

Abstract
Francisella tularensis harbors genes with similarity to genes encoding components of a type VI secretion system (T6SS) recently identified in several gram-negative bacteria. These genes include iglA and iglB encoding IglA and IglB, homologues of which are conserved in most T6SSs. We used a yeast two-hybrid system to study the interaction of the Igl proteins of F. tularensis LVS. We identified a region of IglA, encompassing residues 33 to 132, necessary for efficient binding to IglB, as well as for IglAB protein stability and intramacrophage growth. In particular, residues 103 to 122, overlapping a highly conserved α-helix, played an absolutely essential role. Point mutations within this domain caused modest defects in IglA-IglB binding in the yeast Saccharomyces cerevisiae but markedly impaired intramacrophage replication and phagosomal escape, resulting in severe attenuation of LVS in mice. Thus, IglA-IglB complex formation is clearly crucial for Francisella pathogenicity. This interaction may be universal to type VI secretion, since IglAB homologues of Yersinia pseudotuberculosis, Pseudomonas aeruginosa, Vibrio cholerae, Salmonella enterica serovar Typhimurium, and Escherichia coli were also shown to interact in yeast, and the interaction was dependent on preservation of the same α-helix. Heterologous interactions between nonnative IglAB proteins further supported the notion of a conserved binding site. Thus, IglA-IglB complex formation is clearly crucial for Francisella pathogenicity, and the same interaction is conserved in other human pathogens.
Francisella tularensis is a gram-negative facultative intracellular bacterial pathogen capable of causing a severe disease, tularemia, in many mammalian species (22). Human infections are caused mainly by two subspecies, the more virulent organism F. tularensis subsp. tularensis (type A) found predominantly in North America and the less virulent organism F. tularensis subsp. holarctica (type B) found in North America, Europe, and Asia (34, 43). While little is known about the molecular mechanisms of Francisella pathogenesis, a key strategy appears to be its ability to survive and replicate within macrophages (42, 46). Francisella-containing vacuoles have been reported to evade phagosome-lysosome fusion, followed by bacterial escape into the cytoplasm (8, 16). Several genes necessary for intramacrophage survival, as well as growth within the amoeba Acanthamoeba castellanii, a putative natural reservoir of F. tularensis, have been identified. Many of these genes, including the members of the iglABCD operon, are located in a 34-kb Francisella pathogenicity island (FPI) (reviewed in reference 31), and they are regulated by the global regulator MglA (4, 23). Almost all of the proteins of the FPI are essentially conserved across subspecies. Studies have shown that IglC and IglD are required for F. tularensis to replicate within the cytosol of macrophages (24, 37). While IglC was shown to be essential for bacterial escape from the phagosome into the cytoplasm (24, 38), the requirement for IglD for this process is being debated (3, 37). In contrast to IglC and IglD, which appear to be unique to F. tularensis, there are homologues of iglA and iglB in many bacterial species, most of which are either pathogenic to animals or plants or plant symbionts (9, 32). Together with several of the FPI-encoded proteins, IglA and IglB show homology to proteins thought to be involved in type VI protein secretion (T6S) (2, 11). Functional T6S was recently demonstrated in pathogens like Vibrio cholerae, Pseudomonas aeruginosa, enteroaggregative Escherichia coli, Aeromonas hydrophila, Burkholderia mallei, and Edwardsiella tarda, and in many of these organisms a direct link between protein secretion and virulence has been established (12, 29, 35, 39, 44, 49). In E. tarda, both EvpA and EvpB were shown to be required for secretion of the substrates EvpC, EvpI, and EvpP (36, 49). Similarly, an aaiB mutant of enteroaggregative E. coli failed to secrete AaiC (12). In contrast, although required for virulence, the IglB homologue TssB of B. mallei was dispensable for secretion of Hcp1, leading Schell et al. to speculate that TssB may be an effector protein rather than a component of the T6S apparatus (39). While T6S has yet to be experimentally demonstrated for F. tularensis, IglA was recently shown to be a cytoplasmic protein required for intramacrophage growth and virulence of Francisella novicida, and an interaction with IglB was demonstrated by immunoprecipitation analysis (11). Here we used a yeast two-hybrid system to study the interaction of the Igl proteins of F. tularensis LVS. We identified an α-helical domain of IglA that was required for the interaction with IglB and for stability of the IglAB complex, as well as for intracellular growth and virulence of LVS. A similar domain was identified in the IglA homologues of other pathogens, such as Yersinia pseudotuberculosis, P. aeruginosa, V. cholerae, Salmonella enterica serovar Typhimurium, and E. coli, and was found to be essential for complex formation by the corresponding IglAB homologues.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. F. tularensis was grown on modified GC agar base or in liquid Chamberlain's medium (7) at 37°C, while E. coli, P. aeruginosa, Y. pseudotuberculosis, V. cholerae, and S. Typhimurium were cultivated on Luria-Bertani agar or in Luria-Bertani broth at either 26°C (Y. pseudotuberculosis) or 37°C (E. coli, P. aeruginosa, V. cholerae, S. Typhimurium). When necessary, carbenicillin (100 μg/ml), kanamycin (50 μg/ml for E. coli and 10 μg/ml for F. tularensis), and chloramphenicol (25 μg/ml for E. coli and 2.5 μg/ml for F. tularensis) were used.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype and/or phenotype | Source or reference |
|---|---|---|
| E. coli strains | ||
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 deoR araD139 Δ(ara-leu)7679 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| S17-1λpir | recA thi pro hsdRM+ Smr <RP4:2-Tc:Mu:Ku:Tn7> Tpr | 41 |
| 536 | UPEC isolate (O6:K15:H31), Smr | 1 |
| F. tularensis strains | ||
| LVS | Live vaccine strain | USAMRIIDa |
| ΔiglA mutant | LVS, in-frame deletion of iglA codons 4 to 174 | This study |
| ΔiglB mutant | LVS, in-frame deletion of iglB codons 54 to 346 | This study |
| ΔiglC mutant | LVS, in-frame deletion of iglC codons 28 to 205 | 17 |
| P. aeruginosa PAO1 | Wild-type isolate | 19 |
| Y. pseudotuberculosis IP32953 | Serotype I | E. Carnielb |
| S. Typhimurium SL1344 | Smr, hisG rpsL xyl | CCUGc |
| V. cholerae N16961 | Wild type, serogroup O1 El Tor biotype, Smr | 18 |
| S. cerevisiae strains | ||
| AH109 | MATα trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1UAS-GAL1TATA-HIS3 GAL2UAS-GAL2TATA-ADE2 URA3::MEL1UAS-MEL1TATA-lacZ MEL1 | Clontech Laboratories |
| Y187 | MATα trp1-901 leu2-3,112 ura3-52 his3-200 ade2-101 gal4Δ met−gal80Δ MEL1 URA3::GAL1UAS-GAL1TATA-lacZ | Clontech Laboratories |
| Plasmids | ||
| pCR4-TOPO | TA cloning vector, Kmr Apr | Invitrogen |
| pBluescript SK(+) | Cloning vector, Apr | Stratagene |
| pDM4 | Suicide plasmid carrying sacBR, Cmr | 28 |
| pJEB485 | 2,633-bp XhoI/SacI PCR fragment of ΔiglA4-174 with flanking regions on pDM4, Cmr | This study |
| pPV | Suicide plasmid carrying sacBR, Cmr | 17 |
| pPV-ΔiglB | 2,322-bp SalI/XbaI PCR fragment of ΔiglB54-346 with flanking regions on pPV, Cmr | This study |
| pKK289Km | Expression plasmid carrying a gfp gene under control of the LVS GroESL promoter, Kmr | 3 |
| pJEB415 | pKK289Km with wild-type iglA, Kmr | This study |
| pJEB526 | pKK289Km with mutant iglA (Δ3-22), Kmr | This study |
| pJEB511 | pKK289Km with mutant iglA (Δ23-32), Kmr | This study |
| pJEB512 | pKK289Km with mutant iglA (Δ33-42), Kmr | This study |
| pJEB513 | pKK289Km with mutant iglA (Δ43-52), Kmr | This study |
| pJEB514 | pKK289Km with mutant iglA (Δ53-62), Kmr | This study |
| pJEB515 | pKK289Km with mutant iglA (Δ63-72), Kmr | This study |
| pJEB516 | pKK289Km with mutant iglA (Δ73-82), Kmr | This study |
| pJEB517 | pKK289Km with mutant iglA (Δ83-92), Kmr | This study |
| pJEB486 | pKK289Km with mutant iglA (Δ93-102), Kmr | This study |
| pJEB507 | pKK289Km with mutant iglA (Δ103-112), Kmr | This study |
| pJEB508 | pKK289Km with mutant iglA (Δ113-122), Kmr | This study |
| pJEB487 | pKK289Km with mutant iglA (Δ123-132), Kmr | This study |
| pJEB509 | pKK289Km with mutant iglA (Δ133-142), Kmr | This study |
| pJEB510 | pKK289Km with mutant iglA (Δ143-162), Kmr | This study |
| pJEB527 | pKK289Km with mutant iglA (Δ163-182), Kmr | This study |
| pJEB518 | pKK289Km with mutant iglA (IglAV105A), Kmr | This study |
| pJEB519 | pKK289Km with mutant iglA (IglAV109A), Kmr | This study |
