

Abstract
Nonautonomous retrotransposon subfamilies are often amplified in preference to their coding-competent relatives. However, the mechanisms responsible for such replicative success are poorly understood. Here, we demonstrate that the autonomous MusD long terminal repeat (LTR) retrotransposons are subject to greater epigenetic silencing than their nonautonomous cousins, the early transposons (ETns), which are expressed at a 170-fold-higher level than MusD in mouse embryonic stem (ES) cells. We show that, in ES cells, 5′ LTRs and the downstream region of MusD elements are more heavily methylated and are associated with less-activating and more-repressive histone modifications than the highly similar ETnII sequences. The internal region of MusD likely contributes to their silencing, as transgenes with MusD, compared to those with ETnII sequences, show reduced reporter gene expression and a higher level of repressive histone marks. Genomic distribution patterns of MusD and ETn elements are consistent with stronger selection against MusD elements within introns, suggesting that MusD-associated silencing marks can negatively impact genes. We propose a model in which nonautonomous retrotransposons may gain transcriptional and retrotranspositional advantages over their coding-competent counterparts by elimination of the CpG-rich retroviral sequence targeting the autonomous subfamilies for silencing.
Long terminal repeat (LTR) retrotransposon and endogenous retroviral (ERV) families typically include autonomous members as well as a heterogeneous collection of nonautonomous defective elements with deletions or rearrangements of the coding sequence (2, 26, 30, 42, 52, 64). While most nonautonomous variants are incapable of transposition even in the presence of the necessary retroviral proteins encoded by the autonomous elements, some may form distinct subfamilies as a result of genomic amplification. For reasons yet unclear, some of these nonautonomous subfamilies transcribe and retrotranspose more efficiently than the parental retrotransposons. For example, both the copy number and transcript levels of the barley nonautonomous BARE-2 elements are higher than those of the autonomous BARE-1 elements (61). For other yet-unconfirmed, but highly plausible, pairs of nonautonomous/autonomous plant LTR retrotransposons, the nonautonomous members have comparable or higher copy numbers than their likely autonomous counterparts (25, 29, 63). ERV families in mammals show similar characteristics. In humans, a partially deleted HERV-H subfamily has amplified to greater numbers than the corresponding full-length form (24, 66). In mice, a partially deleted IΔ1 subclass of the retroviral-like intracisternal A-type particle (IAP) family is highly transcribed (3, 17) and is actively involved in insertional mutagenesis (39), suggesting a current retrotransposition rate greater than that of fully coding-competent IAP. The reasons for genomic amplification of defective subfamilies, often in preference to the coding-competent members on which they depend for retrotransposition, are largely unknown.
As a model system to study this phenomenon, we have chosen the mouse early transposon (ETn)/MusD family. While the progenitor of MusD elements was likely an infectious retrovirus, this family has acquired the status of a successful intracellular retrotransposon due to the loss of its envelope gene and a plasma membrane-targeting signal (49). Close to 100 full-length MusD elements exist in the C57BL/6 genome but only a few are still functional for autonomous retrotransposition (48). MusD-derived nonautonomous ETns (10, 37, 60) lack all retroviral genes and instead carry a noncoding nonretroviral sequence (Fig. 1A). ETns are actively transcribed in undifferentiated cells and owe their retrotransposition potential to the proteins encoded by the structurally intact MusD (48). Two subtypes of ETns have been described, with ETnI elements differing from ETnII and MusD in the 3′ end of the LTR and a short segment of the 5′ internal sequence (5, 57) (Fig. 1A). While the ETnI copy number of approximately 200 is greater than that of MusD at 100 and ETnII at 40 (5), the low number of recent ETnI-induced mutations (39) suggests that the transposition activity of this subtype is now relatively low. Being a younger subfamily than MusD, ETnII elements have a higher polymorphic fraction and, therefore, a more variable copy number between mouse strains (4, 71). Furthermore, the level of ETnII transcripts in embryos and undifferentiated cells, such as embryonic stem (ES) and embryonic carcinoma (EC) cells, is much higher than the level of MusD transcripts (5, 38). While over 27 new germ line mutations and strain variants caused by this family have been described, only 2 of them are due to an insertion of a MusD element (27), with all others resulting from integration of ETn elements (39). These facts imply that amplification of the younger ETnII subfamily is ongoing and, according to typical transposable element dynamics (9), will plateau once the forces of transposition and negative selection are in balance.
FIG. 1.

Structural and sequence comparisons of ETn and MusD elements. (A) Structure of ETn and MusD retrotransposon subfamilies. com, ETnII/MusD common region; gag, prot, and pol are retroviral genes. (B) Transcript levels of ETn and MusD retrotransposons. Quantitative RT-PCR on cDNA from J1 ES cells was performed; the MusD transcript number was set as 1. Data are means and standard deviations from three separate experiments each performed in duplicate. (C) Presence of Sp1/Sp3 sites in ETnII and MusD LTRs. The number of ETnII and MusD 5′ LTRs in the C57/BL6 genome with an intact core is shown for each of the three Sp1/Sp3 binding sites important for LTR activity (38). The number of elements with a 100% match to the LTR consensus sequence, with all three intact Sp1/Sp3 sites, is also indicated.
In vitro assays have shown that retrotransposition efficiencies of ETn and MusD elements are similar (48). Additionally, the number of nonmutated Lys3 tRNA primer binding sites used as a docking sequence for tRNAs that prime reverse transcription are also similar, as we found 11 such elements for ETnII and 10 for MusD in the C56BL/6 genome (data not shown). According to these data, the retrotransposition potentials of ETnII and MusD elements should be in a similar range. However, the preponderance of ETnII-induced germ line mutations implies otherwise. Since transcription rate has been shown to correlate with retrotransposition efficiency (12, 19, 20, 45, 53), we hypothesize that the current skewing of retrotranspositional events to the ETnII subfamily is due to the much higher number of ETnII than MusD transcripts available for copackaging with MusD-encoded proteins.
In this study, we investigated the reasons leading to retrotransposition success of nonautonomous ETn retrotransposons. We show that in ES cells, the autonomous MusD elements are transcriptionally repressed to a greater degree than ETns via epigenetic mechanisms. Our data suggest that the high level of ETn transcription is at least in part due to their lack of CpG-rich retroviral sequence targeting the MusD subfamily for silencing. We propose a model according to which nonautonomous retrotransposons may gain transcriptional and retrotranspositional advantages over their coding-competent counterparts through elimination of a CpG-rich retroviral sequence that increases the likelihood of their targeting for suppression.
MATERIALS AND METHODS
Cell culture.
Mouse embryonic teratocarcinoma cell line P19 (ATCC) and mouse erythroleukemia (MEL) cell line clone RL5 (16) (a gift from M. Lorincz) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. J1 wild-type (wt; 129S4/SvJae), 36c/c (Dnmt1−/− (31), 6aa (Dnmt3a−/−), 8bb (Dnmt3b−/−), and 7aabb (Dnmt3a−/− b−/−) (46) cell lines, courtesy of E. Li, were cultured in high-glucose Dulbecco's modified Eagle's medium supplemented with 15% fetal bovine serum, 2 mM glutamine, 0.1 mM nonessential amino acids, 10 ng/ml mouse leukemia inhibitory factor, 0.1 mM monothiolglycidol, penicillin, and streptomycin on gelatinized plates.
Plasmids and constructs.
For luciferase assays, internal sequences of ETnII#7 (AC079540; element position 130750 to 136290) and MusD#3 (AC084696; element position 139824 to 132348) were cloned into the BamHI-SalI restriction site opposite to the ETnII#7 LTR-containing KpnI-BglII cloning site of the pGL3-Basic (pGL3B; Promega) luciferase reporter vector. A plasmid with LTR#7 in pGL3B was designated pGL3B-LTR#7, and this LTR cloning was described previously (38). Internal regions were cloned from C57BL/6 genomic DNA under standard PCR conditions using the following primers: ETnII, first round, 540_1s and ETnIIgr1_3610as; second round, IM_ETn_637s-BamHI and IM_ETn_3593as-SalI for the forward-oriented insert and IM_INT_637s-SalI and IM_ETn_3593as-BamHI for the reverse-oriented insert. For MusD, the first-round primers were 696_1s and MusD_4094as; second-round primers were IM_MusD_636s-SalI and IM_MusD_3992as-SalI, with subsequent selection of correctly oriented clones. For stable transfections into MEL cells, ETnII#7 or MusD#3 internal regions were cloned in reverse orientation into the ClaI restriction site of the L1-CMV-GFP-1L vector (15) upstream of the cytomegalovirus (CMV) promoter driving the expression of the enhanced green fluorescent protein (EGFP) gene. The 3.3-kb nonretroviral internal region of ETnII-β (AC079540), including the common sequence, and the corresponding-sized MusD (AC084696) sequences were cloned from the C57/BL6 genomic DNA using the following primers: for ETnII, first round, 540_1s and ETnIIgr1_3636as; second round, IM_ETnII_318s-ClaI and IM_ETnII_3616as-ClaI. Primers for MusD were the following: first round, 696_1s and MusD_4094as; second round, IM_MusD_320s-ClaI and IM_MusD_3618as-ClaI. L1-CMV-GFP-1L and Cre-expressing CMV-Cre plasmids were generously donated by M. C. Lorincz. All inserts were confirmed by sequencing. For further information, see the primer sequences in Table S1 of the supplemental material.
