

Abstract
In order to understand the impact of zidovudine resistance and thymidine analog mutations (TAMs) on subtype C human immunodeficiency virus type 1, we created mutants in subtype C reverse transcriptase (RT). The subtype B RT was placed in a subtype C backbone to act as a control. Mutants and wild-type (WT) virus were competed in a head-to-head competition assay to determine how different clones grew in the same culture. Different viruses were distinguished by sequence tags in nef and a quantitative-PCR assay. The 67N and 70R accessory mutations gave an advantage over the WT in subtype C, but these mutations in subtype B had replication capacities similar to that of the WT. Of the triple mutants examined, the TAM-1 types, 41L210W215Y, were the most fit in both subtypes, but only in subtype C was the replication capacity the same as that of the WT. The TAM-2 mutants, 67N70R215F, had the slowest replication in both clones. The mixed TAM pathway mutant, 67N70R215Y, in subtype C had a significant advantage over the TAM-2 mutant, but this was not seen in subtype B. When the WT viruses were competed with each other, the subtype B RT had enhanced replication relative to subtype C. The increased capacities of the 67N and 70R mutations may indicate that there will be greater transmitted resistance and persistence in a subtype C setting than what is known for subtype B.
Human immunodeficiency virus type 1 (HIV-1) remains a leading cause of morbidity and mortality worldwide. With the advent of highly active antiretroviral therapy (HAART), the life expectancy for those on treatment has increased dramatically. Given HIV-1's ability to replicate and mutate quickly, drug resistance has been a significant problem since treatment began. While drug resistance has been studied extensively in the developed world, HIV-1 subtypes that affect developing countries are rarely targeted for drug resistance research (9).
Sub-Saharan Africa continues to bear the bulk of the burden of the HIV-1 epidemic, with 22.5 million infections out of the 33.2 million global infections (22). However, the developed world has begun to take notice by contributing unprecedented amounts of money and expertise to combat the epidemic. Much of the support for developing countries has gone toward drug treatment programs for those in need, making research into drug resistance of paramount importance. As more and more people get the treatment they require, drug resistance will increasingly play a major role in the epidemic in these hard-hit areas, where expertise and laboratory tests for clinical monitoring are often not available.
Some in vivo data point to different patterns of resistance between subtypes. Brenner et al. (3) have shown for subtype C that the 65R mutation occurs faster in cell culture in response to tenofovir pressure than it does in subtype B. A study from Brazil looked at 160 sequences from treatment-experienced HIV-1-infected individuals (21). It was found that subtype C viruses accumulated drug resistance mutations at a lower rate than subtype B isolates after at least 4 years of treatment.
Zidovudine (AZT) is a cheap antiretroviral and has relatively few side effects, making it a popular choice for a first-line regimen of HAART worldwide. While AZT drug resistance mutations have been extensively studied in subtype B, few studies have looked at non-B subtypes.
Resistance to AZT is conferred by thymidine analog mutations (TAMs). There are two pathways of TAMs that confer high levels of resistance with mutations in the viral reverse transcriptase (RT). TAM-1 is characterized by the 215Y mutation, along with the 41L and 210W mutations. TAM-2 is defined by the 215F mutation, along with 67N and 70R accessory mutations (14). In subtype B, TAM-1 occurs twice as often as TAM-2 (14, 24), and it is thought that viral fitness of the mutations plays a large part in this difference in frequency (10, 18). After years of treatment and different drug regimens, the mutations in the two pathways often become interchangeable due to the amount of pressure placed on the RT by continued antiretroviral therapy.
Viral fitness is a determinant of drug resistance mutation evolution. This is clearest when looking at the prevalence of nonnucleoside RT inhibitor drug resistance mutations. The most common nonnucleoside RT inhibitor mutations are 103N, 181C, 106 M, 188C, and 190A (6). Collins et al. (5) found that the fittest mutations are found more often on a population level, so that 103N is the fittest and most prevalent mutation. 181C, 190A, 188C, and 106M followed in order of fitness and prevalence. Several studies have looked at the viral fitness in different TAM pathways, as well. A different group found that the 41L215Y double mutant is much more fit than the 70R215Y double mutant, perhaps explaining the distinct separation of the two pathways in subtype B (10).