| pJEB520 | pKK289Km with mutant iglA (IglAI112A), Kmr | This study |
| pJEB521 | pKK289Km with mutant iglA (IglAL115A), Kmr | This study |
| pJEB522 | pKK289Km with mutant iglA (IglAL116A), Kmr | This study |
| pJEB523 | pKK289Km with mutant iglA (IglAL122A), Kmr | This study |
| pJEB524 | pKK289Km with mutant iglA (IglAF125A), Kmr | This study |
| pJEB525 | pKK289Km with mutant iglA (IglAL1155A,F125A), Kmr | This study |
| pJEB416 | pKK289Km with wild-type iglB, Kmr | This study |
| pGADT7 | LEU2, Apr | Clontech Laboratories |
| pJEB393 | pGADT7 with wild-type iglA, LEU2, Apr | This study |
| pJEB450 | pGADT7 with mutant iglA (Δ3-22), LEU2, Apr | This study |
| pJEB473 | pGADT7 with mutant iglA (Δ23-32), LEU2, Apr | This study |
| pJEB474 | pGADT7 with mutant iglA (Δ33-42), LEU2, Apr | This study |
| pJEB475 | pGADT7 with mutant iglA (Δ43-52), LEU2, Apr | This study |
| pJEB476 | pGADT7 with mutant iglA (Δ53-62), LEU2, Apr | This study |
| pJEB477 | pGADT7 with mutant iglA (Δ63-72), LEU2, Apr | This study |
| pJEB478 | pGADT7 with mutant iglA (Δ73-82), LEU2, Apr | This study |
| pJEB479 | pGADT7 with mutant iglA (Δ83-92), LEU2, Apr | This study |
| pJEB480 | pGADT7 with mutant iglA (Δ93-102), LEU2, Apr | This study |
| pJEB481 | pGADT7 with mutant iglA (Δ103-112), LEU2, Apr | This study |
| pJEB482 | pGADT7 with mutant iglA (Δ113-122), LEU2, Apr | This study |
| pJEB483 | pGADT7 with mutant iglA (Δ123-132), LEU2, Apr | This study |
| pJEB484 | pGADT7 with mutant iglA (Δ133-142), LEU2, Apr | This study |
| pJEB457 | pGADT7 with mutant iglA (Δ143-162), LEU2, Apr | This study |
| pJEB458 | pGADT7 with mutant iglA (Δ163-182), LEU2, Apr | This study |
| pJEB498 | pGADT7 with mutant iglA (IglAV105A), LEU2, Apr | This study |
| pJEB499 | pGADT7 with mutant iglA (IglAV109A), LEU2, Apr | This study |
| pJEB500 | pGADT7 with mutant iglA (IglAI112A), LEU2, Apr | This study |
| pJEB501 | pGADT7 with mutant iglA (IglAL115A), LEU2, Apr | This study |
| pJEB502 | pGADT7 with mutant iglA (IglAL116A), LEU2, Apr | This study |
| pJEB503 | pGADT7 with mutant iglA (IglAL122A), LEU2, Apr | This study |
| pJEB504 | pGADT7 with mutant iglA (IglAF125A), LEU2, Apr | This study |
| pJEB505 | pGADT7 with mutant iglA (IglAL1155A,F125A), LEU2, Apr | This study |
| pJEB395 | pGADT7 with wild-type iglB, LEU2, Apr | This study |
| pJEB397 | pGADT7 with wild-type iglC, LEU2, Apr | This study |
| pJEB399 | pGADT7 with wild-type iglD, LEU2, Apr | This study |
| pGBKT7 | TRP1, Kmr | Clontech Laboratories |
| pJEB392 | pGBKT7 with wild-type iglA, TRP1, Kmr | This study |
| pJEB394 | pGBKT7 with wild-type iglB, TRP1, Kmr | This study |
| pJEB396 | pGBKT7 with wild-type iglC, TRP1, Kmr | This study |
| pJEB398 | pGBKT7 with wild-type iglD, TRP1, Kmr | This study |
| pJEB545 | pGADT7 encoding PdpB363-1093, LEU2, Apr | This study |
| pJEB546 | pGBKT7 encoding PdpB363-1093, TRP1, Kmr | This study |
| pJEB536 | pGADT7 encoding YPTB2666, LEU2, Apr | This study |
| pJEB558 | pGBKT7 encoding YPTB2666, TRP1, Kmr | This study |
| pJEB557 | pGADT7 encoding YPTB2665, LEU2, Apr | This study |
| pJEB537 | pGBKT7 encoding YPTB2665, TRP1, Kmr | This study |
| pJEB538 | pGADT7 encoding YPTB1483, LEU2, Apr | This study |
| pJEB560 | pGBKT7 encoding YPTB1483, TRP1, Kmr | This study |
| pJEB582 | pGBKT7 encoding YPTB1483 (Δ105-114), TRP1, Kmr | This study |
| pJEB559 | pGADT7 encoding YPTB1484, LEU2, Apr | This study |
| pJEB539 | pGBKT7 encoding YPTB1484, TRP1, Kmr | This study |
| pJEB540 | pGADT7 encoding PA1657, LEU2, Apr | This study |
| pJEB554 | pGBKT7 encoding PA1657, TRP1, Kmr | This study |
| pJEB553 | pGADT7 encoding PA1658, LEU2, Apr | This study |
| pJEB541 | pGBKT7 encoding PA1658, TRP1, Kmr | This study |
| pJEB542 | pGADT7 encoding PA2365, LEU2, Apr | This study |
| pJEB556 | pGBKT7 encoding PA2365, TRP1, Kmr | This study |
| pJEB584 | pGBKT7 encoding PA2365 (Δ109-118), TRP1, Kmr | This study |
| pJEB555 | pGADT7 encoding PA2366, LEU2, Apr | This study |
| pJEB543 | pGBKT7 encoding PA2366, TRP1, Kmr | This study |
| pJEB576 | pGADT7 encoding SL0267, LEU2, Apr | This study |
| pJEB577 | pGBKT7 encoding SL0267, TRP1, Kmr | This study |
| pJEB578 | pGADT7 encoding SL0268, LEU2, Apr | This study |
| pJEB579 | pGBKT7 encoding SL0268, TRP1, Kmr | This study |
| pJEB564 | pGADT7 encoding VCA0107, LEU2, Apr | This study |
| pJEB607 | pGADT7 encoding VCA0107 (Δ104-113), LEU2, Apr | This study |
| pJEB565 | pGBKT7 encoding VCA0107, TRP1, Kmr | This study |
| pJEB566 | pGADT7 encoding VCA0108, LEU2, Apr | This study |
| pJEB567 | pGBKT7 encoding VCA0108, TRP1, Kmr | This study |
| pJEB568 | pGADT7 encoding ECP_0238, LEU2, Apr | This study |
| pJEB603 | pGADT7 encoding ECP_0238 (Δ108-117), LEU2, Apr | This study |
| pJEB569 | pGBKT7 encoding ECP_0238, TRP1, Kmr | This study |
| pJEB571 | pGADT7 encoding ECP_0237, LEU2, Apr | This study |
| pJEB572 | pGBKT7 encoding ECP_0237, TRP1, Kmr | This study |
USAMRIID, U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, Frederick, MD.
Pasteur Institute, Paris, France.
CCUG, Culture Collection, University of Gothenburg, Gothenburg, Sweden.
Construction of iglA and iglB null mutants of F. tularensis LVS.
Primer combinations used to construct the iglA and iglB null mutants of LVS are listed in Table 2. To create the iglA mutant, upstream and downstream flanking regions were amplified by PCR and sequentially cloned into pBluescript SK(+) (Stratagene, La Jolla, CA) using the XhoI/BamHI and BamHI/SacI sites, respectively, generating a fragment encoding IglA lacking codons 4 to 174, with ∼ 1,300-bp flanking regions joined by a BamHI site. This fragment was cloned into XhoI/SacI-digested pDM4 (28), generating pJEB485. Amplified DNA fragments used for constructing the in-frame iglB deletion mutant were generated by overlap PCR (20). The resulting 2,322-bp product encoding IglB lacking codons 54 to 346, with flanking regions, was cloned into SalI/XbaI-digested pPV (17), resulting in pPV-ΔiglB. Conjugal mating experiments using S17-1λpir as the donor strain allowed allelic exchange of the suicide plasmids pJEB485 and pPV-ΔiglB in regions with complementary sequences on the LVS chromosome, as described previously (17). To remove both copies of the iglA and iglB genes, the procedure was repeated, resulting in the null mutants designated the ΔiglA and ΔiglB mutants.
TABLE 2.
Oligonucleotides used in this study
| Use | Oligonucleotides |
|---|---|
| LVS null mutants | |
| IglA Δ4-174 | IglA_a (5′-CTC GAG AGC TAT AAC ACA TAA ACC AGC-3′) (XhoI) and IglA_b (5′-GGA TCC TTT TGC CAT CTT ATT GTC CTT-3′) (BamHI) |
| IglA_c (5′-GGA TCC GAC TTA AGT AAT CAA CAA GTA G-3′) (BamHI) and IglA_d (5′-GAG CTC CTC GAC ATC CAC AAT ACT AC-3′) (SacI) | |
| IglB Δ54-346 | IglB_a (5′-CCG GTC GAC GGG GTT TAG GAT TTT AAA ACA-3′) (SalI) and IglB_b (5′-AAG TGA TAA CTC CAT AAG GTT TCT AGC ATT GTA-3′) |
| IglB_c (5′-GAA ACC TTA TGG AGT TAT CAC TTG CAA ATA TT-3′) and IglB_d (5′-CAT CTT CCC AAT AAA TCC TTT CTA-3′) | |
| Complementation | |
| IglA | IglA_F (5′-CAT ATG GCA AAA AAT AAA ATC CCA AAT TCA AGG-3′) (NdeI) and IglA_R (5′-GAA TTC CTA CTT ACC ATC TAC TTG TTG ATT-3′) (EcoRI) |
| IglA Δ3-22 | IglA_del1_F (5′-CAT ATG GCA TTA AAG AAA AAA GAG CTA CCT TAC AG-3′) (NdeI) and IglA_R |
| IglA Δ23-32 | IglA_F and IglA_del2_b (5′-GAC ACC ATC AAC ATT AGT TTC-3′) |
| IglA_del10_c (5′-AAT GTT GAT GGT GTC CTA GTT GTT GGC GAT TTA TCA AAA GGA-3′) and IglA_R | |
| IglA Δ33-42 | IglA_F and IglA_del11_b (5′-GAC TCT GTA AGG TAG CTC TT-3′) |
| IglA_del11_c (5′-CTA CCT TAC AGA GTC TCT GTG GAT GCA AAA AAA GAG TTC GCA-3′) and IglA_R | |
| IglA Δ43-52 | IglA_F and IglA_del3_b (5′-TCT TCC TTT TGA TAA ATC GCC-3′) |
| IglA_del12_c (5′-TTA TCA AAA GGA AGA AGA GAG GTC AGA AGA GTA AAT AAT GGT-3′) and IglA_R | |
| IglA Δ53-62 | IglA_F and IglA_del13_b (5′-ATA TGC GAA CTC TTT TTT TGC AT-3′) |
| IglA_del13_c (5′-AAA GAG TTC GCA TAT GAT AGG GTT TTA GAA GAG ATG AAT ATA TC-3′) and IglA_R | |
| IglA Δ63-72 | IglA_F and IglA_del_4b (5′-AAC ACC ATT ATT TAC TCT TCT GAC-3′) |
| IglA_del14_c (5′-GTA AAT AAT GGT GTT TTT GAT TTT GAG GCA CCA AAC TTT GTT TC-3′) and IglA_R | |
| IglA Δ73-82 | IglA_F and IglA_del15_b (5′-AGA TAT ATT CAT CTC TTC TAA AAC-3′) |
| IglA_del15_c (5′-GAG ATG AAT ATA TCT AAA GAT CCT AGT AAT TTA AAA GTT AAT TAT AG-3′) and IglA_R | |
| IglA Δ83-92 | IglA_F and IglA_del5_b (5′-AGA AAC AAA GTT TGG TGC CTC-3′) |
| IglA_del16_c (5′-CCA AAC TTT GTT TCT AGA ATT GAA AGT GTC AAA GAT TTT AGA CC-3′) and IglA_R | |
| IglA Δ93-102 | IglA_F and IglA_del17_b (5′-ATA ATT AAC TTT TAA ATT ACT AGG A-3′) |
| IglA_del17_c (5′-TTA AAA GTT AAT TAT GAT GCT GTT GCT AAA AAA GTT CCT GAA-3′) and IglA_R | |
| IglA Δ103-112 | IglA_F and IglA_del6_b (5′-AGG TCT AAA ATC TTT GAC ACT T-3′) |