In vitro patch methylation.
Constructs with ETnII and MusD LTRs cloned into the KpnI-BglII restriction site of the pGL3B luciferase reporter vector were described previously (38). Two ETnII LTRs, LTR#7 and LTR#13 (AC079497; element position 103995 to 96963) and two MusD LTRs, LTR#3 and LTR#9 (AF132039; element position 79690 to 87160), were used. ETnII and MusD LTR-containing pGL3B vectors were methylated in vitro with SssI methyltransferase according to the recommendations of the manufacturer (New England Biolabs); mock methylation was performed similarly, with the exclusion of SssI. Complete CpG methylation, or its absence, was confirmed by digestion with a methylation-sensitive enzyme, ApaLI, which has a restriction site in all LTRs. Methylated and mock-methylated LTRs were released from the respective plasmids by double KpnI/BglII digestion and gel isolated using a MinElute gel extraction kit (Qiagen). In parallel, an unmethylated pGL3B vector was double digested with KpnI and BglII, purified with a QIAquick PCR purification kit (Qiagen), treated with calf intestinal phosphatase (New England Biolabs), and again purified with a PCR purification kit (Qiagen). Each methylated and mock-methylated LTR was ligated into the unmethylated vector backbone in a 1:1 molar ratio with a high concentration of T4 DNA ligase (New England Biolabs) for 2 days at 16°C. As a promoterless control, pGL3B digested with a single KpnI enzyme, purified, and then religated was used.
Transient transfections and luciferase assays.
With large DNA fragments introduced into plasmid DNA, we saw evidence of reduced plasmid DNA uptake via lipofection. Such effects have been described previously and shown to account for greater-than-10-fold reductions in luciferase expression, inversely correlating with insert size (69). We cloned a 3-kb and a 3.7-kb lambda phage “stuffer” DNA fragment into the SalI restriction site of the pGL3B vector to serve as a promoterless size-adjusted control and normalized pGL3B-LTR#7 luciferase activity to nonmodified pGL3B, since the LTR is only 317 bp long, while the luciferase activities of the 3.15-kb ETnII internal region-containing constructs were normalized to the 3-kb lambda phage-containing pGL3B and those of the 3.68-kb MusD internal region-containing constructs were normalized to the 3.7-kb lambda phage-containing pGL3B.
For luciferase assays, P19 cells were seeded into 24-well plates at a density of 3 × 104 cells per well 24 h prior to transfection. Cells grown in monolayers were transfected with 500 ng of test plasmid DNA and 50 ng of the Renilla luciferase vector pRL-TK using 0.5 μl of Lipofectamine 2000 (Invitrogen) per well for the analysis of the internal ETnII and MusD regions or 1 μl of Lipofectamine 2000 per well for the analysis of the methylated LTR activity. Cells were washed 24 h after transfection in phosphate-buffered saline and harvested in 100 μl of 1× passive lysis buffer (Promega). Firefly and Renilla luciferase activities were measured using the dual-luciferase reporter assay system (Promega).
The data were first standardized to the internal Renilla luciferase control and expressed relative to the activity of the promoterless cut and religated pGL3B vector for the methylated LTR analysis or the pGL3B vector with a λ-phage stuffer DNA of the corresponding size for the ETnII and MusD internal region analysis. The results are means and standard deviations of three separate experiments each performed in duplicate.
Preparation of RNA and RT.
Total RNA was isolated using an RNeasy minikit (Qiagen) according to the manufacturer's protocol. For quantitative reverse transcription-PCR (RT-PCR), RNA was treated with DNase I and reverse transcribed using SuperScript III (Invitrogen) according to the manufacturer's protocol. A pool of two separate cDNA preparations was used for quantitative RT-PCR.
Preparation of genomic DNA and Southern blotting.
Genomic DNA was extracted using DNAzol (Invitrogen) according to the manufacturer's protocol. DNA was dissolved in 200 μl of 8 mM NaOH per 1 ml of DNAzol. Southern blotting and hybridizations were performed as described previously (38), with the probe specific for both ETnII and MusD elements synthesized using primers ETnII/MusD_465-s and ETnII/MusD_652-as.
Bisulfite treatment, T/A cloning, and sequencing.
Bisulfite conversion of DNA was performed using the EZ DNA methylation kit (Zymo Research) according to the manufacturer's protocol with modifications (51). The ∼750-bp region containing the LTR and the common region was amplified by PCR from J1 and Dnmt1−/− converted DNA using Platinum Taq (Invitrogen) with the following primers: forward, ETn/MusD_bis-40s (same for ETn and MusD); reverse, ETnI-ETnII-bis-rv for ETn and MusD-bis-rv for MusD. The larger ETnII fragment was separated from ETnI by using a Purelink quick gel extraction kit (Invitrogen). The common region from ETnII- or MusD-containing transgenes was amplified with vector primer L1_insert-bis-as and either ETnII_insert-bis-as or MusD_insert-bis-as, respectively. PCR products were purified using a Purelink quick gel extraction kit. PCR products from three separate PCRs for each sample were cloned using the pGEM-T Easy kit (Promega). Sequencing was performed by McGill University with the Genome Québec Innovation Centre sequencing platform. Only unique sequences (with either a unique CpG methylation pattern, a unique nonconversion of non-CpG cytosines, or polymorphisms) are shown; all sequences had a conversion rate of >98%.
ChIPs.
Chromatin immunoprecipitations (ChIPs) were performed with a ChIP assay kit (Upstate) according to the manufacturer's instructions. The following antibodies were used for immunoprecipitations: polyclonal anti-acetyl-histone H3 (Lys9; 07-352), polyclonal anti-trimethyl-histone H3 (Lys9; 07-442), and normal rabbit immunoglobulin G (IgG; 12-370) from Upstate Biotechnology (Millipore). The DNA was purified using a QIAquick PCR purification kit (Qiagen) and eluted in 50 μl of elution buffer.
Quantitative real-time PCR for ChIP and RT.
For analysis of immunoprecipitated ETnII and MusD sequences from J1 DNA, a primer in the common region, ETn-MusD_514-s, together with either an ETnII-specific primer, ETnII_662-as, or a MusD-specific primer, MusD_690-as, were used in quantitative real-time PCRs to amplify a 218- or a 175-bp sequence in the common region of the population of ETnII or MusD elements in the genome, respectively. For analysis of immunoprecipitated ETnII and MusD DNA in MEL cells with ETn or MusD transgenes, a primer in the common region, ChIP_E/M-comm-as, and a primer in the L1-CMV-GFP-1L vector, ChIP_L1-34as, were used to amplify a 166-bp common region sequence. The promoter region of the housekeeping gene Hprt1 was amplified as an example of an active gene, and a CpG-rich region of mouse Nkg2a was amplified as an example of an inactive gene (see reference 50 for primer sequences).
For quantitative RT-PCR (qRT-PCR) to determine relative ETnI, ETnII, and MusD expression levels, cDNA from J1 ES cells was amplified with primers specific for ETnI (ETnI-s plus ETnI-as), ETnII (ETn-MusD_514-s plus ETnII_662-as), and MusD (ETn-MusD_514-s plus MusD_690-as). To correct for possible differences in primer efficiencies, serial dilution of plasmids containing the respective amplicons was used for standards. For expression analysis in J1 versus Dnmt1−/− ES cells, the same primer pairs were used for ETnII and MusD. For Gapdh, GAPex6F plus GAPex7R were used; for IAP, IAP_3400s plus IAP_4400as were used. Forty-five rounds of amplification with Fast SYBR green PCR master mix (Applied Biosystems) were performed. The default Fast 7500 Fast System SDS software version 1.4.0.25 (7500 Fast RealTime PCR system; Applied Biosystems) cycle was used with the exception that amplification was performed at 65°C in a total volume of 20 μl. Data are presented as means and standard deviations of three biological replicates each performed in duplicate. Dissociation curve analysis was performed after the end of the PCR to confirm the presence of a single and specific product.
RMCE, transfection, and transgene selection.
For targeting of the ETnII or MusD internal region-containing constructs into the genome, a Cre recombinase-mediated cassette exchange (RMCE) method (7, 16) was used. Briefly, we used an MEL cell line clone with a cassette containing the hygromycin B and herpes simplex virus thymidine kinase (HyTK) fusion gene at the RL5 locus (15). This selectable marker allows for positive selection through resistance to hygromycin B and for negative selection through sensitivity to ganciclovir. The RL5 locus has been shown to be permissive for long-term expression of the integrated cassette (15). MEL cells were cultured in 750 μg/ml hygromycin B for 14 days before transfection to select for cells expressing the fusion gene; 4.5 × 105 cells were seeded per well of a six-well plate the day of transfection. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Briefly, 4 μg of L1-CMV-GFP-1L vector with an ETnII or a MusD insert was cotransfected with 1 μg CMV-Cre plasmid using 4 μl Lipofectamine 2000 per well. After 3 days, ganciclovir was added to the medium at a concentration of 10 μM per ml to select against cells still expressing the HyTK fusion gene. Five separate transfections were carried out to avoid clonal effects; cells were cultured in ganciclovir-containing medium for 10 days, subculturing when necessary. The presence of a single colony was confirmed under the microscope, and genomic PCR was carried out to confirm the presence and the direction of the transgene.
