vaginal health and disease, susceptibility to sexually transmitted disease, reproductive fecundity, and pregnancy outcome. Notably, standard clinical practice today mandates a single swab of the vaginal area, but protocols are poorly defined. The data presented here indicate that the bacterial diversity detected by combining swab samples only from three different locations in the vaginal canal is lower than that detected using both swab and scrape samples from multiple locations within an individual. The broader implication of this demonstration of the differences between samples within individuals is that it highlights the importance of the sampling site and sampling method in determining the composition of vaginal microbial communities within individuals.
The methods utilized in this study are applicable to both clinical and laboratory settings. Our clinical partners have been implementing the specimen collection methods described here with relative ease. Clinical laboratory techniques are changing rapidly and exploring the use of PCR and DNA gene-chip technologies for diagnostics. Consequently, it is very likely that culture-independent bacterial detection techniques such as 16S sequencing could be utilized in a clinical laboratory setting in the near future.
These findings could significantly impact specimen collection protocols and sampling techniques for experimental studies and for clinical diagnostic evaluation and treatment of vaginal disease, as microbes indicative of an unhealthy vaginal state may be absent or underrepresented in one sampling site or by one sampling method but may be predominant in another. This niche variation is particularly important with regard to obstetric and neonatal care in considering the appropriate collection technique for the determination of group B streptococcal colonization in pregnant women, a critical risk factor for adverse postpartum outcome (15, 23). Thus, based on the results of this study, an overall scoop of the entire vaginal tract that includes collection of adherent bacteria might provide a more representative view of the vaginal microbial composition and may be important for accurate clinical evaluation of individuals, especially those with highly heterogeneous microbiota or those who may be transitioning into disease.
Supplementary Material
Acknowledgments
This work was supported in part by the Research Board and the Institute for Genomic Biology of the University of Illinois at Urbana-Champaign and the Carle Foundation Hospital.
We thank Michelle Hughes, Barbara Hall, Cindy Fraser, and Ann Benefiel for their clinical and administrative assistance with sample collection. We thank Brian Ho, Rachel Whitaker, Angela Kent, and Abigail Salyers for helpful discussions.
Footnotes
Published ahead of print on 21 January 2009.
Supplemental material for this article may be found at http://jcm.asm.org/.
REFERENCES
- 1.Antonio, M. A., S. E. Hawes, and S. L. Hillier. 1999. The identification of vaginal Lactobacillus species and the demographic and microbiologic characteristics of women colonized by these species. J. Infect. Dis. 1801950-1956. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett, J. G., N. E. Moon, P. R. Goldstein, B. Goren, A. B. Onderdonk, and B. F. Polk. 1978. Cervical and vaginal bacterial flora: ecologic niches in the female lower genital tract. Am. J. Obstet. Gynecol. 130658-661. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett, J. G., and B. F. Polk. 1984. Bacterial flora of the vagina: quantitative study. Rev. Infect. Dis. 6(Suppl. 1)S67-S72. [DOI] [PubMed] [Google Scholar]
- 4.Bradshaw, C. S., A. N. Morton, S. M. Garland, M. B. Morris, L. M. Moss, and C. K. Fairley. 2005. Higher-risk behavioral practices associated with bacterial vaginosis compared with vaginal candidiasis. Obstet. Gynecol. 106105-114. [DOI] [PubMed] [Google Scholar]
- 5.Bray, J. R., and J. T. Curtis. 1957. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27325-349. [Google Scholar]
- 6.Burton, J. P., and G. Reid. 2002. Evaluation of the bacterial vaginal flora of 20 postmenopausal women by direct (Nugent score) and molecular (polymerase chain reaction and denaturing gradient gel electrophoresis) techniques. J. Infect. Dis. 1861770-1780. [DOI] [PubMed] [Google Scholar]
- 7.Cole, J. R., B. Chai, R. J. Farris, Q. Wang, A. S. Kulam-Syed-Mohideen, D. M. McGarrell, A. M. Bandela, E. Cardenas, G. M. Garrity, and J. M. Tiedje. 2007. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35D169-D172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney, M. L., and A. B. Onderdonk. 2001. Nugent score related to vaginal culture in pregnant women. Obstet. Gynecol. 9879-84. [DOI] [PubMed] [Google Scholar]
- 9.Domingue, P. A., K. Sadhu, J. W. Costerton, K. Bartlett, and A. W. Chow. 1991. The human vagina: normal flora considered as an in situ tissue-associated, adherent biofilm. Genitourin. Med. 67226-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donders, G. G. 2007. Definition and classification of abnormal vaginal flora. Best Pract. Res. Clin. Obstet. Gynaecol. 21355-373. [DOI] [PubMed] [Google Scholar]
- 11.Felsenstein, J. 1989. PHYLIP—phylogeny inference package (version 3.2). Cladistics 5164-166. [Google Scholar]