| IglA_del18_c (5′-AAA GAT TTT AGA CCT AGA GCG CTG CTT GAA ATG AAA GAG ATA-3′) and IglA_R | |
| IglA Δ113-122 | IglA_F and IglA_del19_b (5′-GAT TTC AGG AAC TTT TTT AGC AAC-3′) |
| IglA_del19_c (5′-AAA GTT CCT GAA ATC GCA TCC TTT GCT AAG GAC ATT GAA AAT-3′) and IglA_R | |
| IglA Δ123-132 | IglA_F and IglA_del7_b (5′-TAA TAT CTC TTT CAT TTC AAG CAG-3′) |
| IglA_del20_c (5′-ATG AAA GAG ATA TTA CGT AAT CTC AAG AAA ACC ATA GAT ATG AT-3′) and IglA_R | |
| IglA Δ133-142 | IglA_F and IglA_del21_b (5′-ATT ATT TTC AAT GTC CTT AGC AA-3′) |
| IglA_del21_c (5′-GAC ATT GAA AAT AAT TTT TCA GAT AGT AAC GAA TTA GAA TCA TTA-3′) and IglA_R | |
| IglA Δ143-162 | IglA_F and IglA_del8_b (5′-AAT CAT ATC TAT GGT TTT CTT GAG-3′) |
| IglA_del8_c (5′-ACC ATA GAT ATG ATT ACG ATT AAA GAC TCT TGT GAT GCT GCT-3′) and IglA_R | |
| IglA Δ163-182 | IglA_F and IglA_del9_R (5′-GAA TTC CTA CTT ACC ATA ATT TGT CAA AGC AGG AAT CTT AC-3′) (EcoRI) |
| IglAV105A | IglA_F and IglA_V105A_b (5′-AGC AGC AGC ATC AGG TCT AAA ATC TT-3′) |
| IglA_V105A_c (5′-CCT GAT GCT GCT GCT AAA AAA GTT CCT GAA ATC A-3′) and IglA_R | |
| IglAV109A | IglA_F and IglA_V109A_b (5′-AGG AGC TTT TTT AGC AAC AGC ATC AGG T-3′) |
| IglA_V109A_c (5′-GCT AAA AAA GCT CCT GAA ATC AGA GCG CTG C-3′) and IglA_R | |
| IglAI112A | IglA_F and IglA_I112A_b (5′-TCT AGC TTC AGG AAC TTT TTT AGC AAC AGC-3′) |
| IglA_I112A_c (5′-GTT CCT GAA GCT AGA GCG CTG CTT GAA ATG AAA GA-3′) and IglA_R | |
| IglAL115A | IglA_F and IglA_L115A_b (5′-AAG AGC CGC TCT GAT TTC AGG AAC TTT-3′) |
| IglA_L115A_c (5′-ATC AGA GCG GCT CTT GAA ATG AAA GAG ATA TTA GCA T-3′) and IglA_R | |
| IglAL116A | IglA_F and IglA_L116A_b (5′-TTC AGC CAG CGC TCT GAT TTC AGG AAC-3′) |
| IglA_L116A_c (5′-AGA GCG CTG GCT GAA ATG AAA GAG ATA TTA GCA TCC-3′) and IglA_R | |
| IglAL122A | IglA_F and IglA_L122A_b (5′-TGC TGC TAT CTC TTT CAT TTC AAG CAG CG-3′) |
| IglA_L122A_c (5′-AAA GAG ATA GCA GCA TCC TTT GCT AAG GAC ATT G-3′) and IglA_R | |
| IglAF125A | IglA_F and IglA_F125A_b (5′-AGC AGC GGA TGC TAA TAT CTC TTT CAT TTC-3′) |
| IglA_F125A_c (5′-TTA GCA TCC GCT GCT AAG GAC ATT GAA AAT AAT CG-3′) and IglA_R | |
| IglB | IglB_F (5′-CAT ATG ACA ATA AAT AAA TTA AG-3′) (NdeI) and IglB_SacI_R (5′-GAG CTC TTA GTT ATT ATT TGT ACC GAA TAA TTC-3′) (SacI) |
| Yeast two-hybrid interaction studies | |
| IglA | IglA_F and IglA_R |
| IglB | IglB_F and IglB_R (5′-GGA TCC TTA GTT ATT ATT TGT ACC GAA TAA TTC-3′) (BamHI) |
| IglC | IglC_F (5′-CAT ATG AGT GAG ATG ATA ACA AGA CAA CAG GTA-3′) (NdeI) and IglC_R (5′-GAA TTC CTA TGC AGC TGC AAT ATA TCC TAT-3′) (EcoRI) |
| IglD | IglD_F (5′-CAT ATG TTT CTA GAA AGG ATT TAT TGG GAA GAT-3′) (NdeI) and IglD_R (5′-GAA TTC TTA AGA AAA GGC TAT AAA GAA ATC A-3′) (EcoRI) |
| PdpB363-1093 | PdpB_F (5′-CCC GGG TGA TAC ATA TGA CTT ATC AAT GAT T-3′) (XmaI) and PdpB_R (5′-CTG CAG CTC GAG TTA TTG TAC ATT GAC TTC TCC TTG-3′) (XhoI and PstI) |
| YPTB2666 | IP2666_F (5′-CAT ATG ATG GTT AGC AAA AGT AAT TCT CA-3′) (NdeI) and IP2666_R (5′-GAA TTC TTA CTG TGG AGT ATC AAC AGC-3′) (EcoRI) |
| YPTB2665 | IP2665_F (5′-CAT ATG ATG TCC ACT CAA GAC GCA A-3′) (NdeI) and IP2665_R (5′-GGA TCC TTA TGC TAC GTC TTT CAG CG-3′) (BamHI) |
| YPTB1483 | IP1483_F (5′-CAT ATG ATG TCC TCT TCA AGC TTC CAA-3′) (NdeI) and IP1483_R (5′-GGA TCC TTA ACC CTC TTT TGG CGC CA-3′) (BamHI) |
| YPTB1483 (Δ105-114) | IP1483_F and IP1483_del1_b (5′-TGG CTC GAA ATC TTT CAT GTC G-3′) |
| IP1483_del1_c (5′-AAA GAT TTC GAG CCA CGT GCC TTG CTG GCC ATG C-3′) and IP1483_R | |
| YPTB1484 | IP1484_F (5′-GAA TTC ATG CTG ATG TCT GTA CAG GAA-3′) (EcoRI) and IP1484_R (5′-GGA TCC TTA TGC CTT AGC CTT TGG CAT-3′) (BamHI) |
| PA1657 | PA1657_F (5′-CAT ATG ATG GCC AAA GAA GGC TCG GTA-3′) (NdeI) and PA1657_R (5′-GAA TTC TCA GGC GTC CTG GGA GGG G-3′) (EcoRI) |
| PA1658 | PA1658_F (5′-CAT ATG ATG AGC ACC AGT GCC GCA CAG-3′) (NdeI) and PA1658_R (5′-GAA TTC TTA CTC TTT GTC CAG CTT GCC GA-3′) (EcoRI) |
| PA2365 | PA2365_F (5′-CAT ATG ATG GCC GAG AGT ACG CAG CAC-3′) (NdeI) and PA2365_R (5′-GAA TTC TCA GGC CGG CTG GTC GGC C-3′) (EcoRI) |
| PA2365 (Δ109-118) | PA2365_F and PA2365_del1_b (5′-CGG GTC GAA GTC CTC GAT GT-3′) |
| PA2365_del1_c (5′-GAG GAC TTC GAC CCG CGT CGC CTG TTC GAA GCG C-3′) and PA2365_R | |
| PA2366 | PA2366_F (5′-CAT ATG ATG CCC AAG TCA TCC GCC GC-3′) (NdeI) and PA2366_R (5′-GGA TCC TCA CGC CGC TAC CGG CGG C-3′) (BamHI) |
| SL0267 | SL0267_F (5′-CAT ATG ATG GCT ATC AAC AAT AGC GCG-3′) (NdeI) and SL0267_R (5′-GAA TTC TTA TCC GCT GAC ACA TCT TGC-3′) (EcoRI) |
| SL0268 | SL0268_F (5′-CAT ATG ATG GCA AAC AGT AAT ATG CAG G-3′) (NdeI) and SL0268_R (5′-GAA TTC TCA GGC ATT GCC CTG CTT CA-3′) (EcoRI) |
| VCA0107 | VCA0107_F (5′-CAT ATG TCT AAA GAA GGA AGT GTA G-3′) (NdeI) and VCA0107_R (5′-GAA TTC TTA CGC TTG TGG CTC TTC TTG-3′) (EcoRI) |
| VCA0107 (Δ104-113) | VCA0107_EcoRI_F (5′- GAA TTC ATG TCT AAA GAA GGA AGT GTA G-3′) (EcoRI) and VCA0107_del1_b (5′-AGG AGC GAA GTC GGC TAA G-3′) |
| VCA0107_del1_c (5′-GCC GAC TTC GCT CCT AAA AAA TTG ATT GAG TTG CGT GAA G-3′) and VCA0107_BamHI_R (5′-GGA TCC TTA CGC TTG TGG CTC TTC TTG-3′) (BamHI) | |
| VCA0108 | VCA0108_F (5′-CAT ATG ATG TCT ACG ACT GAA AAG-3′) (NdeI) and VCA0108_R (5′-GGA TCC TCA GGC TTG ATC AAG ACG TC-3′) (BamHI) |
| ECP_0238 | ECP_0238_F (5′-CAT ATG AGC AAA ATG AAC AAC AAT GG-3′) (NdeI) and ECP_0238_R (5′-GAA TTC TTA TTT CTG AAC GGC GAT ACC-3′) (EcoRI) |
| ECP_0238 (Δ108-117) | ECP0238_EcoRI_F (5′-GAA TTC ATG AGC AAA ATG AAC AAC AAT GG-3′) (EcoRI) and ECP0238_del1_b (5′-AGG AGA AAG GTC ATC CAT AGA-3′) |
| ECP0238_del1_c (5′-GAT GAC CTT TCT CCT AAA CGT CTG CTC GAA TTG CGT G-3′) and ECP0238_BamHI_R (5′-GGA TCC TTA TTT CTG AAC GGC GAT ACC-3′) (BamHI) | |
| ECP_0237 | ECP_0237_F (5′-CAT ATG TCA GTA AAG GAA GAA ATT GC-3′) (NdeI) and ECP_0237_R (5′-GAA TTC TTA TTC CTT ATC CAG TCG TCC-3′) (EcoRI) |
| Quantitative PCR | |
| iglA | FTT1359c-F (5′-TTA GCA ACA GCA TCA GGT CTA AA-3′) and FTT1359c-R (5′-GGC ACC AAA CTT TGT TTC TAA AG-3′) |
| iglB | FTT1358c-F (5′-ACA AAC TCA TTA AAG CCT TCC ATA-3′) and FTT1358c-R (5′-GTT GCT GCA TAT ATA GGT TTG ACC-3′) |
| iglC | FTT1357c-F (5′-TTT CAT ATC TGT AGC ACT TGC TTG-3′) and FTT1357c-R (5′-CCA GGC TCT ATA AAT CCA ACA ATA-3′) |
| tul4 | 17kD-F (5′-GTG CCA TGA TAC AAG CTT CC-3′) and 17kD-R (5′-GCT GTC CAC TTA CCG CTT CA-3′) |
a Double underlining indicates the incorporated NdeI, EcoRI, BamHI, SacI, XmaI, XhoI, and PstI restriction sites used for cloning of the PCR-amplified DNA fragments (the restriction endonucleases are indicated in parentheses). Underlining indicates the complementary overlap between corresponding primers in the overlap PCRs. In primers used to generate alanine substitutions, the nucleotides substituted are indicated by boldface type. To optimize expression, alanine substitutions were adapted based on the codon usage preferences of F. tularensis (http://www.kazusa.or.jp/codon/).
Construction of complementation expression plasmids in trans.
For studies of complementation in trans, PCR-amplified iglA and iglB were introduced into plasmid pKK289Km to allow constitutive expression from the GroEL promoter (3). In-frame deletions and alanine substitutions of iglA were constructed by overlap PCR (20). Primer combinations and restriction sites used for vector construction are listed in Table 2. To express the IglAV109A mutant protein and green fluorescent protein (GFP) from the same plasmid, the GFP gene from pKK289Km was introduced into the EcoRI site downstream of iglA on pJEB519, resulting in pJEB587. Plasmids were transferred into F. tularensis by electroporation.
Yeast plasmid construction.
To facilitate protein-protein interaction studies with Saccharomyces cerevisiae yeast cells, PCR-amplified fragments encoding IglA or IglA mutant derivatives constructed by overlap PCR and cognate IglB proteins from F. tularensis (IglA and IglB), Y. pseudotuberculosis (YPTB2666 and YPTB2665; YPTB1483 and YPTB1484), P. aeruginosa (PA1657 and PA1658; PA2365 and PA2366), V. cholerae (VCA0107 and VCA0108), uropathogenic E. coli (UPEC) (ECP_0238 and ECP_0237), and S. Typhimurium (SL0267 and SL0268) were ligated into the GAL4 activation domain plasmid pGADT7 or the GAL4 DNA-binding domain plasmid pGBKT7 (Clontech Laboratories, Palo Alto, CA) to allow native as well as heterologous IglA-IglB interactions to be assessed. Similarly, iglC, iglD, and a fragment encoding the soluble domain (amino acids 363 to 1093) of pdpB from F. tularensis were also introduced into pGADT7 and pGBKT7. All primer combinations and restriction sites used to generate the yeast plasmids are listed in Table 2. Most sequences were obtained from GenBank (accession numbers AM233362, AE003853, AE004091, NC_006155, and CP000247), while sequences encoding SL0267 and SL0268 were obtained from the Sanger Institute home page (http://www.sanger.ac.uk/).