Fluorescence-activated cell sorting (FACS) and analysis of cassette integration.
Transfected cells were sorted into 96-well plates at a density of 1 cell per well after 10 days of ganciclovir selection. Cells from five independent transfections were mixed before sorting. Cell sorting was performed on BD FACSDiva and flow analysis was performed on a benchtop BD FACSCalibur. Nonviable cells were gated on the basis of propidium iodide exclusion; 20,000 propidium iodide-negative events were analyzed; untransfected cells were used as a control for EGFP fluorescence. The direction of the integrated cassette was determined by PCR and clones in orientation B (15), identified using the common primer Chr4_RL5-as and either ETn_INT_2045s or MusD_int_3263s for ETnII or MusD internal region constructs, respectively, were selected. Mean EGFP fluorescence of the clones was graphed. We consider the mean to be a sufficiently good representation of each clonal population, as cells within each clone formed a tight peak with respect to EGFP fluorescence, and EGFP-negative cells were never observed.
RESULTS
Potential explanations for higher levels of ETnII than MusD transcripts.
We have previously found that in mouse embryos, ETnII transcripts are 30-fold more abundant than MusD transcripts, despite the higher genomic copy number of MusD and high similarities of their LTRs (5, 38) (Fig. 1A, ETn and MusD structures). To assess whether this is also true for ES cells, we performed qRT-PCR on cDNA from the J1 ES cell line, with primers capturing 139 ETnI, 25 ETnII, and 115 MusD genomic sequences according to the UCSC in silico PCR engine. Notably, we found that the ETnII transcript level was 172-fold higher than that of MusD (Fig. 1B). ETnI elements, with a copy number higher than that of both MusD and ETnII, had transcript levels only 11-fold higher than MusD. These results were confirmed by Northern blotting (data not shown). It should be pointed out here that, for most of our analyses, we focused on comparing MusD and ETnII subfamilies, since ETnI is substantially different from ETnII/MusD in the LTR sequence (5) (Fig. 1A) and has low levels of current retrotransposition activities (39). Such a dramatic difference in ETnII and MusD transcript levels could result from several factors, including (i) the presence of one or a few highly active ETnII elements, (ii) functional differences in LTR sequences, (iii) different enhancer/suppressor sequences within the elements themselves, or (iv) differential epigenetic recognition of the two subfamilies.
The first possibility, namely, that ETnII transcript abundance is due to a single, highly transcribed copy, is unlikely given that different ETnII sequences were recovered from cDNA (5, 38; unpublished observations). The second possibility listed above, that ETnII/MusD transcript level differences may stem from small LTR differences, is partially supported by our previous transient-transfection assays. Those results suggested that on average, promoter activity of four ETnII LTRs was threefold higher than the promoter activity of four MusD LTRs (38). Because only a few, randomly chosen LTRs of each type were tested, the results may not reflect the average promoter activity of the whole ETnII or MusD populations. However, it is possible that a greater proportion of LTRs of the slightly older MusD family have acquired mutations that result in lower promoter activity. Even if that were the case, we believe that such differences in promoter activities of ETnII and MusD LTRs would be insufficient to explain the 170-fold difference in their transcript levels.
As another way of assessing potentially significant sequence differences in LTRs, we analyzed 68 MusD and 33 ETnII elements recovered from the assembled C57BL/6 genome. We had previously demonstrated that the first 76 bp of the LTR, and especially three Sp1/Sp3 binding sites, are critical for LTR promoter activity in EC cells (38). Indicative of the synergistic effect of the sites, mutations of all three of them reduced the LTR promoter activity to 9% of the original level (38). We aligned the 5′ LTRs of ETnII and MusD elements (see Fig. S1 in the supplemental material) to analyze the presence of these binding sites in genomic copies (a detailed alignment of the first 93 bp is shown in Fig. S2 of the supplemental material). As summarized in Fig. 1C, we detected few differences in the core regions of Sp1/Sp3 binding sites. In agreement with a higher copy number of MusD elements in the genome, more MusD than ETnII elements matched a consensus sequence in the first 93 bp of the LTR, which encompasses all three Sp1/Sp3 sites (see Fig. S2 in the supplemental material). While regulatory motifs other than the described Sp1/Sp3 binding sites may play a role in ETn/MusD transcription, we believe the most important ones are located within the first 93 bp shown, as deletion of the 5′-most 76 bp reduced the promoter activity of the LTR to 5% of the wild-type levels (38). Since no dramatic differences between ETnII and MusD LTRs are apparent in this region, it is unlikely that LTR sequence differences play a major role in ETnII elements having higher transcript levels.
To investigate the third possibility, that the internal ETnII-specific or MusD-specific sequences contain cis-acting elements, such as enhancers or suppressors, we tested these regions in transient-transfection assays. Reporter constructs included either a 3.15-kb ETnII or a 3.58-kb MusD internal sequence on the same plasmid as the ETnII LTR#7 promoter driving the expression of the firefly luciferase reporter gene (Fig. 2A). For this study, we cloned internal sequences from a “young” coding-competent MusD (48) and the most abundant and transcriptionally active ETnII-β subtype (5). As shown in Fig. 2B, neither ETnII nonretroviral nor MusD retroviral sequences possessed strong enhancer or suppressor properties when assessed in both orientations, and these data were confirmed by deletion analysis of the ETnII internal region (data not shown). Therefore, distinct internal regions of ETnII and MusD elements do not appear to harbor cis-acting motifs that significantly affect LTR activity.
FIG. 2.

Effects of ETnII and MusD internal regions on LTR promoter activity. (A) Cloning scheme of ETnII and MusD internal regions. IntETn and intMusD regions were cloned into the reporter constructs in forward (Fw) and reverse (Rv) orientations. (B) Relative luciferase activities of the ETnII and MusD internal region-containing LTR-promoted constructs. The data were standardized to the internal Renilla luciferase control and expressed relative to the activity of the promoterless pGL3B vector with a λ-phage “stuffer” DNA of corresponding size (see Materials and Methods for details). Results are means and standard deviations of three separate experiments performed in duplicate.
The MusD LTR and the adjacent common region have higher DNA methylation levels than ETnII in ES cells.
Given the above analyses, for the remainder of this study we have focused on the fourth possibility listed above, namely, that of differential epigenetics. Because large sections of the internal regions of ETnII and MusD elements are completely unrelated (5), it is possible that these elements are recognized differently by factors establishing epigenetic marks. To examine this possibility, we performed genomic Southern blotting using the strategy described below. Two methylation-sensitive enzymes, NotI and AfeI, cut DNA only if it is unmethylated (Fig. 3A). The recognition sites of these enzymes in the LTR and the adjacent 5′ internal sequence, which we refer to as the “common” region, are present in the majority of ETnII and MusD elements by virtue of the relatively young age of these subfamilies, allowing us to assay a large fraction of ETnII and MusD sequences in the genome. The second, methylation-insensitive enzyme, PstI, cuts at a different distance in ETnII and MusD elements and permits detection of both ETnII and MusD in one lane when using a single probe that recognizes both subfamilies. A methylation-insensitive enzyme, XbaI, cutting in the LTR, is used as a control for genomic ETnII and MusD copy numbers. The unique setup of this Southern blot assay allows for analysis of DNA methylation levels across the broad population of ETnII and MusD elements at CpG dinucleotides within sites recognized by methylation-sensitive restriction enzymes.
FIG. 3.

Methylation of MusD and ETnII elements in ES cells. (A) Digestion scheme of genomic Southern blotting. Expected fragment sizes and methylation sensitivity of the enzymes are shown. (B) Genomic Southern blotting of the digests. DNA from J1 ES cells was used, and lane numbers correspond to enzyme numbers in panel A. Arrows indicate bands expected to result from digestion of MusD elements. (C) Bisulfite analysis of ETnII and MusD LTRs and common regions. Each horizontal line represents a single clone. Unmethylated CpGs are shown as open circles and methylated CpGs are shown as solid circles. The percentage of methylated CpGs is shown above each of the regions. CpGs in NotI and AfeI sites in ETnII and MusD sequences are shown below. Note that NotI encompasses two CpGs. For reference, a MusD AC084696 sequence was chosen. Because the population of ETn and MusD sequences is heterogeneous, some CpG dinucleotides are lacking in select copies. In those cases, circles representing the CpG were omitted. Conversely, if a CG was present in the bisulfite-treated clone but not the reference sequence, we added a closed circle depicting a methylated CG, which corresponds to a gap in the reference sequence. The number of unmethylated CpGs is an underrepresentation, since methylated CpGs were plotted regardless of their presence in the reference sequence, while we had no means of determining whether a TG identical to the reference sequence was an unmethylated CG or a true TG in the genomic copy.