- 12.Field, J. G., K. R. Clarke, and R. M. Warwick. 1982. A practical strategy for analyzing multispecies distribution patterns. Mar. Ecol. Prog. Ser. 837-52. [Google Scholar]
- 13.Frank, J. A., C. I. Reich, S. Sharma, J. S. Weisbaum, B. A. Wilson, and G. J. Olsen. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 742461-2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fredricks, D. N., T. L. Fiedler, and J. M. Marrazzo. 2005. Molecular identification of bacteria associated with bacterial vaginosis. N. Engl. J. Med. 3531899-1911. [DOI] [PubMed] [Google Scholar]
- 15.Heath, P. T., and A. Schuchat. 2007. Perinatal group B streptococcal disease. Best Pract. Res. Clin. Obstet. Gynaecol. 21411-424. [DOI] [PubMed] [Google Scholar]
- 16.Hugenholtz, P. 2002. Exploring prokaryotic diversity in the genomic era. Genome Biol. 3REVIEWS0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyman, R. W., M. Fukushima, L. Diamond, J. Kumm, L. C. Giudice, and R. W. Davis. 2005. Microbes on the human vaginal epithelium. Proc. Natl. Acad. Sci. USA 1027952-7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leone, F. C., R. B. Nottingham, and L. S. Nelson. 1961. The folded normal distribution. Technometrics 3543-550. [Google Scholar]
- 19.Ley, R. E., R. Knight, and J. I. Gordon. 2007. The human microbiome: eliminating the biomedical/environmental dichotomy in microbial ecology. Environ. Microbiol. 93-4. [DOI] [PubMed] [Google Scholar]
- 20.Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier, Yadhukumar, A. Buchner, T. Lai, S. Steppi, G. Jobb, W. Forster, I. Brettske, S. Gerber, A. W. Ginhart, O. Gross, S. Grumann, S. Hermann, R. Jost, A. Konig, T. Liss, R. Lussmann, M. May, B. Nonhoff, B. Reichel, R. Strehlow, A. Stamatakis, N. Stuckmann, A. Vilbig, M. Lenke, T. Ludwig, A. Bode, and K. H. Schleifer. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 321363-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald, S. W., F. R. Manuel, and J. A. Embil. 1979. Localization of group B beta-hemolytic streptococci in the female urogenital tract. Am. J. Obstet. Gynecol. 13357-59. [DOI] [PubMed] [Google Scholar]
- 22.Nugent, R. P., M. A. Krohn, and S. L. Hillier. 1991. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of Gram stain interpretation. J. Clin. Microbiol. 29297-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettersson, K. 2007. Perinatal infection with group B streptococci. Semin. Fetal Neonatal Med. 12193-197. [DOI] [PubMed] [Google Scholar]
- 24.Schloss, P. D., B. R. Larget, and J. Handelsman. 2004. Integration of microbial ecology and statistics: a test to compare gene libraries. Appl. Environ. Microbiol. 705485-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwebke, J. R. 2005. Abnormal vaginal flora as a biological risk factor for acquisition of HIV infection and sexually transmitted diseases. J. Infect. Dis. 1921315-1317. [DOI] [PubMed] [Google Scholar]
- 26.Schwebke, J. R., and R. Desmond. 2005. Risk factors for bacterial vaginosis in women at high risk for sexually transmitted diseases. Sex. Transm. Dis. 32654-658. [DOI] [PubMed] [Google Scholar]
- 27.Smart, S., A. Singal, and A. Mindel. 2004. Social and sexual risk factors for bacterial vaginosis. Sex. Transm. Infect. 8058-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swidsinski, A., V. Loening-Baucke, H. Verstraelen, S. Osowska, and Y. Doerffel. 2008. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 135568-579. [DOI] [PubMed] [Google Scholar]
- 29.Swidsinski, A., W. Mendling, V. Loening-Baucke, A. Ladhoff, S. Swidsinski, L. P. Hale, and H. Lochs. 2005. Adherent biofilms in bacterial vaginosis. Obstet. Gynecol. 1061013-1023. [DOI] [PubMed] [Google Scholar]
- 30.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 254876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verhelst, R., H. Verstraelen, G. Claeys, G. Verschraegen, J. Delanghe, L. Van Simaey, C. De Ganck, M. Temmerman, and M. Vaneechoutte. 2004. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis, and bacterial vaginosis. BMC Microbiol. 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verstraelen, H. 2008. Cutting edge: the vaginal microflora and bacterial vaginosis. Verh. K. Acad. Geneeskd. Belg. 70147-174. [PubMed] [Google Scholar]
- 33.Verstraelen, H., R. Verhelst, G. Claeys, M. Temmerman, and M. Vaneechoutte. 2004. Culture-independent analysis of vaginal microflora: the unrecognized association of Atopobium vaginae with bacterial vaginosis. Am. J. Obstet. Gynecol. 1911130-1132. [DOI] [PubMed] [Google Scholar]
- 34.Witkin, S. S., I. M. Linhares, and P. Giraldo. 2007. Bacterial flora of the female genital tract: function and immune regulation. Best Pract. Res. Clin. Obstet. Gynaecol. 21347-354. [DOI] [PubMed] [Google Scholar]
- 35.Zhou, X., S. J. Bent, M. G. Schneider, C. C. Davis, M. R. Islam, and L. J. Forney. 2004. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 1502565-2573. [DOI] [PubMed] [Google Scholar]
- 36.Zhou, X., C. J. Brown, Z. Abdo, C. C. Davis, M. A. Hansmann, P. Joyce, J. A. Foster, and L. J. Forney. 2007. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 1121-133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.