Yeast two-hybrid assay.
Transformation of the S. cerevisiae reporter strains AH109 and Y187, protein expression analysis of yeast lysates, and analysis of protein-protein interactions were performed using previously established methods (15). Specifically, interactions were determined by growing yeast on synthetic dropout minimal agar (Clontech Laboratories) lacking tryptophan, leucine, and adenine resulting from ADE2 reporter gene activation. The interactive potential was confirmed by comparing growth at 25°C, 30°C, and 37°C to obtain insight into the relative energy required for each interaction and by inducing two independent reporter genes, HIS3 and lacZ, by growing yeast on synthetic dropout minimal agar lacking tryptophan, leucine, and histidine and in liquid culture using o-nitrophenyl-β-d-galactopyranoside (Sigma-Aldrich, St. Louis, MO) as the substrate, respectively. Due to intrinsic leakiness with the HIS3 reporter, 3 mM 3-aminotriazole was added to histidine dropout medium to suppress false positives (21). Protein expression was verified using antibodies recognizing the activation or DNA-binding domain of GAL4 (Clontech Laboratories). Strain AH109 was used for all yeast analyses with the exception of the β-galactosidase assay, for which strain Y187 was used.
Igl protein production.
Levels of Igl proteins in pellet fractions of F. tularensis grown on modified GC agar base were analyzed by Western blotting using polyclonal antibodies recognizing IglA (BEI Resources, Manassas, VA) or IglD (Agrisera, Vännäs, Sweden) or monoclonal antibodies specific for IglB (BEI Resources) or IglC (3). Proteins were visualized using the enhanced chemiluminescence system (Amersham Biosciences, Uppsala, Sweden).
Protein stability.
The intrabacterial protein stability assay of Feldman and colleagues (13) was used, with some modifications. In short, F. tularensis was grown overnight at 37°C in liquid Chamberlain medium, diluted twofold in fresh medium, and grown for 1 h before protein synthesis was stopped by addition of 10 μg/ml chloramphenicol (corresponding to time zero). Samples were taken at different time points and analyzed by Western blotting using antisera recognizing IglA or IglB in combination with the enhanced chemiluminescence system.
Quantitative real-time PCR.
Bacterial RNA was isolated using Trizol reagent (Invitrogen Life Technologies, Paisley, United Kingdom) and subjected to DNase I (Ambion, Austin, TX) treatment to eliminate genomic DNA contaminants. To generate cDNA from 1-μg RNA templates, an iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) was used. RNA was degraded by adding 2 μl 2.5 M NaOH to each 20-μl reaction mixture, followed by incubation at 42°C for 10 min. Upon neutralization using 5 μl 1 M HCl, the cDNA was diluted to 100 μl, diluted another 10-fold, and used for quantitative PCR with SYBR green PCR master mixture (Applied Biosystems, Foster City, CA). Each 25-μl reaction mixture consisted of 5 μl template, 12.5 μl SYBR green PCR master mixture, and 0.2 μM of each primer. Primers were designed using Primer Express software (Applied Biosystems) and are listed in Table 2. For all samples, controls were prepared, in which either the template or Superscript reverse transcriptase was omitted during cDNA synthesis. All reactions were performed in triplicate with five independent RNA preparations, using a 7900HT sequence detection system (Applied Biosystems), the sequence detection system software, and a program consisting of one cycle of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. Samples were normalized against the F. tularensis 17-kDa housekeeping gene tul4 (FTL0421) and compared to corresponding genes in LVS. Serial dilutions of templates were used to determine the amplification efficiencies of the target and housekeeping genes, which were found to be approximately the same. Results were analyzed using the ΔΔCt method and converted to a relative expression ratio (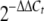 ) for statistical analysis (25). Paired two-tailed t tests were used to compare means.
) for statistical analysis (25). Paired two-tailed t tests were used to compare means.
Cultivation and infection of macrophages.
To determine the ability of F. tularensis to grow within macrophages, J774A.1 cells were infected using our established methods and plated on modified GC agar base plates for determination of viable counts (17). To assess phagosomal escape, infection was performed using the same protocol, with the following modifications. Cells were seeded onto glass coverslips in 24-well culture plates and infected with GFP-expressing F. tularensis or with green fluorescent latex beads (Sigma-Aldrich). After infection and subsequent washing, cells were incubated in culture medium without gentamicin for 3 h to allow full phagosomal escape of the positive control LVS.
Intracellular immunofluorescence assay.
Cells infected as described above were stained for the LAMP-1 glycoprotein as described previously (3). Colocalization of GFP-labeled F. tularensis and LAMP-1 was analyzed with an epifluorescence microscope (Zeiss Axioskop 2; Carl Zeiss MicroImaging GmbH, Germany) and a confocal microscope (Leica SP2; Leica Microsystems, Bensheim, Germany). In two separate experiments, each with a total of four glass slides per strain, 50 bacteria per slide were scored. To verify that the colocalization level was significantly different from that of LVS, a Student two-tailed t test with unequal variance was used.
Mouse infection.
Five C57BL/6 female mice were infected intradermally with 5 × 106 CFU of F. tularensis LVS or the ΔiglA mutant expressing wild-type IglA (pJEB415), while 5 × 108 CFU was used for the noncomplemented ΔiglA mutant or a mutant expressing IglAV109A (pJEB519), IglAL115A (pJEB521), or IglAF125A (pJEB524). Aliquots of the diluted cultures were also plated on GC agar to determine the numbers of CFU injected, which were 4.7 × 106 CFU for LVS, 4.5 × 106 CFU for the ΔiglA mutant containing pJEB415, 4.4 × 108 CFU for the ΔiglA mutant, 3.6 × 108 CFU for the ΔiglA mutant containing pJEB519, 4.7 × 108 CFU for the ΔiglA mutant containing pJEB521, and 3.3 × 108 CFU for the ΔiglA mutant containing pJEB524. Mice were examined twice daily for signs of severe infection and euthanized by CO2 asphyxiation as soon as they displayed signs of irreversible morbidity. In our experience, such mice were at most 24 h from death, and the time to death of these animals was estimated based on this premise. All animal experiments were approved by the Local Ethical Committee on Laboratory Animals, Umeå, Sweden.
RESULTS
IglA and IglB depend on each other for stability.
Most type VI secretion systems (T6SSs) identified so far by genome comparisons have homologues of iglA and iglB of F. tularensis. These genes are always linked and in the same orientation, suggesting that they are likely to perform their function as an intimate unit. To determine the biological function of IglA and IglB in F. tularensis LVS, we constructed in-frame deletion mutants, mutating both copies of each gene on the chromosome, resulting in the ΔiglA and ΔiglB mutants. To compare the levels of Igl protein synthesis in these strains with that in wild-type LVS, bacterial pellets were analyzed by Western blotting using antisera recognizing either IglA, IglB, IglC, or IglD. As expected, the ΔiglA mutant failed to produce IglA, in contrast to LVS (Fig. 1, lanes a and b). While wild-type levels of IglC and IglD were observed, this mutant produced ∼16-fold less IglB, as estimated using dilution series of the protein samples (Fig. 1, lanes a and b, and data not shown). This was not a polar effect caused by the iglA deletion, since introducing wild-type IglA in trans (pJEB415) efficiently restored IglA production, as well as IglB production, in the ΔiglA mutant (Fig. 1, lanes a and c). This suggested that the levels of IglA influenced the IglB levels. To investigate whether IglB similarly influenced the amount of IglA in the cell, pellet fractions from the ΔiglB mutant and the strain complemented in trans (pJEB416) were analyzed. As expected, the ΔiglB mutant produced wild-type levels of both IglC and IglD, while IglB was not produced (Fig. 1, lanes a and d). Intriguingly, barely detectable levels of IglA (levels ∼60-fold less than the level for LVS) were seen in the absence of iglB (Fig. 1, lanes a and d, and data not shown). By expressing IglB in trans, IglA production was partially restored (Fig. 1, lanes a and e), clearly demonstrating that the presence of IglB influenced the level of IglA. Similar results have been reported for F. novicida; however, here no IglB could be found in the iglA mutant and no IglA could be found in the iglB mutant (11, 26). To determine whether the low levels of IglA and IglB in iglB and iglA mutants, respectively, were due to effects at the transcriptional level, we performed quantitative real-time PCR. While no changes in the levels of the iglC transcript were seen for any of the mutants, the iglA mutant exhibited a ∼2-fold decrease in the iglB transcript level compared to the level in parental strain LVS (P < 0.001), while the levels of the iglA transcript in the iglB mutant were not affected (Table 3). Since these results did not provide a reasonable explanation for the low levels of IglA in the iglB mutant and the low levels of IglB in the iglA mutant, we performed an intrabacterial protein stability assay (13). LVS and the iglA and iglB mutants with or without plasmids complementing the missing gene in trans were grown in Chamberlain's medium overnight and subcultured into fresh medium. After addition of chloramphenicol to stop de novo protein synthesis, bacteria were collected at different time points and subjected to immunoblotting with antisera recognizing IglA or IglB. In LVS, IglB was very stable over a period of 180 min (Fig. 2, top panel). In contrast, very little IglB was detected in the iglA mutant after 20 min, suggesting that IglB was more susceptible to endogenous proteases in the absence of IglA. When IglA was supplied in trans, stable levels of IglB were restored (Fig. 2, top panel). Similarly, IglA was very stable over time in LVS (Fig. 2, lower panel). However, the IglA levels were extremely low in the iglB mutant and could barely be detected in the time zero sample if ∼20-fold more sample was loaded, suggesting that it was rapidly degraded (Fig. 2, lower panel, and data not shown). These results clearly demonstrate that the absence of either of IglA or IglB markedly shortens the half-life of the other protein.
FIG. 1.

Analysis of Igl protein synthesis by F. tularensis strains. Igl proteins in the pellet fraction were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and identified by immunoblotting using antiserum specific for IglA, IglB, IglC, or IglD. Lane a, wild-type strain LVS; lane b, ΔiglA null mutant; lane c, complemented ΔiglA mutant (pJEB415); lane d, ΔiglB null mutant; lane e, complemented ΔiglB mutant (pJEB416). The asterisk indicates a protein band that cross-reacts with the anti-IglD antiserum. The experiment was repeated at least three times, and the results of a representative experiment are shown. α-IglA, anti-IglA; α-IglB, anti-IglB; α-IglC, anti-IglC; α-IglD, anti-IglD.
TABLE 3.
Quantitative real-time PCR analysis of iglA and iglB mutants
| Strain | Level of expression of genea
|
||
|---|---|---|---|
| iglA | iglB | iglC | |
| ΔiglA mutant | NA | 0.48 ± 0.08** | 1.00 ± 0.34* |
| ΔiglB mutant | 0.88 ± 0.23* | NAb | 0.94 ± 0.29* |
The differential expression of each target gene in the ΔiglA or ΔiglB mutant compared to the relative expression of the same gene in LVS was defined as 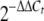 (25). As described previously (40), the mRNA expression was defined by the change, as follows: overexpressed, ≥2.0-fold change; and underexpressed, ≤0.5-fold change. The data are the means ± standard deviations of five independent experiments. The ΔiglA mutant was shown to produce less iglB transcript according to a two-sided paired Student t test (*, P < 0.01; **, P < 0.001).