We analyzed the methylation status of ETnII and MusD LTRs and common regions in J1 ES cells (32, 46). The Southern blot results suggest that about half of the ETnII elements are unmethylated at the examined CpG dinucleotides (Fig. 3B). In contrast, MusD elements are predominantly methylated at the same sites, judged by the reduced intensity of the specific band, since the fragment lengths of methylated copies will vary depending on the location of the nearest PstI site in the upstream flanking genomic sequence. Similar patterns were seen in experiments with methylation-sensitive restriction enzymes ApaLI and NarI (data not shown).
To more quantitatively assess the difference in the levels of DNA methylation at the LTR and the common region of ETnII and MusD retrotransposons, we performed bisulfite analysis on these sequences in J1 ES cells (Fig. 3C). In support of our Southern blotting results, MusD elements showed considerable methylation levels, with no completely unmethylated sequences. In contrast, roughly half of the ETnII copies were predominantly or completely unmethylated at sites homologous to those of MusD. In addition, the MusD subfamily has a twofold-higher average level of CpG methylation in the examined region (59.4% methylation for MusD versus 32% methylation for ETnII). Therefore, while the majority of MusD elements are methylated, about half of the ETnII elements are unmethylated in the homologous regions.
More-repressive and less-activating histone marks on MusD compared with ETnII elements.
Since dense CpG methylation frequently correlates with repressive histone modifications (14, 36, 55), we analyzed the state of histone acetylation and methylation in the common region of ETnII and MusD retrotransposons. ES cell line J1 was used for ChIP experiments with antibodies against histone 3 acetylated at Lys9 (H3K9Ac), a modification consistent with active chromatin, and an antibody against H3 trimethylated at Lys9 (H3K9me3), a hallmark of silent chromatin (reviewed in reference 6). Precipitation reactions with normal rabbit IgG and the input fraction before immunoprecipitation were used as negative and positive controls, respectively. After immunoprecipitation and reversal of the cross-links, enrichment for modifications in ETnII and MusD was analyzed by real-time qPCR relative to the input. Due to the high similarity of the ETn and MusD LTRs and their common regions, it is impossible to distinguish the promoters of these subfamilies by qPCR following ChIP. The sequence closest to the promoter that can be analyzed using a primer pair differentiating between ETnII and MusD is the 3′ end of the common region, where a reverse primer can be placed in the subfamily-specific flanking sequence. Therefore, to differentiate between ETnII and MusD, we amplified an approximately 150-bp sequence at the 3′ end of the ∼300-bp ETnII/MusD common region using a common forward primer and an ETnII- or a MusD-specific reverse primer just downstream of the common region (Fig. 4A). A promoter region of the housekeeping gene Hprt was amplified as an example of an active gene, and a GC-rich region of the NK cell-specific gene Nkg2a, which is deacetylated in nonexpressing cells (50), was amplified as an example of an inactive gene.
FIG. 4.

Chromatin marks on the ETnII/MusD common region in ES cells. (A) Positions of primers used in ChIP analyses of ETnII and MusD elements. (B) Amplification of ETnII, MusD, Hprt, and Nkg2a promoter regions from J1 DNA material immunoprecipitated with antibodies against acetylated H3K9 and trimethylated H3K9. The results are presented as the relative amount of acetylated or methylated histones associated with the target relative to input material. Quantitative PCRs on DNA from three separate immunoprecipitations were each performed in triplicate; error bars are standard deviations between biological replicates.
In line with our expectations, ETnII elements, carrying less DNA methylation in the common region (Fig. 3B and C), displayed higher levels of H3K9Ac and lower levels of H3K9me3, while MusD, methylated to a greater extent, was associated with less active and more repressive histone marks in ES cells (Fig. 4B). In three separate immunoprecipitations, ETnII sequences were associated on average with 5.4-fold-higher levels of H3K9Ac. Conversely, MusD sequences showed a twofold-greater enrichment for H3K9me3.
Because the examined regions are highly similar between ETnII and MusD elements, we propose that the difference in CpG methylation and repressive histone modification levels between these two subfamilies is at least partially directed by the distinctly different adjacent internal regions: the nonretroviral ETnII and the retroviral MusD sequences.
Internal region of ETnII has a lower density of CpG dinucleotides than MusD.
In this study, we noticed that methylation in MusD elements tends to increase toward the internal retroviral sequence (Fig. 3C). In fact, only 50.5% of CpGs are methylated within the first 222 bp of the LTR, which include the enhancer and most transcription start sites (38), and 53% of CpGs are methylated in the 3′ end of the MusD LTR. In contrast, 69.3% CpGs are methylated in the ETnII/MusD common region (Fig. 3C). While the lower level of methylation in the 5′ end of the LTR is likely the result of an open chromatin state in the vicinity of transcription factor binding sites, the higher level of DNA methylation in the common region of MusD may be the result of repressive influence of the flanking internal retroviral sequence.
To examine whether internal regions of ETnII and MusD elements have dissimilar sequence compositions which may affect upstream regions, we analyzed two representative elements from the genome with MethPrimer (http://www.urogene.org) (Fig. 5A). The ETnII nonretroviral region has on average 0.9 CpGs per 100 bp, compared with 2.9 CpGs per 100 bp in the MusD retroviral region. Moreover, the same MusD sequence has an overall GC content of approximately 55%, whereas an ETnII nonretroviral region has a GC content of around 35%. This difference suggests that a large block of CpG-rich sequence in MusD elements may be involved in the recruitment of proteins that promote DNA methylation and/or a repressive chromatin structure. Such modifications may result in suppressed transcription from the upstream LTR promoter.
FIG. 5.

CpG composition of ETnII and MusD retrotransposons and promoter activity of in vitro patch-methylated LTRs. (A) CpG composition of ETnII and MusD retrotransposons. The y axis represents overall GC content. CpG dinucleotides, depicted by black vertical bars below, are mapped to a representative ETnII, element 7 (AC079540), and a representative MusD, element 3 (AC084696). Fragments cloned for stable transfection via RMCE (see Fig. 7) are also shown. (B) Relative luciferase activities of methylated and mock-methylated (produced by omitting SssI methyltransferase) LTR-promoted constructs. The data were first standardized to the internal Renilla luciferase control and are expressed relative to the activity of the promoterless cut and religated pGL3B vector. Results are means and standard deviations of three separate experiments performed in duplicate.
ETnII and MusD LTRs are silenced by CpG methylation.
To determine if ETnII and MusD LTR promoter activity may indeed be suppressed by CpG methylation, we performed transient-transfection assays with in vitro-methylated LTR reporter constructs. Only the LTR sequence was methylated, to avoid gene silencing due to downstream reporter gene methylation. The methyltransferase SssI, which adds a methyl group to every cytosine in a CpG context, was used. ETnII and MusD LTRs that showed the highest and the lowest promoter activities in the previous studies (38) were chosen for patch methylation in vitro. Two ETnII LTRs, LTR#7 and LTR#13, and two MusD LTRs, LTR#3 and LTR#9, were subjected to in vitro methylation and reporter assays. The results in Fig. 5B show that DNA methylation causes a 3.1- to 5.6-fold reduction in promoter activity of all ETnII and MusD LTRs, suggesting that DNA methylation of these LTRs in vivo is sufficient to suppress their promoter activity.
ETnII and MusD elements show a threefold decrease in DNA methylation but only mild upregulation in the DNMT1 knockout (KO) ES cells.
In Fig. 5B, we show that DNA methylation is capable of reducing promoter activity of ETnII and MusD LTRs. To determine if the reverse is true, namely, if demethylation is capable of derepressing this family of ERVs, we made use of Dnmt1−/− cells, which lack the major maintenance methyltransferase and have reduced levels of genomic DNA methylation (32, 46). We analyzed the methylation status of LTRs and common regions of ETnII and MusD elements in J1 wild-type and DNMT-deficient ES cell lines by Southern blotting experiments (Fig. 6A) similar to those described for Fig. 3A. Both ETnII and MusD elements were mildly demethylated in the Dnmt3a−/− 3b−/− cell line and highly demethylated in the Dnmt1−/− cell line at the examined CpG dinucleotides (Fig. 6B). Bisulfite analysis on DNA from the Dnmt1−/− ES cells showed demethylation of ETnII (32% in the wild type and 9.7% in Dnmt1−/−) and MusD elements (59.4% in the wild type and 19.1% in Dnmt1−/−) (Fig. 6C; compare to Fig. 3C).
FIG. 6.