(25). As described previously (40), the mRNA expression was defined by the change, as follows: overexpressed, ≥2.0-fold change; and underexpressed, ≤0.5-fold change. The data are the means ± standard deviations of five independent experiments. The ΔiglA mutant was shown to produce less iglB transcript according to a two-sided paired Student t test (*, P < 0.01; **, P < 0.001).
NA, not applicable.
FIG. 2.

Intrabacterial stability of IglA and IglB in strains of F. tularensis. The intrabacterial stability of IglB produced by LVS, the ΔiglA mutant, and the ΔiglA mutant complemented in trans (pJEB415) (upper panel) and the intrabacterial stability of IglA produced by LVS, the ΔiglB mutant, and the ΔiglB mutant complemented in trans (pJEB416) grown in Chamberlain's medium (lower panel) were examined. At time zero, chloramphenicol was added to stop protein synthesis. Samples of pelleted bacteria were taken at different times, and the amount of proteins was detected by Western blotting. The experiment was repeated at least two times, and the results of a representative experiment are shown. α-IglA, anti-IglA; α-IglB, anti-IglB.
IglA and IglB interact in the yeast two-hybrid system.
The mutual dependence on the other protein for stability suggests that IglA and IglB may interact in F. tularensis. Indeed, this was recently shown to be the case in F. novicida (11). To study the IglA-IglB interaction in more detail using the yeast two-hybrid assay, we expressed iglA from the GAL4 activation domain plasmid pGADT7 and iglB from the GAL4 DNA-binding domain plasmid pGBKT7. Transformation of either of these plasmids into the reporter yeast strain S. cerevisiae AH109 did not result in ADE2 or HIS3 reporter gene activation (data not shown). In contrast, when the plasmids were cotransformed, the ADE2 and HIS3 reporter genes were activated, allowing growth of the yeast on minimal media devoid of adenine and histidine, respectively (Fig. 3A). Importantly, these reporter genes were activated irrespective of the growth temperature (25, 30, or 37°C), indicating that there is a strong interaction between IglA and IglB (data not shown). We also confirmed the interaction by using a semiquantitative enzymatic assay measuring the production of β-galactosidase activity. When IglA and IglB were coexpressed, high levels of β-galactosidase were produced (68.8 ± 4.9 Miller units, compared to 0.042 ± 0.007 Miller unit when IglA was expressed and 0.067 ± 0.012 Miller unit when IglB was expressed), indicating that there was strong activation of the lacZ reporter gene. Thus, the yeast two-hybrid system is suitable for studying the interaction between IglA and IglB. We also investigated putative interactions between IglA and IglB and the other products of the igl operon, namely IglC and IglD. However, no interaction between IglA and IglC, between IglA and IglD, between IglB and IglC, between IglB and IglD, or between IglC and IglD was detected regardless of the vector orientation or the growth temperature, nor did any of the Igl proteins form homodimers (data not shown).
FIG. 3.

Schematic representation of the IglA in-frame deletions (A) and point mutants (B) fused to the GAL4 activation domain of plasmid pGADT7. All constructs were cotransformed with IglB in the GAL4 DNA-binding domain plasmid pGBKT7 into the S. cerevisiae two-hybrid system reporter strain AH109. The ability of each mutant to bind IglB was recorded as the degree of activation of two independent reporter genes, ADE2 and HIS3, that permitted growth of the yeast on minimal medium devoid of adenine and histidine, respectively, after day 5 with incubation at 30°C, which was expressed using a scale from ++++ (wild-type growth) to − (no growth). The results reflect the trends in growth based on three independent experiments in which several individual transformants were tested on each occasion. To measure activation of the lacZ reporter, constructs were introduced into the S. cerevisiae reporter strain Y187. The mean ± standard deviation β-galactosidase (β-gal) activities produced by mutants compared to wild-type IglA in two independent experiments in which several transformants were tested on each occasion are indicated. In panel A the relative positions in the full-length IglA construct of the four α-helices, H1 (amino acids 62 to 68), H2 (amino acids 102 to 128), H3 (amino acids 133 to 143), and H4 (amino acids 146 to 157), are indicated. In panel B the amino acid sequence for residues 102 to 128 predicted to form α-helix H2 is shown. Amino acids that were replaced with alanine are indicated by filled triangles.
A central region of IglA is required for efficient binding to IglB.
To determine the structural mechanism of the interaction between IglA and IglB, we constructed small sequential internal deletions in IglA (Fig. 3A). To investigate the ability of the mutant proteins to bind to IglB in yeast, the altered alleles were individually cloned into pGADT7 and transformed into AH109 containing the wild-type iglB allele on pGBKT7 (pJEB394). Yeast cells containing pJEB394 and plasmids harboring genes encoding IglA mutant proteins Δ3-22 aa, Δ23-32 aa, Δ133-142 aa, Δ143-162 aa, and Δ163-182 aa all expressed the ADE2 and HIS3 reporter genes, implying that an IglA-IglB protein-protein interaction had occurred (Fig. 3A). At 25°C and 30°C, the extent of growth of these strains was similar to that of yeast containing wild-type iglA and iglB, while at 37°C, the Δ23-32 aa, Δ133-142 aa, and Δ143-162 aa mutants showed slightly reduced growth, suggesting that there was a minor binding defect (Fig. 3A and data not shown). These mutants also produced less β-galactosidase activity (∼44 to 60% of wild-type levels), while the Δ3-22 aa and Δ163-182 aa mutants, which displayed no visible defect for ADE2 or HIS3 reporter gene activation, produced ∼76 and 89% of the wild-type activity, respectively (Fig. 3A). Interestingly, the Δ33-42 aa, Δ43-52 aa, Δ53-62 aa, Δ63-72 aa, Δ73-82 aa, Δ83-92 aa, Δ93-102 aa, and Δ123-132 aa mutant proteins were also able to activate the ADE2 and HIS3 reporter genes, albeit at a very reduced level compared to wild-type IglA. These weak interactions were detected at 25°C and to a small extent at 30°C but not at all at 37°C (Fig. 3A and data not shown), suggesting that there was a major defect in IglB binding. In support of this, these mutants did not activate or only poorly activated the lacZ reporter, resulting in β-galactosidase activity that was ∼0.1 to 7% of the activity produced by wild-type IglA (Fig. 3A). Significantly, the Δ103-112 aa and Δ113-122 aa mutant proteins were completely unable to activate any of the reporter genes regardless of the temperature, suggesting that a region of IglA encompassing amino acids 103 to 122 is essential for the interaction with IglB (Fig. 3A).
Importantly, the loss of IglB binding observed for some IglA mutant proteins was not the result of increased proteolysis in the yeast, as the relative abundance of each deletion mutant protein in yeast protein extracts was essentially the same (data not shown). Taken together, these results clearly demonstrate that a central domain including residues 33 to 132 of IglA is required for the ability of this protein to interact with IglB and that residues 103 to 122 play an absolutely essential role. Using Psipred V2.5 (http://bioinf.cs.ucl.ac.uk/psipred/), this region was predicted to overlap with an α-helix (residues 102 to 128) that, together with three smaller α-helices, is highly conserved in IglA homologues from other species (Fig. 3A; see Fig. S1 in the supplemental material). In fact, this secondary structure was predicted to be conserved for IglA homologues from 50 bacterial species (a total of 65 homologues) analyzed in silico despite sometimes modest levels of sequence identity (see Table S1 in the supplemental material). In contrast, the extreme N and C termini show greater variability in IglA homologues and were found to be of minor importance for the IglA-IglB interaction (Fig. 3A; see Fig. S1 in the supplemental material).
Residues in a conserved α-helix of IglA contribute to the interaction.
To analyze the contribution of individual residues to the IglA-IglB interaction and to determine whether hydrophobicity plays a role, some of the hydrophobic residues in the 27-residue α-helix were replaced with alanine (Fig. 3B). Alanine is only mildly hydrophobic and is commonly the amino acid of choice in site-directed mutagenesis studies (48). In addition, its high helix-forming propensity prevents the mutations from destabilizing the α-helix (27, 33). Indeed, none of the mutations were predicted to alter the structure of the α-helix according to Psipred V2.5 (data not shown). Each altered variant was expressed from pGADT7 and transformed into AH109 containing the wild-type iglB allele on pGBKT7 (pJEB394). Intriguingly, when grown on medium lacking adenine or histidine, all of the mutants were found to display defects in IglB binding compared to wild-type IglA. For the V105A, I112A, L116A, and L122A mutants, a minor, but reproducible, reduction in the ability to activate the ADE2 and HIS3 reporter genes was observed (for the L116A mutant this defect was observed only at 37°C) (Fig. 3B and data not shown). In contrast, the V109A and L115A mutants and the L115A F125A double mutant (a spontaneous mutant generated during cloning of the F125A mutation) showed more pronounced defects in the ability to activate these reporter genes at both high and low temperatures (Fig. 3B and data not shown). Interestingly, one mutant, the F125A mutant, actually appeared to be even better than wild-type IglA at activating the ADE2 reporter (Fig. 3B). To confirm these findings, all point mutants were analyzed using the β-galactosidase assay. Here, mutants that showed more pronounced defects in ADE2 and HIS3 reporter activation (the V109A, L115A, and L115A F125A mutants) were also less efficient at activating the lacZ reporter, resulting in levels of β-galactosidase activity that were ∼10 to 35% of the wild-type levels (Fig. 3B). In contrast, the point mutants displaying only minor defects in ADE2 and HIS3 reporter activation (the V105A, I112A, L116A, and L122A mutants) resulted in levels of β-galactosidase activity that were ∼40 to 74% of the wild-type levels (Fig. 3B), while the F125A mutant produced ∼108% of the β-galactosidase activity produced by wild-type IglA, confirming the slightly enhanced IglB binding of this mutant protein. Since the L115A F125A double mutant showed less activation of ADE2, HIS3, and lacZ than the original single mutants (Fig. 3B), hydrophobic forces from neighboring amino acids may have an additive effect in the interaction.
In the absence of a strong interaction, IglA and IglB become less stable.
The IglA mutant collection generated for the yeast two-hybrid assay provided a powerful tool to investigate the biological consequence of a diminished IglA-IglB interaction in F. tularensis. We hypothesized that IglA variants that interacted strongly with IglB would be more stable and able to promote stable IglB based on the phenotype of the ΔiglA null mutant. In this scenario, we expected to find large intracellular pools of IglA and IglB when complex formation was strong and small intracellular pools of IglA and IglB when complex formation was weak for poorly interacting mutants. To test this hypothesis, iglA mutant alleles were introduced in trans into the ΔiglA mutant, and bacterial pellet fractions were subjected to immunoblotting. As predicted, the levels of IglA mutant proteins that interacted strongly with IglB in yeast were all similar to wild-type IglA levels, suggesting that the proteins were stable. These mutants corresponded to the Δ3-22 aa, Δ23-32 aa, Δ133-142 aa, Δ143-162 aa, and Δ163-182 aa mutants (Fig. 4A, compare lanes b to f with lane a). With the exception of the Δ23-32 aa and Δ133-142 aa mutants, which synthesized somewhat less IglB than the wild-type strain, these variants produced high levels of IglB, suggesting that they could bind and stabilize IglB efficiently (Fig. 4A, compare lanes b and f with lane a). IglA deletion mutant proteins with deletions in the central domain that were found to interact poorly with IglB in yeast (i.e., the Δ33-42 aa, Δ43-52 aa, Δ53-62 aa, Δ63-72 aa, Δ73-82 aa, Δ83-92 aa, Δ93-102 aa, and Δ123-132 aa mutants) were all less abundant when they were expressed in the ΔiglA mutant and resulted in low IglB levels, except for the Δ33-42 aa mutant (Fig. 4A, compare lanes g to n with lane a). This was likely due to a specific reduction in IglA-IglB stability and not to a general defect in protein stability since all mutants produced high levels of IglC (Fig. 4A, compare lanes g to n with lane a). Furthermore, since C-terminally His-tagged versions of these mutants displayed the same pattern upon detection with anti-His antiserum, the low IglA levels were not merely an artifact of the failure of the anti-IglA serum to recognize the variants (data not shown). The Δ103-112 aa and Δ113-122 aa mutant proteins, which failed to bind IglB in yeast and which carried deletions overlapping a large conserved α-helix, were also presumably rapidly degraded (Fig. 4A, compare lanes o and p with lane a). Not surprisingly, IglB was barely detectable in the strains containing these mutants, while the levels of IglC were not affected (Fig. 4A, compare lanes o and p with lane a). Taken together, these results demonstrated the importance of IglA-IglB complex formation in F. tularensis LVS in order to prevent the degradation of the proteins by endogenous proteases. Of the eight mutant proteins with point mutations in the large conserved α-helix, only the L115A F125A double mutant, which showed the strongest defect in IglB binding in yeast, appeared to be less stable and resulted in less IglB (Fig. 4B, compare lanes i and a). The modest defect in IglB binding observed for the remaining mutant proteins was apparently too small to have an impact on the IglA-IglB stability in F. tularensis, since these proteins appeared to be indistinguishable from the wild-type control (Fig. 4B).
FIG. 4.

Analysis of Igl protein synthesis for an iglA null mutant expressing wild-type or mutated IglA in trans. Igl proteins in the pellet fraction were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and identified by immunoblotting using antiserum specific for IglA, IglB, or IglC. (A) Expression profiles for IglA deletions mutants, divided into three phenotypic groups (strong, poor, or abolished) based on their ability to interact with IglB in yeast. (B) Expression profiles for alanine substitution mutants. Mutants with more pronounced defects in IglB binding are indicated by an asterisk. The experiment was repeated at least two times, and the results of a representative experiment are shown. α-IglA, anti-IglA; α-IglB, anti-IglB; α-IglC, anti-IglC.
IglA-IglB complex formation impacts the ability of LVS to grow in macrophages and escape from the phagolysosome.
IglA is required for F. novicida to grow in macrophages (11). To determine whether IglA plays a similar role in LVS, J774 cells were infected with the ΔiglA null mutant and the mutant expressing iglA from pKK289Km (pJEB415). To assess the role of IglB in intramacrophage growth, cells were infected with the ΔiglB mutant and the mutant complemented in trans (pJEB416). While the ΔiglA and ΔiglB mutants were essentially unable to grow in J774 cells, the growth of the complemented mutants was indistinguishable from the growth of parental strain LVS (Fig. 5). In contrast, an iglA mutant expressing any of the IglA variants that were markedly defective for IglB binding in yeast (i.e., Δ33-42 aa, Δ43-52 aa, Δ53-62 aa, Δ63-72 aa, Δ73-82 aa, Δ83-92 aa, Δ93-102 aa, Δ103-112 aa, Δ113-122 aa, and Δ123-132 aa) behaved like the iglA null mutant, suggesting that these variants cannot support intracellular growth (data not shown). The low IglA-IglB stability observed for the majority of these mutants is likely to contribute to the null phenotype. To our surprise, however, the Δ3-22 aa, Δ23-32 aa, Δ133-142 aa, Δ143-162 aa, and Δ163-182 aa mutants, all of which interacted efficiently with IglB in yeast and promoted its stable production in LVS, also failed to complement the iglA mutant for growth (data not shown). While we cannot rule out the possibility that this phenotype may be due to the very subtle defects in IglB binding observed for these mutants in the yeast two-hybrid assay, an alternative explanation is that IglA has another crucial function besides binding and stabilizing IglB. An interaction between the IglA homologue EvpA of E. tarda and the soluble domain of EvpO (residues 363 to 1093 of PdpB in F. tularensis) was recently demonstrated in yeast (49); however, we did not detect the homologous interaction in yeast, suggesting that IglA of F. tularensis may play a different role than EvpA (data not shown). When the IglA substitution mutants with mutations located in the 27-residue α-helix were used in cell infections, two distinct groups were distinguished. Mutants belonging to the first group (i.e., the V105A, I112A, L116A, and L122A mutants) all efficiently complemented the iglA null mutant for replication in J774 cells (Fig. 5). This was no surprise, since they all displayed strong IglB binding and promoted stable IglB expression (Fig. 3B and 4B). In contrast, the V109A, L115A, F125A, and L115A F125A mutant proteins failed to support growth (Fig. 5). This was not due to plasmid instability, since 100% of the CFU were kanamycin resistant (data not shown). Instead, a possible explanation for this phenotype was the more pronounced IglB-binding defect observed for the V109A, L115A, and L115A F125A mutants in yeast, although none of the mutations were severe enough to cause a reduction in IglA-IglB stability in LVS (the L115A F125A mutant was an exception to this) (Fig. 3B and 4B). The null mutant phenotype of the F125A mutant is an enigma, however, since this mutant showed no apparent defect in IglB binding or stability (Fig. 3B and 4B). In fact, this mutant activated the ADE2 and lacZ reporter genes even better than wild-type IglA did (Fig. 3B). Apparently, an interaction that was too strong somehow prevented functioning of the IglA-IglB complex.
FIG. 5.

Intracellular growth of strains of F. tularensis. J774 cells were infected with various strains of F. tularensis at a multiplicity of infection of 200 for 2 h. After gentamicin treatment, cells were allowed to recover for 30 min, after which they were lysed immediately (corresponding to 0 h) (gray bars) or after 24 h (black bars) with a phosphate-buffered saline-buffered 0.1% sodium deoxycholate solution and plated to determine the number of viable bacteria (log10). All infections were repeated three times with triplicate data sets. The results of a representative experiment are shown. The bars indicate the means, and the error bars indicate the standard deviations.
Like highly virulent strains of F. tularensis, LVS escapes from phagosomes to replicate in the permissive cytoplasm (8, 16). This raises the possibility that the nonreplicating iglA and iglB mutants are defective for phagosomal escape. To investigate this, cells were infected with LVS, with the ΔiglA mutant, the ΔiglB mutant, or the ΔiglC mutant expressing GFP from pKK289Km (3), or with the ΔiglA mutant coexpressing IglAV109A and GFP from pJEB587. The percentage of bacteria colocalizing with the late endosomal and lysosomal marker LAMP-1 was determined by microscopy. At 3 h postinfection, only 17.0% ± 5.2% of the LVS bacteria colocalized with LAMP-1. In contrast, 79.5% ± 6.3% (P<0.0001) of the ΔiglA mutant bacteria and 80.0% ± 4.0% (P<0.0001) of the ΔiglB mutant bacteria colocalized with LAMP-1 (Fig. 6). Similar results were obtained for the IglA V109A point mutant (91.3% ± 3.7% [P<0.0001]) or the ΔiglC mutant (92.3% ± 3.5% [P<0.0001]) or when latex beads were added to the cells (98.8% ± 2.1% [P<0.0001]) (Fig. 6). Altogether, these results clearly demonstrate that IglA and IglB are required for phagosomal escape and subsequent multiplication in the cytosol. Mutations that prevent or alter the strength of the IglA-IglB interaction, even to a minor extent, have a major impact on the ability of F. tularensis to survive within macrophages. Thus, IglA-IglB complex formation is a key virulence mechanism for this important pathogen.
FIG. 6.

Colocalization of GFP-expressing F. tularensis strains and LAMP-1. J774 cells were infected for 2 h with F. tularensis strains expressing GFP at a multiplicity of infection of 200 or with green fluorescent latex beads (LB) at a multiplicity of infection of 10 and, after washing, incubated for 3 h. Fixed specimens were labeled for the late endosomal and lysosomal marker LAMP-1. Confocal images were acquired with a Leica SP2 confocal microscope (Leica Microsystems, Bensheim, Germany) and were assembled using Adobe Photoshop CS2 (Adobe Systems, San Jose, CA). In the representative images shown, green indicates bacteria or latex beads and red indicates the endocytic marker.
A functional IglAB complex is required for virulence.
To determine whether the inability of the IglA V109A, L115A, and F125A substitution mutants to grow in macrophages correlated with decreased virulence, mice were infected by the intradermal route. After an infection dose containing 5 × 106 CFU, parental LVS caused 100% mortality (mean time to death, 4.6 ± 0.55 days), while 80% of the mice died after infection with the ΔiglA mutant expressing wild-type IglA in trans (pJEB415) (mean time to death, 5.0 ± 0.0 days). In contrast, the ΔiglA mutant strain or the mutant expressing either IglAV109A (pJEB519), IglAL115A (pJEB521), or IglAF125A (pJEB524) caused no mortality (length of the follow-up period, 28 days) with an infection dose containing 5 × 108 CFU. This was not due to plasmid instability in animals, since at day 6 postinfection 100% of the mutant bacteria isolated from spleens, although less than the number of LVS bacteria, were kanamycin resistant, suggesting that there was retention of the plasmids (data not shown). Thus, the specific mutants showed at least 100-fold attenuation, and the possibility that they are essentially avirulent cannot be ruled out. These findings agree with previous demonstrations that other F. tularensis mutants incapable of escaping from the phagolysosome and of replicating intracellularly are avirulent in a mouse model (17, 47).
The IglA-IglB interaction may be a universal feature of T6S.
Most T6SSs involve homologues of iglA and iglB of F. tularensis, suggesting that the IglA-IglB interaction may be conserved in a wide range of pathogens. To analyze this, we used V. cholerae, S. Typhimurium, P. aeruginosa, UPEC, and Y. pseudotuberculosis as model organisms. For the first three organisms, the importance of T6S in pathogenicity has been demonstrated. V. cholerae uses its T6SS (vas) to cause cytotoxicity in amoebas and macrophages (35), while S. Typhimurium relies on the sci system to enter eukaryotic cells (14). Furthermore, an active role of T6S during cystic fibrosis infections was recently established for P. aeruginosa HSI-I (29, 30). To date, the functions of the putative T6SSs encoded in the genomes of UPEC and Y. pseudotuberculosis have not been investigated (5, 6). Intriguingly, P. aeruginosa and Y. pseudotuberculosis harbor three and four T6SSs, respectively, which is likely to reflect the need for different systems during various stages of infection (6, 29). To investigate the putative IglA-IglB interaction in these pathogens, we cloned VCA0107 and VCA0108 from V. cholerae El Tor N16961, SL0267 and SL0268 from S. Typhimurium SL1344, and ECP_0238 and ECP_0237 from UPEC strain 536 into yeast vectors pGADT7 and pGBKT7. Since P. aeruginosa PAO1 and Y. pseudotuberculosis IP32953 contain multiple IglAB homologues (see above), two pairs of sequences from each pathogen were cloned, corresponding to proteins PA1657 and PA1658 and proteins PA2365 and PA2366 for P. aeruginosa PAO1 and to proteins YPTB1483 and YPTB1484 and proteins YPTB2666 and YPTB2665 for Y. pseudotuberculosis IP32953. In the presence of their IglB partners, VCA0107, SL0267, ECP_0238, PA1657, PA2365, YPTB1483, and YPTB2666 all caused activation of the ADE2 and HIS3 reporter genes at 30°C (all combinations) and 37°C (all combinations except PA2365 and PA2366), implying that strong protein-protein interactions had occurred (see Table S2 in the supplemental material and data not shown). From these experiments we concluded that the IglA-IglB interaction is conserved in at least six pathogens causing widely different forms of disease.
Promiscuous binding between IglAB homologues of different species suggests that there is a common mode of recognition.