Methylation and expression of MusD and ETnII elements in Dnmt1−/− ES cells. (A) Digestion scheme of genomic Southern blotting. Expected fragment sizes for unmethylated elements and methylation sensitivity of the enzymes are shown. (B) Genomic Southern blotting of the digests. Lane numbers correspond to the enzyme numbers in panel A. DNA from J1 wild type (wt), Dnmt3a−/− (3a−/−), Dnmt3b−/− (3b−/−), Dnmt3a−/− b−/− (3a−/− 3b−/−), and Dnmt1−/− (1−/−) was used. In lanes 2 and 3, arrows indicate bands expected to result from unmethylated MusD elements. (C) Bisulfite analysis of ETnII and MusD LTRs and common regions. Each horizontal line represents a single clone. Unmethylated CpGs are shown as open circles and methylated CpGs are shown as solid circles. The percentage of methylated CpGs is shown above. CpGs in NotI and AfeI sites in ETnII and MusD sequences are shown below. Note that NotI encompasses two CpGs. Because the population of ETn and MusD sequences is heterogeneous, some CpG dinucleotides are lacking in select copies. In those cases, we omitted circles representing the CpG. Conversely, if a CG was present in the bisulfite-treated clone but not the reference sequence, we added a closed circle, depicting a methylated CG, which corresponds to a gap in the reference sequence. (D) ETnII, MusD, and IAP expression in Dnmt1−/− ES cells. Quantitative RT-PCR results with cDNA from Dnmt J1 (wt) and Dnmt1−/− (1−/−) cells are shown. Results for each cell line were normalized to Gapdh; expression in wild-type cells was set as 1. Data are means and standard deviations between three separate experiments each performed in triplicate.
Since IAP ERVs are known to be upregulated in response to Dnmt1−/− KO in embryos (65) and ES cells (62), we assessed whether the same is true for the ETn/MusD family. Despite substantial demethylation of multiple ETnII and MusD copies, our qRT-PCR analysis demonstrated that these subfamilies are only mildly upregulated in the Dnmt1−/− ES cell line (Fig. 6D), while the higher-copy-number IAP retrotransposons showed a fivefold upregulation, similar to results reported by others (62). It should be noted that in Dnmt1−/− ES cells, residual methylation persists on both ETn/MusD and IAP elements due to the action of de novo DNMTs, with IAPs maintaining approximately 20% of DNA methylation (62). However, the expression level of IAP elements is similar between Dnmt1−/− and the triple KO (TKO) ES cells that lack DNMT1, DNMT3A, and DNMT3B (62). Lack of a further increase in IAP expression despite complete removal of DNA methylation in the TKO cells suggests that residual methylation in Dnmt1−/− ES cells of IAP and ETn/MusD elements is not the reason for the low upregulation of ETn/MusD. It is likely that repressive chromatin modifications, such as H3K9me3 deposited by SETDB1, as proposed by Dong et al. (13), or specific repressor proteins, such as Trim28 (67, 68), are involved in suppression of the ETn/MusD family in the context of reduced DNA methylation. This is supported by the fact that substantial demethylation of MusD elements in the G9a−/− ES cells does not result in their upregulation (13). Experiments are under way to identify the histone marks and proteins associated with such repression.
Internal MusD sequence reduces reporter gene expression in stable transgenes.
According to our hypothesis, the retroviral region of MusD may negatively affect the promoter activity of its LTR by functioning as a target for DNA methylation and repressive histone modifications. To directly assess the influence of internal MusD and ETnII regions on promoters, we performed stable transfections using the RMCE method. The constructs described below were introduced into the RL5 site in the genome of a MEL cell line shown permissive for long-term expression of a transgene (15).
The 3.3-kb nonretroviral internal region of ETnII-β (AC079540), including the common sequence, and the corresponding sized MusD (AC084696) sequences were cloned from the C57BL/6 genomic DNA (cloned sequences are indicated in Fig. 5A). These were inserted in reverse orientation upstream of the CMV promoter driving the expression of the EGFP gene in the L1-CMV-EGFP-1L plasmid (Fig. 7A). The CMV promoter was chosen over the native LTR promoter because the latter demonstrated very low activity in this differentiated cell line (data not shown). ETnII and MusD sequences were inserted in reverse orientation to more closely resemble the natural situation where the common sequence is adjacent to the promoter. They were cloned upstream as opposed to downstream of the CMV promoter to avoid a significant reduction in EGFP expression (data not shown), likely due to the abundance of natural and cryptic stop signals in these sequences.
FIG. 7.

Effect of ETnII and MusD internal regions on stably integrated transgenes. (A) Scheme of an integrated transgene. Internal and common sequences indicated in Fig. 5A were cloned in the reverse orientation upstream of the CMV promoter. Common region fragments amplified after ChIP or bisulfite treatment are shown. (B) EGFP expression of ETnII and MusD internal region-containing transgenes during single-cell sorting 13 days after transfection. (C) EGFP expression of ETnII and MusD internal region-containing transgenes monitored as separate clones until day 110 posttransfection, after which they were pooled and subjected to ChIP. A total of 10,000 to 20,000 propidium iodine-negative cells were analyzed. (D) ChIP results for cells with either an ETnII or a MusD internal region transgene. The results are presented as the relative amount of acetylated or methylated histones associated with target samples with respect to input material. Quantitative PCRs on DNA from three separate immunoprecipitations were performed in triplicate; error bars are standard deviations between biological replicates. (E) Bisulfite analysis of the ETnII/MusD common region of the transgene. Each horizontal line represents a single clone. Unmethylated CpGs are shown as open circles and methylated CpGs are shown as solid circles.
Cre-lox-mediated site-specific integration was performed. Cells from five separate transfections were selected with ganciclovir and pooled, and EGFP-positive cells were single-cell sorted into 96-well plates for cloning. Notably, even at the time of cell sorting, 13 days after transfection, a substantial difference in EGFP intensity was evident between the bulk ETn and MusD internal sequence-containing populations (Fig. 7B), indicative of the dissimilar influences of ETn and MusD sequences on the adjacent CMV promoter. Six of each ETnII and MusD internal region-containing clones in orientation B (15) were maintained without selection and monitored by FACS for EGFP expression approximately every 10 days, and mean EGFP fluorescence was graphed (Fig. 7C). The majority of MusD transgenes displayed considerably lower EGFP intensities compared to ETnII transgenes, each group forming a tight cluster (Fig. 7C). This difference persisted throughout the 110 days of the experiment duration. The lower EGFP intensity of clones with MusD sequence unequivocally points to its suppressing factor in transcription or, conversely, to the enhancing effect of the ETnII sequence due to possible hyperacetylation (47). The latter, however, is unlikely, given that the expression from the CMV promoter in transgenes without the upstream ETnII or MusD sequences was most similar to the ETnII-containing clones (data not shown).
Since ETnII and MusD internal regions lacked evident cis-acting elements in the reporter assays (Fig. 2B), we propose that lower transcription of MusD retrotransposons may result from a greater likelihood of transcriptional suppression via repressive chromatin and/or DNA methylation initiating from the retroviral sequence.
Higher level of activating and lower level of repressive chromatin modifications in ETnII compared with MusD internal region-containing transgenes.
It was reported previously that, in an early erythroid cell line, transgene silencing is accompanied by rapid H3 deacetylation and H3K9 dimethylation preceding DNA methylation (44). To determine whether the observed difference in EGFP expression between ETnII and MusD internal region transgenes (Fig. 7B and C) may be due to different chromatin states, we performed ChIP on pooled ETnII- and MusD-containing clones (Fig. 7D), analyzing the common region with the same primer pair (Fig. 7A). In three independent immunoprecipitation experiments, the common region of ETnII transgenes showed a 2.7-fold-greater enrichment for H3K9Ac modification over the common region of MusD transgenes. Conversely, the same sequence of MusD had an H3K9me3 enrichment 10.9-fold higher than that of ETnII. Despite higher association with H3K9me3, MusD transgenes still expressed EGFP, albeit at a lower level than ETnII transgenes, with no negative colonies detected. Possibly, only a subset of histone tails, not sufficient for complete transcriptional silencing, carries this modification. The results of ChIP suggest that dissimilar internal sequences of ETn and MusD may direct establishment of different chromatin modifications on sequences that are similar between the two subfamilies—the common region and the promoter.
De novo DNA methylation on MusD and ETnII internal region-containing transgenes.
Bisulfite analysis of the ETn/MusD common region of the transgenes at day 115 after transfection showed limited de novo methylation predominantly in the MusD-containing clones (2.9% methylation for ETnII and 9.5% for MusD) (Fig. 7E). While the average methylation level for each clone (means of 0.4 methylated CpGs for ETnII and 1.33 for MusD) between the two populations was not significantly different (P = 0.064), the difference in the total number of methylated CpGs in each population (6 of 210 for ETnII versus 20 of 210 for MusD) is highly significant (P = 0.0032). These data suggest that at least limited de novo methylation is inducible at the RL5 locus in MEL cells and, given the time it takes to be established, is likely a secondary event appearing in the context of repressive chromatin, predominantly on MusD-specific sequences. While randomly integrated proviruses are readily methylated in MEL cells (35), the RL5 locus is permissive for long-term expression of the transgene (15), which may explain the low level of de novo methylation. Alternatively, and more likely, the presence of the strong CMV promoter immediately downstream may prevent active de novo methylation events.
Presumably, H3K9me3 and other repressive modifications recruited to the MusD elements may act as a scaffold for recruitment of histone-modifying proteins and DNMTs, whose actions lead to stable repression of the endogenous MusD retrotransposons and induction of de novo methylation in germ cells, embryos, and ES cells that possess high de novo DNMT activities. However, since demethylation of ETnII and MusD elements (Fig. 6C) does not result in their upregulation (Fig. 6D), it is likely that DNA methylation is a secondary event in repression and does not play a major role in the establishment of ETn/MusD silencing.
Support for stronger selection against intronic MusD elements.
The above analyses indicate that MusD elements in general have higher levels of repressive chromatin modifications and DNA methylation compared with ETn elements. Therefore, one might expect that MusD elements that integrate within gene introns or near genes could be subject to stronger negative selection since, compared with ETns, the presence of such an element might have a higher probability of silencing gene expression due to spreading of repressive chromatin. Since ETn elements require MusD integrase and other MusD-encoded proteins to retrotranspose (48), both subgroups of elements should have the same initial insertion site preferences. Hence, any differences in genomic distributions will likely reflect differences in selective pressures against these elements. To look for evidence of such a phenomenon, we examined the prevalence of MusD and ETn elements with respect to genes. To increase numbers for the purpose of statistics, we combined the ETnI and ETnII subgroups, but the same result was obtained when both subgroups were treated separately. As shown in Fig. 8, the distribution of ETn and MusD elements is similar across distances of 1 kb to 1 Mb to the nearest gene. Strikingly, 12.5% of 164 ETn elements were found within gene introns, but none of the 69 MusD elements was located in an intron, suggesting differential selection pressure on these subfamilies. Moreover, a greater percentage of MusDs were located more than 1 Mb away from an annotated gene. While we cannot rule out other explanations for the lack of MusD elements within introns, this finding suggests that intronic MusD elements are more detrimental to genes than ETn elements, possibly due to epigenetic effects they have on the adjacent sequences.
FIG. 8.
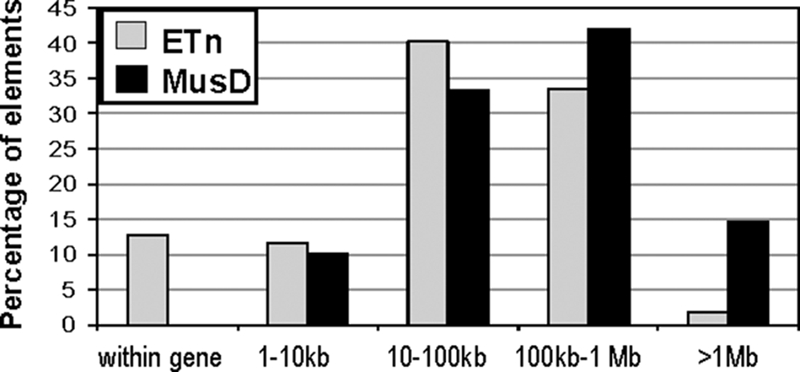
ETn and MusD distributions with respect to genes. ETn and MusD elements in the mm9 release of the C57BL/6 genome were binned based on the distance to the closest RefSeq gene. The number of ETn or MusD elements in each bin is shown as a percentage relative to the total number of elements in each subfamily.
DISCUSSION
DNA methylation and chromatin modifications as mechanisms of MusD suppression.
Eukaryotic genomes have evolved multiple lines of defense against active retrotransposons and retroviruses (see reference 18 for review). DNA methylation has long been regarded as a major means of transcriptional suppression. It is prevalent on repetitive sequences, including tandem repeats, such as pericentromeric major and minor satellites, and interspersed repeats, which constitute DNA transposons, LTR retrotransposons or ERVs, long interspersed nuclear elements, and short interspersed nuclear elements. In fact, it has been suggested that most of the genome's methylated CpGs lie within transposable elements (70). Moreover, some ERVs are transcriptionally reactivated in response to demethylation (8, 21, 65), supporting the notion that DNA methylation is indeed an important defense mechanism against transposable elements in general and endogenous retroviruses in particular (70). More recent studies have demonstrated that histone structure and modifications also play an important role in silencing of transposable elements and ERVs in mammals (23, 40, 43) and model organisms (see references 28, 33, and 59 for reviews).
In this study, we showed that transcription of MusD, in contrast to the related nonautonomous subfamily of ETn retrotransposons, is largely suppressed in ES cells. We found that a higher level of DNA methylation is accompanied by a higher level of repressive and lower level of activating histone marks associated with MusD. We propose that such repression, resulting in the observed lower transcription level of MusD compared with ETnII elements in early embryos (5) and ES cell lines (this study), stems from a higher probability of repressive chromatin and DNA methylation originating in the downstream CpG-rich retroviral region. A similar case was reported for the rho globin gene in chicken embryonic erythroid cells, in which, during normal developmental silencing, de novo methylation originates in the downstream CpG-dense region and then spreads into the promoter, silencing transcription and resulting in complete DNA methylation in adult erythroid cells (58). Notably, roughly half of the ETnII copies in the genome have methylation levels similar to MusD, but the other half are essentially unmethylated (Fig. 3C). Thus, some ETnII copies appear to be silenced as effectively as MusD. However, we suggest that the different internal region of ETnII elements increases the probability that an individual genomic copy will escape silencing. Presumably, the high levels of ETnII transcripts in ES cells are being produced from the unmethylated copies. Overall, MusD elements are associated with a twofold-higher level of H3K9me3 than are ETn elements. However, in a genome-wide chromatin screen, sequences found to be enriched for H3K9me3 and H4K20me3 in ES cells were annotated as ETn and IAP elements (43). Since REPBASE (http://www.girinst.org), used in that study to identify these classes of repeats, does not distinguish between ETn and MusD and designates both subfamilies as ETn, it is likely that most of the elements picked up by the screen were, in fact, the more numerous MusD retrotransposons. Moreover, because we only detected mild upregulation of MusD elements in Dnmt1−/− ES cells and because MusD and IAP ERVs maintain silencing despite their demethylation and reduced levels of H3K9me2 in the G9A KO ES cells (13), we propose that H3K9me3 and possibly other histone modifications and/or repressor proteins may play an important role in establishing silencing of these ERV families, with DNA methylation being a secondary event. Alternatively, a lower level of MusD derepression compared with that of IAP in Dnmt1−/− ES cells may be due to more efficient targeting of IAP by DNA methylation compared with MusD, as IAP elements possess much higher levels of methylation (95%) (56) than MusD (60%) in wild-type ES cells. Transcriptional suppression of silenced ETn/MusD copies may occur via several mechanisms. First, the elongation efficiency of the transcript may be directly influenced by CpG methylation downstream of the promoter (34). In addition, histone hypoacetylation associated with dense CpG methylation (14, 36, 55) may cause a reduction in transcription rate (47). Downstream methylation, prominent in MusDs (Fig. 3C), may virtually abolish transcription even in the context of an unmethylated LTR (22). Moreover, downstream DNA methylation and hypoacetylated chromatin may spread into the promoter, inhibiting transcription factor binding (41) or polymerase II loading (55). Since transcription level correlates with the degree of nucleosome acetylation (47), the lower transcription level of MusD compared with ETnII (Fig. 1B) may result from the lower acetylation and higher CpG methylation level of this subfamily.
Interestingly, we found that EGFP expression in RMCE-derived MusD transgenes, while lower than expression in ETnII transgenes, still persisted despite a higher association with H3K9me3 modification. It is possible that this mark is only present on a few histone tails, preventing complete silencing, or on rare nonexpressing clones. Alternatively, a heavier load of DNA methylation or a higher concentration of repressive proteins in ES, germ cells, and preimplantation embryos compared with MEL cells may be required for stable transgene and MusD repression.
Implications for retrotransposon evolution.
A common feature of the evolution of retrotransposon families involves the emergence of nonautonomous retrotransposon variants that gain advantage over their coding-competent counterparts by different mechanisms. These mechanisms may include, but are not limited to the following: (i) acquisition of stronger enhancer and promoter motifs; (ii) translation of defective products that aid in recruitment of functional proteins encoded by the autonomous members for enhanced reverse transcription and retrotransposition, as recently shown for IΔ1 IAP retrotransposons (54); (iii) reduced binding of a repressive histone variant, such as macroH2A, as has been reported for a deletion variant of an MLV ERV (11); or (iv) an increased probability of evading epigenetic suppression, as proposed here.
It has been shown that elevated expression of LTR retrotransposons can lead to their genomic expansion via retrotransposition (12, 19, 20, 45, 53). We suggest that, in some cases, the abundance of transcripts may be a limiting factor in the retrotransposition process. Supporting this hypothesis is the evidence of dramatic ETnII genomic amplification in the EC P19 cell line, which exhibits exceptionally high ETnII expression (38). We propose that active transcription of ETnII elements which, in contrast to MusD, are more successful in circumventing epigenetic suppression, may lead to higher retrotransposition levels and as a result, expansion of this subfamily in the mouse genome.