Since IglA proteins are likely to have a highly conserved secondary structure (see Fig. S1 in the supplemental material), we wanted to determine whether this is sufficient to allow IglA and IglB proteins from different systems and/or species to interact. Thus, we cotransformed yeast with all possible combinations of the IglA and IglB homologous proteins from F. tularensis, V. cholerae, S. Typhimurium, UPEC, P. aeruginosa, and Y. pseudotuberculosis (see above) and analyzed their ability to activate the ADE2 and HIS3 reporter genes (the results are summarized in Table S2 in the supplemental material). All IglA homologues were able to form nonnative interactions, except for IglA from F. tularensis and PA2365 from P. aeruginosa, whose IglB partners also failed to establish efficient interactions with nonnative IglA (see Table S2 in the supplemental material). At least for F. tularensis, a likely explanation for this specificity is the low levels of sequence identity between the IglA and IglB proteins and their homologues (24 to 29% for IglA and 31 to 34% for IglB) (see Table S3 in the supplemental material). These low levels of identity were not an artifact resulting from the small number of sequences analyzed, since a large-scale BLAST analysis revealed similar levels (see Table S1 in the supplemental material). In contrast, the IglAB homologues SL0267 and SL0268 of S. Typhimurium and YPTB2666 and YPTB2665 of Y. pseudotuberculosis share unusually high sequence identity (77 and 78%) (see Table S3 in the supplemental material), which is likely to facilitate efficient formation of heterologous SL0267-YPTB2665 and YPTB2666-SL0268 complexes (see Table S2 in the supplemental material). Similarly, the IglA homologues PA1657, VCA0107, and ECP_0238, which share between 40 and 52% sequence identity (see Table S3 in the supplemental material), also formed heterologous complexes with each other's IglB partners, which are 63 to 69% identical (see Tables S2 and S3 in the supplemental material). Interestingly, interactions between IglA and IglB homologues did not always occur in a reciprocal fashion. Thus, while YPTB1483 efficiently formed complexes with PA1658, as well as VCA0108, no PA1657-YPTB1484 or VCA0107-YPTB1484 complexes were formed (see Table S2 in the supplemental material). Similar patterns were also seen for other combinations of IglA-IglB homologues (see Table S2 in the supplemental material). These findings suggest that there are unique features that characterize the interaction of IglA and IglB homologues. On the one hand, there is an overall conserved domain, presumably encompassing two of the four α-helices, leading to promiscuous interactions (i.e., strong cross-species binding); on the other hand, there are additional constraints in some interactions that are delicate and require a high degree of amino acid conservation in order to provide strong binding.
To further prove that the mode of recognition between IglA and IglB homologous proteins from different systems is conserved, we engineered PA2365, YPTB1483, VCA0107, and ECP_0238 mutant proteins, which carried deletions in the large α-helix shown to be essential for binding of F. tularensis IglA to IglB. These variants (PA2365Δ109-118, YPTB1483Δ105-114,VCA0107Δ104-113, and ECP_0238Δ108-117) were equivalent to IglA protein Δ103-112 aa of F. tularensis that failed to bind IglB in yeast (Fig. 3A). Indeed, mutating these homologous regions not only prevented the ability of the IglA homologues to form complexes with their native IglB partners but also prevented the heterologous interactions previously observed for YPTB1483 (with PA1658 and VCA0108), VCA0107 (with PA1658 and ECP_0237), and ECP_0238 (with PA1658 and VCA0107) (see Table S4 in the supplemental material). Again, these findings point to a conserved mode of recognition between IglA and IglB homologous proteins from different systems.
DISCUSSION
Since their discovery in 2006, T6SSs have been identified in almost 100 different bacterial species, and a recent phylogenetic analysis suggested that they can be divided into four major groups, designated groups A to D, with F. tularensis forming an evolutionarily distinct fifth group (2). Intriguingly, many genomes harbor more than one T6SS gene cluster, and as many as six gene clusters are present in Burkholderia pseudomallei (39). Moreover, phylogenetic analysis has demonstrated that in genomes with multiple clusters, some clusters have not necessarily resulted from duplications but have presumably been acquired by horizontal gene transfer (e.g., in P. aeruginosa PAO1) (29). This emphasizes the ubiquitous nature of T6SSs and indicates that they can be adapted to perform multiple functions in host-pathogen interactions and likely in completely different types of ecological niches as well. In support of this, while there has been ample evidence that T6SSs play important roles in virulence, they are also found in nonpathogenic soil bacteria and bacteria with no known associations with eukaryotes.
Although many of the T6SSs described so far indeed appear to function as secretion systems, there is no compelling evidence that F. tularensis harbors a secretion system; however, this is an attractive possibility based on our findings that mutations in the IglAB proteins rendered the bacterium incapable of escaping from the phagosome and of replicating intracellularly. This is the first formal demonstration of an attenuated phenotype of an iglB mutant. We have recently demonstrated that iglC and iglD mutants of LVS have identical phenotypes and that there seems to be a direct correlation between the inability of F. tularensis to escape from the phagosome and the lack of intracellular replication, indicating that the bacterium is not capable of intraphagosomal replication, at least in most types of macrophages (3). Moreover, these phenotypic characteristics correlated to a lack of virulence in the mouse model (17). Importantly, as shown by Western blotting and quantitative real-time PCR analysis of iglA and iglB mutants, we did not see any effects on expression of iglC, which is transcribed downstream of iglB. Thus, the phenotypes of these mutants are not caused by polar effects on iglC. This was expected since they were generated by in-frame deletions and could be efficiently complemented for macrophage growth when wild-type IglA or IglB was supplied in trans.
Although the present study produced no indication of direct binding of IglC or IglD to any other Igl protein, the evidence collectively implies that there are functional interactions between the four proteins and that the function of each of the proteins is directly or indirectly dependent on effective expression of all three other proteins. An attractive hypothesis to explain this interdependence is that the IglA-IglB complex, like some of its homologues (e.g., EvpAB of E. tarda and AaiAB of enteroadhesive E. coli), is part of a secretion apparatus and that IglC and IglD are secreted effectors, although evidence substantiating this hypothesis has not been obtained. In support of this hypothesis, genes encoding proteins found to be secreted by the T6SSs of B. pseudomallei, P. aeruginosa PAO1, and E. tarda have been shown to be located immediately downstream of their iglAB homologues (29, 39, 49). One role of the IglA-IglB complex may be to guide effector proteins to the secretion channel, analogous to what has been suggested for chaperones of type III secretion systems (10, 45). In this scenario, the two-hybrid assay would fall short of being able to provide the substrate needed for analysis of a putative tripartite protein complex. Thus, a yeast three-hybrid assay might provide further insights into the interactions between the members of the igl operon. Since the igl operon is conserved in virulent strains of F. tularensis, it is likely that the Igl proteins of these strains have roles identical to those of LVS proteins. Indeed, we have shown that a SCHU S4 (F. tularensis subsp. tularensis) iglC mutant is avirulent (47), and the phenotype of a SCHU S4 iglB mutant appears to be identical (unpublished). Thus, importantly, all evidence indicates that LVS is a relevant model for studies aimed at dissecting the roles of the Igl proteins.
An intriguing finding was the marked instability of IglA and IglB in the ΔiglB and ΔiglA mutants, respectively. Previously, the IglA and IglB proteins were shown to be absent in F. novicida iglB and iglA mutants, respectively, which was assumed to be the result of instability (11, 26). Importantly, by investigating protein decay over time, we have shown that both proteins are rapidly degraded in the absence of their partners. In the case of IglA, our observations revealed extreme instability since the amounts of samples had to be increased 20-fold in order to detect any IglA in the iglB mutant. Whether this phenomenon is unique to F. tularensis or can be applied to IglAB homologues in other systems is not known, since no other study has addressed this question to date. Although the mechanism for the rapid degradation is unknown, it could be a tool used by F. tularensis, and possibly other bacteria, to control assembly of the two-component complex and thereby the function of the T6SS in rapid response to environmental signals that may trigger T6S.
Our strategy to identify functionally relevant regions by analyzing the phenotypes of IglA mutants was successful. By introducing short deletions that together encompassed the whole gene, we were able to map the major domain required for the Ig1A-Ig1B complex formation to residues 33 to 133 of IglA. By analyzing the interaction in yeast, we could overcome the stability problem observed for noninteracting IglA variants when they are expressed in Francisella, which rendered pulldowns with bacterial extracts difficult to perform. By creating specific mutations in this large domain, we were able to perform a detailed analysis of the requirements for individual amino acids for the function of the complex. This is the first time that this type of systematic mapping has been carried out in F. tularensis. Importantly, the mutants provide a powerful tool for further dissecting the role of IglAB in the putative T6SS of F. tularensis. The same comprehensive strategy was not applied to IglB; however, six amino acids (E101, S144, R235, I309, Y398, and V461), which are highly conserved among IglB homologues, were mutated with no apparent effect on the IglA interaction in yeast or on the ability of F. tularensis to grow in macrophages (data not shown). Thus, the nature of the IglA-IglB interaction makes an in silico approach to identifying regions in IglB important for the interaction insufficient, and the same comprehensive strategy that was used for IglA is needed to fully reveal the structure(s) of IglB required for this crucial interaction.
By using an in silico approach, we identified four distinct α-helices (H1 to H4) in IglA. Interestingly, these α-helices were all shown to be essential for growth of F. tularensis in macrophages. While H3 and H4 were both located in regions that have little importance for IglB binding, H1 and H2 overlapped with regions crucial for the interaction. As mentioned above, the F. tularensis T6SS is phylogenetically only distantly related to other T6SSs, and this is exemplified by the fact that IglA shows at most 29% identity to the homologues included in the present study. The low levels of identity were not an artifact due to the small number of sequences analyzed, since a large-scale BLAST analysis also produced similar results (the closest homologue appears to be YPTB3638 from Y. pseudouberculosis IP32953, with ∼33% identity). For this reason, we were somewhat surprised to predict domains structurally very similar to H1 to H4 and with the same specific locations in the seven non-Francisella IglA homologues investigated in this study. In fact, an in silico analysis of 65 IglA homologues from various species showed that all of them harbored the four α-helices. These structural similarities also correlated to a functional relationship, as evidenced by our demonstration of both native and heterologous interactions between the IglA and IglB homologues tested. All seven IglA homologues interacted with their cognate IglB partners. Moreover, in 17 of 56 heterologous interactions investigated (∼30%), binding was observed. This was not anticipated since the average levels of amino acid identity were only ∼34% for the IglA homologues, although a few of them did show unusually high levels of identity. Even more surprising was the finding that despite the rather modest overall amino acid similarity to IglA of F. tularensis, the conservation of the second helix, H2, appears to be required for the formation of the two-component complex not only in F. tularensis but also in P. aeruginosa, Y. pseudotuberculosis, V. cholerae, and UPEC. Further corroborating our hypothesis that H2, and possibly also H1, is essential for the formation of the two-component complex is the strong preservation of these α-helical structures despite different evolutionary origins. The previously published phylogenetic analysis of B. pseudomallei indicated that its six T6SSs have a common origin and most likely developed to perform specialized functions during different stages of the bacterium's life cycle (39). Despite this evolutionary divergence, our analysis indicated that all of the copies harbor the four α-helical structures. The same applies to all of the copies in P. aeruginosa, Y. pseudotuberculosis, and Yersinia pestis, although some of them were acquired by horizontal gene transfer. The conservation of the domains most likely is related to a key function of the two-component complex for T6S. Thus, we postulate that our finding of an essential role for the second α-helical domain (H2) in F. tularensis, P. aeruginosa, Y. pseudotuberculosis, V. cholerae, and UPEC can be generalized to encompass all T6SSs containing IglAB homologues.
Altogether, our findings indicate that the two-component complex that we have characterized has unique functional constraints. There are common features that include the presence of four α-helices, the second of which is crucial for complex formation. Since IglAB homologues are present in such a wide variety of pathogens, this interaction offers a unique and attractive target for the development of novel antibacterials. Future investigations to identify drugs that block the IglA-IglB interaction could lead to the development of therapeutics effective against a wide range of infectious diseases.