The ETn/MusD family illustrates an intriguing way for a new retrotransposon variant to gain a transcriptional advantage and increase its likelihood of retrotransposition. This route involves evolution of deleted or mutated elements which retain structural features necessary for retrotransposition assisted in trans by coding-competent members of the same family but which have lost some retroviral sequence that increases their probability of being silenced (Fig. 9). In the “arms race” between parasitic transposable elements and their host (39), the newly emerged ETnII elements, devoid of most retroviral sequences that may act as foci of acquisition of repressive epigenetic marks, may have found a weakness in the defense systems of the mouse genome. The exploitation of this weakness by ETnII elements is ongoing, resulting in numerous germ line mutations (39).
FIG. 9.

Model for amplification of nonautonomous LTR retrotransposons. (1) Transcription of a coding-competent LTR retrotransposon is partially suppressed by epigenetic machinery. (2) Viral mRNA is translated to produce matrix, capsid and nucleocapsid, reverse transcriptase, and integrase proteins. (3) Reverse transcription of viral RNA in a retroviral particle. (4) Integration of a fully functional ERV into a new genomic locus. (5) Mutations and deletions, frequently acquired during reverse transcription, render most newly integrated retroelements nonfunctional. (6) Some deletions or mutations offer the newly integrated provirus an advantage with respect to evasion of epigenetic (or other) host restriction mechanisms. Although noncoding, these proviruses are transcribed at a much higher level than their coding-competent predecessor. (7) The deleted/mutated viral RNA is packaged and reverse transcribed. (8) Due to the higher transcription rate of the deleted/mutated variant, a greater number of viral RNAs are reverse transcribed and integrated into new genomic loci, resulting in amplification of the nonautonomous retrotransposon.
Multiple routes may lead to the establishment of different epigenetic states of ETnII and MusD elements. These may include small RNA-mediated methylation in germ cells as shown for other ERVs (1) and retroviral sequence-induced suppression during early embryonic development, as suggested by extrapolation from the results of this ES cell study. The nonautonomous ETnII subfamily may continue its amplification in the genome until the disappearance of the last source of MusD proteins required for retrotransposition or until development by the host of a more effective epigenetic and/or genetic silencing machinery targeting this subfamily.
Supplementary Material
Acknowledgments
We are grateful to E. Li for J1 and Dnmt KO cells and M. Lorincz for the RMCE system. We thank Liane Gagnier for technical assistance and S. Rogers, N. Tabatabaei, D. Leung, S. Lee, and K. Dong for maintaining ES cells. We also thank D. Reiss and M. Lorincz for critical reading and suggestions on the manuscript and L. van de Lagemaat for early bioinformatics analysis.
This work was supported by grant 10825 from the Canadian Institutes of Health Research to D.L.M. with core support provided by the BC Cancer Agency. I.A.M. was supported by a University of British Columbia Graduate Fellowship, and Y.Z. is supported by an Alexander Graham Bell Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada.
I.A.M. and D.L.M. designed the study and analyzed the data. I.A.M. performed most experiments; Y.Z. performed multiple alignments and analyzed ETn/MusD distance to genes. I.A.M. and D.L.M. wrote the paper.
Footnotes
Published ahead of print on 9 March 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Aravin, A. A., R. Sachidanandam, D. Bourc'his, C. Schaefer, D. Pezic, K. F. Toth, T. Bestor, and G. J. Hannon. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 31785-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bannert, N., and R. Kurth. 2006. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genomics Hum. Genet. 7149-173. [DOI] [PubMed] [Google Scholar]
- 3.Barbot, W., A. Dupressoir, V. Lazar, and T. Heidmann. 2002. Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res. 302365-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baust, C., G. J. Baillie, and D. L. Mager. 2002. Insertional polymorphisms of ETn retrotransposons include a disruption of the wiz gene in C57BL/6 mice. Mamm. Genome 13423-428. [DOI] [PubMed] [Google Scholar]
- 5.Baust, C., L. Gagnier, G. J. Baillie, M. J. Harris, D. M. Juriloff, and D. L. Mager. 2003. Structure and expression of mobile ETnII retroelements and their coding-competent MusD relatives in the mouse. J. Virol. 7711448-11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger, S. L. 2007. The complex language of chromatin regulation during transcription. Nature 447407-412. [DOI] [PubMed] [Google Scholar]
- 7.Bouhassira, E. E., K. Westerman, and P. Leboulch. 1997. Transcriptional behavior of LCR enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood 903332-3344. [PubMed] [Google Scholar]
- 8.Bourc'his, D., and T. H. Bestor. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 43196-99. [DOI] [PubMed] [Google Scholar]
- 9.Brookfield, J. F. Y. 2005. The ecology of the genome: mobile DNA elements and their hosts. Nat. Rev. Genet. 6128-136. [DOI] [PubMed] [Google Scholar]
- 10.Brûlet, P., H. Condamine, and F. Jacob. 1985. Spatial distribution of transcripts of the long repeated ETn sequence during early mouse embryogenesis. Proc. Natl. Acad. Sci. USA 822054-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Changolkar, L. N., G. Singh, and J. R. Pehrson. 2008. MacroH2A1-dependent silencing of endogenous murine leukemia viruses. Mol. Cell. Biol. 282059-2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng, C., M. Daigen, and H. Hirochika. 2006. Epigenetic regulation of the rice retrotransposon Tos17. Mol. Genet. Genomics 276378-390. [DOI] [PubMed] [Google Scholar]
- 13.Dong, K. B., I. A. Maksakova, F. Mohn, D. Leung, R. Appanah, S. Lee, H. W. Yang, L. L. Lam, D. L. Mager, D. Schübeler, M. Tachibana, Y. Shinkai, and M. C. Lorincz. 2008. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 272691-2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eden, S., T. Hashimshony, I. Keshet, H. Cedar, and A. W. Thorne. 1998. DNA methylation models histone acetylation. Nature 394842. [DOI] [PubMed] [Google Scholar]
- 15.Feng, Y.-Q., M. C. Lorincz, S. Fiering, J. M. Greally, and E. E. Bouhassira. 2001. Position effects are influenced by the orientation of a transgene with respect to flanking chromatin. Mol. Cell. Biol. 21298-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng, Y.-Q., J. Seibler, R. Alami, A. Eisen, K. A. Westerman, P. Leboulch, S. Fiering, and E. E. Bouhassira. 1999. Site-specific chromosomal integration in mammalian cells: highly efficient CRE recombinase-mediated cassette exchange. J. Mol. Biol. 292779-785. [DOI] [PubMed] [Google Scholar]
- 17.Gaubatz, J. W., B. Arcement, and R. G. Cutler. 1991. Gene expression of an endogenous retrovirus-like element during murine development and aging. Mech. Ageing Dev. 5771-85. [DOI] [PubMed] [Google Scholar]
- 18.Goff, S. P. 2004. Retrovirus restriction factors. Mol. Cell 16849-859. [DOI] [PubMed] [Google Scholar]
- 19.Hirochika, H. 1993. Activation of tobacco retrotransposons during tissue culture. EMBO J. 122521-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirochika, H., K. Sugimoto, Y. Otsuki, H. Tsugawa, and M. Kanda. 1996. Retrotransposons of rice involved in mutations induced by tissue culture. Proc. Natl. Acad. Sci. USA 937783-7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard, G., R. Eiges, F. Gaudet, R. Jaenisch, and A. Eden. 2008. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 27404-408. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh, C. L. 1997. Stability of patch methylation and its impact in regions of transcriptional initiation and elongation. Mol. Cell. Biol. 175897-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang, J., T. Fan, Q. Yan, H. Zhu, S. Fox, H. J. Issaq, L. Best, L. Gangi, D. Munroe, and K. Muegge. 2004. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 325019-5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jern, P., G. O. Sperber, and J. Blomberg. 2004. Definition and variation of human endogenous retrovirus H. Virology 32793-110. [DOI] [PubMed] [Google Scholar]
- 25.Jiang, N., I. K. Jordan, and S. R. Wessler. 2002. Dasheng and RIRE2. A nonautonomous long terminal repeat element and its putative autonomous partner in the rice genome. Plant Physiol. 1301697-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalendar, R., C. M. Vicient, O. Peleg, K. Anamthawat-Jonsson, A. Bolshoy, and A. H. Schulman. 2004. Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 1661437-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kano, H., H. Kurahashi, and T. Toda. 2007. Genetically regulated epigenetic transcriptional activation of retrotransposon insertion confers mouse dactylaplasia phenotype. Proc. Natl. Acad. Sci. USA 10419034-19039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavi, H. H., H. R. Fernandez, W. Xie, and J. A. Birchler. 2005. RNA silencing in Drosophila. FEBS Lett. 5795940-5949. [DOI] [PubMed] [Google Scholar]
- 29.Kejnovsky, E., Z. Kubat, J. Macas, R. Hobza, J. Mracek, and B. Vyskot. 2006. Retand: a novel family of gypsy-like retrotransposons harboring an amplified tandem repeat. Mol. Genet. Genomics 276254-263. [DOI] [PubMed] [Google Scholar]
- 30.Kuff, E. L., and K. K. Lueders. 1988. The intracisternal A-particle gene family: structure and functional aspects. Adv. Cancer Res. 51183-276. [DOI] [PubMed] [Google Scholar]
- 31.Lei, H., S. P. Oh, M. Okano, R. Juttermann, K. A. Goss, R. Jaenisch, and E. Li. 1996. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1223195-3205. [DOI] [PubMed] [Google Scholar]
- 32.Li, E., T. H. Bestor, and R. Jaenisch. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69915-926. [DOI] [PubMed] [Google Scholar]
- 33.Lisch, D. 13 November 2008, posting date. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. doi: 10.1146/annurev.arplant.59.032607.092744. [DOI] [PubMed]
- 34.Lorincz, M. C., D. R. Dickerson, M. Schmitt, and M. Groudine. 2004. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 111068-1075. [DOI] [PubMed] [Google Scholar]
- 35.Lorincz, M. C., D. Schübeler, S. C. Goeke, M. Walters, M. Groudine, and D. I. Martin. 2000. Dynamic analysis of proviral induction and de novo methylation: implications for a histone deacetylase-independent, methylation density-dependent mechanism of transcriptional repression. Mol. Cell. Biol. 20842-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorincz, M. C., D. Schübeler, and M. Groudine. 2001. Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol. Cell. Biol. 217913-7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mager, D. L., and J. D. Freeman. 2000. Novel mouse type D endogenous proviruses and ETn elements share long terminal repeat and internal sequences. J. Virol. 747221-7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maksakova, I. A., and D. L. Mager. 2005. Transcriptional regulation of early transposon elements, an active family of mouse long terminal repeat retrotransposons. J. Virol. 7913865-13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maksakova, I. A., M. T. Romanish, L. Gagnier, C. A. Dunn, L. N. van de Lagemaat, and D. L. Mager. 2006. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martens, J. H., R. J. O'Sullivan, U. Braunschweig, S. Opravil, M. Radolf, P. Steinlein, and T. Jenuwein. 2005. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 24800-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuo, K., J. Silke, O. Georgiev, P. Marti, N. Giovannini, and D. Rungger. 1998. An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J. 171446-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayer, J., and E. Meese. 2005. Human endogenous retroviruses in the primate lineage and their influence on host genomes. Cytogenet. Genome Res. 110448-456. [DOI] [PubMed] [Google Scholar]
- 43.Mikkelsen, T. S., M. Ku, D. B. Jaffe, B. Issac, E. Lieberman, G. Giannoukos, P. Alvarez, W. Brockman, T.-K. Kim, R. P. Koche, W. Lee, E. Mendenhall, A. O/'Donovan, A. Presser, C. Russ, X. Xie, A. Meissner, M. Wernig, R. Jaenisch, C. Nusbaum, E. S. Lander, and B. E. Bernstein. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448553-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mutskov, V., and G. Felsenfeld. 2004. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 23138-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okamoto, H., and H. Hirochika. 2000. Efficient insertion mutagenesis of Arabidopsis by tissue culture-induced activation of the tobacco retrotransposon Tto1. Plant J. 23291-304. [DOI] [PubMed] [Google Scholar]
- 46.Okano, M., D. W. Bell, D. A. Haber, and E. Li. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99247-257. [DOI] [PubMed] [Google Scholar]
- 47.Protacio, R. U., G. Li, P. T. Lowary, and J. Widom. 2000. Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol. Cell. Biol. 208866-8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribet, D., M. Dewannieux, and T. Heidmann. 2004. An active murine transposon family pair: retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res. 142261-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribet, D., F. Harper, M. Dewannieux, G. Pierron, and T. Heidmann. 2007. Murine MusD retrotransposon: structure and molecular evolution of an “intracellularized” retrovirus. J. Virol. 811888-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogers, S. L., A. Rouhi, F. Takei, and D. L. Mager. 2006. A role for DNA hypomethylation and histone acetylation in maintaining allele-specific expression of mouse NKG2A in developing and mature NK cells. J. Immunol. 177414-421. [DOI] [PubMed] [Google Scholar]
- 51.Rouhi, A., L. Gagnier, F. Takei, and D. L. Mager. 2006. Evidence for epigenetic maintenance of Ly49a monoallelic gene expression. J. Immunol. 1762991-2999. [DOI] [PubMed] [Google Scholar]
- 52.Sabot, F., and A. H. Schulman. 2006. Parasitism and the retrotransposon life cycle in plants: a hitchhiker's guide to the genome. Heredity 97381-388. [DOI] [PubMed] [Google Scholar]
- 53.Sacerdot, C., G. Mercier, A.-L. Todeschini, M. Dutreix, M. Springer, and P. Lesage. 2005. Impact of ionizing radiation on the life cycle of Saccharomyces cerevisiae Ty1 retrotransposon. Yeast 22441-455. [DOI] [PubMed] [Google Scholar]
- 54.Saito, E.-S., V. W. Keng, J. Takeda, and K. Horie. 2008. Translation from nonautonomous type IAP retrotransposon is a critical determinant of transposition activity: implication for retrotransposon-mediated genome evolution. Genome Res. 18859-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schübeler, D., M. C. Lorincz, D. M. Cimbora, A. Telling, Y. Q. Feng, E. E. Bouhassira, and M. Groudine. 2000. Genomic targeting of methylated DNA: influence of methylation on transcription, replication, chromatin structure, and histone acetylation. Mol. Cell. Biol. 209103-9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharif, J., M. Muto, S. Takebayashi, I. Suetake, A. Iwamatsu, T. A. Endo, J. Shinga, Y. Mizutani-Koseki, T. Toyoda, K. Okamura, S. Tajima, K. Mitsuya, M. Okano, and H. Koseki. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450908-912. [DOI] [PubMed] [Google Scholar]
- 57.Shell, B. E., J. T. Collins, L. A. Elenich, P. F. Szurek, and W. A. Dunnick. 1990. Two subfamilies of murine retrotransposon ETn sequences. Gene 86269-274. [DOI] [PubMed] [Google Scholar]
- 58.Singal, R., and J. M. vanWert. 2001. De novo methylation of an embryonic globin gene during normal development is strand specific and spreads from the proximal transcribed region. Blood 983441-3446. [DOI] [PubMed] [Google Scholar]
- 59.Slotkin, R. K., and R. Martienssen. 2007. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8272-285. [DOI] [PubMed] [Google Scholar]
- 60.Sonigo, P., S. Wain-Hobson, L. Bougueleret, P. Tiollais, F. Jacob, and P. Brûlet. 1987. Nucleotide sequence and evolution of ETn elements. Proc. Natl. Acad. Sci. USA 843768-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanskanen, J. A., F. Sabot, C. Vicient, and A. H. Schulman. 2007. Life without GAG: the BARE-2 retrotransposon as a parasite's parasite. Gene 390166-174. [DOI] [PubMed] [Google Scholar]
- 62.Tsumura, A., T. Hayakawa, Y. Kumaki, S.-I. Takebayashi, M. Sakaue, C. Matsuoka, K. Shimotohno, F. Ishikawa, E. Li, H. R. Ueda, J.-I. Nakayama, and M. Okano. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11805-814. [DOI] [PubMed] [Google Scholar]
- 63.Vitte, C., C. Chaparro, H. Quesneville, and O. Panaud. 2007. Spip and Squiq, two novel rice non-autonomous LTR retro-element families related to RIRE3 and RIRE8. Plant Sci. 1728-19. [Google Scholar]
- 64.Voisset, C., O. Bouton, F. Bedin, L. Duret, B. Mandrand, F. Mallet, and G. Paranhos-Baccala. 2000. Chromosomal distribution and coding capacity of the human endogenous retrovirus HERV-W family. AIDS Res. Hum. Retrovir. 16731-740. [DOI] [PubMed] [Google Scholar]
- 65.Walsh, C. P., J. R. Chaillet, and T. H. Bestor. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20116-117. [DOI] [PubMed] [Google Scholar]
- 66.Wilkinson, D. A., N. L. Goodchild, T. M. Saxton, S. Wood, and D. L. Mager. 1993. Evidence for a functional subclass of the RTVL-H family of human endogenous retrovirus-like sequences. J. Virol. 672981-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolf, D., and S. P. Goff. 2007. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 13146-57. [DOI] [PubMed] [Google Scholar]
- 68.Wolf, D., K. Hug, and S. P. Goff. 2008. TRIM28 mediates primer binding site-targeted silencing of Lys1,2 tRNA-utilizing retroviruses in embryonic cells. Proc. Natl. Acad. Sci. USA 10512521-12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin, W., P. Xiang, and Q. Li. 2005. Investigations of the effect of DNA size in transient transfection assay using dual luciferase system. Anal. Biochem. 346289-294. [DOI] [PubMed] [Google Scholar]
- 70.Yoder, J. A., C. P. Walsh, and T. H. Bestor. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13335-340. [DOI] [PubMed] [Google Scholar]
- 71.Zhang, Y., I. A. Maksakova, L. Gagnier, L. N. van de Lagemaat, and D. L. Mager. 2008. Genome-wide assessments reveal extremely high levels of polymorphism of two active families of mouse endogenous retroviral elements. PLoS Genet. 4e1000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.