Supplementary Material
Acknowledgments
This work was supported by grants 2006-3426 (J.E.B.) and 2006-2877 (A.S.) from the Swedish Research Council and by a grant from the Medical Faculty, Umeå University, Umeå, Sweden. The work was performed in part at the Umeå Centre for Microbial Research (UCMR).
We thank Lena Lindgren and Helena Lindgren for technical assistance with the mouse infections, Linda Bönqvist and Roland Rosqvist for technical advice concerning the microscopy studies, Igor Golovliov for ΔiglB mutant construction, Marie Lindgren for helpful advice concerning the quantitative PCR analysis, and Lena Lindgren for performing the β-galactosidase assay. V. cholerae N16961, E. coli 536, and S. Typhimurium SL1344 were kindly provided by Sun Wai Nyunt, Bernt-Eric Uhlin, and Charlotta Sundin, respectively, while Y. pseudotuberculosis IP32953 and P. aeruginosa PAO1 were provided by Åke Forsberg.
Footnotes
Published ahead of print on 6 February 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Berger, H., J. Hacker, A. Juarez, C. Hughes, and W. Goebel. 1982. Cloning of the chromosomal determinants encoding hemolysin production and mannose-resistant hemagglutination in Escherichia coli. J. Bacteriol. 1521241-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bingle, L. E., C. M. Bailey, and M. J. Pallen. 2008. Type VI secretion: a beginner's guide. Curr. Opin. Microbiol. 113-8. [DOI] [PubMed] [Google Scholar]
- 3.Bönquist, L., H. Lindgren, I. Golovliov, T. Guina, and A. Sjöstedt. 2008. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 763502-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brotcke, A., D. S. Weiss, C. C. Kim, P. Chain, S. Malfatti, E. Garcia, and D. M. Monack. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 746642-6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brzuszkiewicz, E., H. Bruggemann, H. Liesegang, M. Emmerth, T. Olschlager, G. Nagy, K. Albermann, C. Wagner, C. Buchrieser, L. Emody, G. Gottschalk, J. Hacker, and U. Dobrindt. 2006. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc. Natl. Acad. Sci. USA 10312879-12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chain, P. S., E. Carniel, F. W. Larimer, J. Lamerdin, P. O. Stoutland, W. M. Regala, A. M. Georgescu, L. M. Vergez, M. L. Land, V. L. Motin, R. R. Brubaker, J. Fowler, J. Hinnebusch, M. Marceau, C. Medigue, M. Simonet, V. Chenal-Francisque, B. Souza, D. Dacheux, J. M. Elliott, A. Derbise, L. J. Hauser, and E. Garcia. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 10113826-13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain, R. E. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13232-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clemens, D. L., B. Y. Lee, and M. A. Horwitz. 2004. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 723204-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das, S., and K. Chaudhuri. 2003. Identification of a unique IAHP (IcmF associated homologous proteins) cluster in Vibrio cholerae and other proteobacteria through in silico analysis. In Silico Biol. 3287-300. [PubMed] [Google Scholar]
- 10.Day, J. B., and G. V. Plano. 1998. A complex composed of SycN and YscB functions as a specific chaperone for YopN in Yersinia pestis. Mol. Microbiol. 30777-788. [DOI] [PubMed] [Google Scholar]
- 11.de Bruin, O. M., J. S. Ludu, and F. E. Nano. 2007. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudley, E. G., N. R. Thomson, J. Parkhill, N. P. Morin, and J. P. Nataro. 2006. Proteomic and microarray characterization of the AggR regulon identifies a pheU pathogenicity island in enteroaggregative Escherichia coli. Mol. Microbiol. 611267-1282. [DOI] [PubMed] [Google Scholar]
- 13.Feldman, M. F., S. Muller, E. Wuest, and G. R. Cornelis. 2002. SycE allows secretion of YopE-DHFR hybrids by the Yersinia enterocolitica type III Ysc system. Mol. Microbiol. 461183-1197. [DOI] [PubMed] [Google Scholar]
- 14.Folkesson, A., S. Löfdahl, and S. Normark. 2002. The Salmonella enterica subspecies I specific centisome 7 genomic island encodes novel protein families present in bacteria living in close contact with eukaryotic cells. Res. Microbiol. 153537-545. [DOI] [PubMed] [Google Scholar]
- 15.Francis, M. S., M. Aili, M. L. Wiklund, and H. Wolf-Watz. 2000. A study of the YopD-lcrH interaction from Yersinia pseudotuberculosis reveals a role for hydrophobic residues within the amphipathic domain of YopD. Mol. Microbiol. 3885-102. [DOI] [PubMed] [Google Scholar]
- 16.Golovliov, I., V. Baranov, Z. Krocova, H. Kovarova, and A. Sjöstedt. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 715940-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golovliov, I., A. Sjöstedt, A. Mokrievich, and V. Pavlov. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett. 222273-280. [DOI] [PubMed] [Google Scholar]
- 18.Heidelberg, J. F., J. A. Eisen, W. C. Nelson, R. A. Clayton, M. L. Gwinn, R. J. Dodson, D. H. Haft, E. K. Hickey, J. D. Peterson, L. Umayam, S. R. Gill, K. E. Nelson, T. D. Read, H. Tettelin, D. Richardson, M. D. Ermolaeva, J. Vamathevan, S. Bass, H. Qin, I. Dragoi, P. Sellers, L. McDonald, T. Utterback, R. D. Fleishmann, W. C. Nierman, O. White, S. L. Salzberg, H. O. Smith, R. R. Colwell, J. J. Mekalanos, J. C. Venter, and C. M. Fraser. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holloway, B. W. 1969. Genetics of Pseudomonas. Bacteriol. Rev. 33419-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton, R. M., and L. R. Pease. 1991. Recombination and mutagenesis of DNA sequences using PCR, p. 217-247. In M. McPherson (ed.), Directed mutagenesis: a practical approach. Oxford University Press, New York, NY.
- 21.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1441425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keim, P., A. Johansson, and D. M. Wagner. 2007. Molecular epidemiology, evolution, and ecology of Francisella. Ann. N. Y. Acad. Sci. 110530-66. [DOI] [PubMed] [Google Scholar]
- 23.Lauriano, C. M., J. R. Barker, S. S. Yoon, F. E. Nano, B. P. Arulanandam, D. J. Hassett, and K. E. Klose. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoeba and intramacrophage survival. Proc. Natl. Acad. Sci. USA 1014246-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindgren, H., I. Golovliov, V. Baranov, R. K. Ernst, M. Telepnev, and A. Sjöstedt. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53953-958. [DOI] [PubMed] [Google Scholar]
- 25.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 26.Ludu, J. S., O. M. de Bruin, B. N. Duplantis, C. L. Schmerk, A. Y. Chou, K. L. Elkins, and F. E. Nano. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 1904584-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyu, P. C., M. I. Liff, L. A. Marky, and N. R. Kallenbach. 1990. Side chain contributions to the stability of alpha-helical structure in peptides. Science 250669-673. [DOI] [PubMed] [Google Scholar]
- 28.Milton, D. L., R. O'Toole, P. Hörstedt, and H. Wolf-Watz. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 1781310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mougous, J. D., M. E. Cuff, S. Raunser, A. Shen, M. Zhou, C. A. Gifford, A. L. Goodman, G. Joachimiak, C. L. Ordonez, S. Lory, T. Walz, A. Joachimiak, and J. J. Mekalanos. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 3121526-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mougous, J. D., C. A. Gifford, T. L. Ramsdell, and J. J. Mekalanos. 2007. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nat. Cell Biol. 9797-803. [DOI] [PubMed] [Google Scholar]
- 31.Nano, F. E., and C. Schmerk. 2007. The Francisella pathogenicity island. Ann. N. Y. Acad. Sci. 1105122-137. [DOI] [PubMed] [Google Scholar]
- 32.Nano, F. E., N. Zhang, S. C. Cowley, K. E. Klose, K. K. Cheung, M. J. Roberts, J. S. Ludu, G. W. Letendre, A. I. Meierovics, G. Stephens, and K. L. Elkins. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 1866430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Neil, K. T., and W. F. DeGrado. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250646-651. [DOI] [PubMed] [Google Scholar]
- 34.Petersen, J. M., and M. E. Schriefer. 2005. Tularemia: emergence/re-emergence. Vet. Res. 36455-467. [DOI] [PubMed] [Google Scholar]
- 35.Pukatzki, S., A. T. Ma, D. Sturtevant, B. Krastins, D. Sarracino, W. C. Nelson, J. F. Heidelberg, and J. J. Mekalanos. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. USA 1031528-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao, P. S., Y. Yamada, Y. P. Tan, and K. Y. Leung. 2004. Use of proteomics to identify novel virulence determinants that are required for Edwardsiella tarda pathogenesis. Mol. Microbiol. 53573-586. [DOI] [PubMed] [Google Scholar]
- 37.Santic, M., M. Molmeret, J. R. Barker, K. E. Klose, A. Dekanic, M. Doric, and Y. Abu Kwaik. 2007. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell. Microbiol. 92391-2403. [DOI] [PubMed] [Google Scholar]
- 38.Santic, M., M. Molmeret, K. E. Klose, S. Jones, and Y. A. Kwaik. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7969-979. [DOI] [PubMed] [Google Scholar]
- 39.Schell, M. A., R. L. Ulrich, W. J. Ribot, E. E. Brueggemann, H. B. Hines, D. Chen, L. Lipscomb, H. S. Kim, J. Mrazek, W. C. Nierman, and D. Deshazer. 2007. Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol. Microbiol. 641466-1485. [DOI] [PubMed] [Google Scholar]
- 40.Shou, J. Z., N. Hu, M. Takikita, M. J. Roth, L. L. Johnson, C. Giffen, Q. H. Wang, C. Wang, Y. Wang, H. Su, L. H. Kong, M. R. Emmert-Buck, A. M. Goldstein, S. M. Hewitt, and P. R. Taylor. 2008. Overexpression of CDC25B and LAMC2 mRNA and protein in esophageal squamous cell carcinomas and premalignant lesions in subjects from a high-risk population in China. Cancer Epidemiol. Biomark. Prev. 171424-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilisation system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Bio/Technology 1787-796. [Google Scholar]
- 42.Sjöstedt, A. 2006. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 8561-567. [DOI] [PubMed] [Google Scholar]
- 43.Sjöstedt, A. 2007. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann. N. Y. Acad. Sci. 11051-29. [DOI] [PubMed] [Google Scholar]
- 44.Suarez, G., J. C. Sierra, J. Sha, S. Wang, T. E. Erova, A. A. Fadl, S. M. Foltz, A. J. Horneman, and A. K. Chopra. 2008. Molecular characterization of a functional type VI secretion system from a clinical isolate of Aeromonas hydrophila. Microb. Pathog. 44344-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun, P., J. E. Tropea, B. P. Austin, S. Cherry, and D. S. Waugh. 2008. Structural characterization of the Yersinia pestis type III secretion system needle protein YscF in complex with its heterodimeric chaperone YscE/YscG. J. Mol. Biol. 377819-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tärnvik, A. 1989. Nature of protective immunity to Francisella tularensis. Rev. Infect. Dis. 11440-451. [PubMed] [Google Scholar]
- 47.Twine, S., M. Bystrom, W. Chen, M. Forsman, I. Golovliov, A. Johansson, J. Kelly, H. Lindgren, K. Svensson, C. Zingmark, W. Conlan, and A. Sjöstedt. 2005. A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect. Immun. 738345-8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wells, J. A. 1991. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 202390-411. [DOI] [PubMed] [Google Scholar]
- 49.Zheng, J., and K. Y. Leung. 2007. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 661192-1206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.