In subtype C, there is some evidence that the two pathways are not as definitive. Studies in our laboratory have shown that in a didanosine-containing regimen (along with AZT and efavirenz or nevirapine), 67N, 70R, and 215Y were the most common mutations seen together (17). The same study showed that the 41L mutation was rare compared to what was expected from the subtype B data; in fact, it did not occur in this study at all. In Brazil, where subtypes B and C cocirculate in the same population, a study found that the 41L and 210W TAM mutations occurred more frequently in subtype B than in subtype C (21).
These data led us to hypothesize that differences seen in the frequencies of TAMs in subtypes B and C are caused by different replicative costs associated with the pathways. We tested this by using Nef-tagged competing viruses in the same culture to determine the relative replication abilities of wild-type (WT) and mutant viruses. It was found that in subtype C, the 67N and 70R mutations enhanced HIV-1 replication compared to the WT, but no such enhancement was seen in subtype B. The TAM-1 double and triple mutants had no replicative cost for subtype C, but in subtype B, the TAM-1 mutations all had replication capacities similar to that of the single 215Y mutation alone. The TAM-2 pathway was the weakest set of mutations, with the triple mutant being the slowest virus in both subtypes. In subtype C, the mixed TAM pathway mutations, 67N70R215Y, were less fit than the WT but similar to the 215Y mutation alone; however, in subtype B, the triple mutant was less fit than the single mutation.
MATERIALS AND METHODS
DNA clones and mutagenesis.
The subtype C HIV-1 molecular clone, MJ4, was the backbone for all viruses used (16). The HXB2 rt was added to the MJ4 backbone by adding an MfeI restriction site at nucleotide 2561 (HXB2 numbering). The new site was 3 amino acids, which are identical between the two clones, into RT. A BbsI site was added at 3603 in HXB2, which is 17 identical residues after RT ends. All mutagenesis reactions were carried out with the Quickchange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) in RT subclones.
Cell culture.
Peripheral blood mononuclear cells (PBMCs) were obtained from HIV-negative blood donors and isolated with a Ficoll gradient (MP Biomedicals, Solon, OH). The PBMCs were stimulated with phytohemagglutinin A (Sigma-Aldrich, St. Louis, MO) at a concentration of 10 μg/ml for 3 days in RPMI medium (Invitrogen, Carlsbad, CA) supplemented with 20% fetal bovine serum (FBS) (Invitrogen), 5 half-maximal units/ml human delectinized interleukin-2 (ABi, Columbia, MD), 100 U/ml penicillin G (Invitrogen), 100 U/ml streptomycin (Invitrogen), and 0.25 μg/ml amphotericin B (Fungizone) (Invitrogen). The PBMCs were maintained in RPMI medium supplemented with 20% FBS, 5 half-maximal units/ml interleukin-2, 100 U/ml penicillin G, 100 U/ml streptomycin, and 0.25 μg/ml amphotericin B.
293 cells were used to produce virus from DNA clones. The cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS, 100 U/ml penicillin G, 100 U/ml streptomycin, and 0.25 μg/ml amphotericin B. Superfect transfection reagent (Qiagen, Valencia, CA) was used for DNA transfection with a modified protocol. Fifteen micrograms of plasmid DNA was diluted in a total volume of 150 μl of Dulbecco's modified Eagle's medium. Superfect (110 μl) was added to the DNA mixture and incubated for approximately 10 min; 1.25 ml of 293 maintenance medium was combined with the DNA, and the entire mixture was added to 5.28 × 106 cells in a T75 flask. The cells were incubated at 37°C for 3 hours and washed with Dulbecco's phosphate-buffered saline (Invitrogen). Fresh maintenance medium was added, and after 3 or 4 days, the viral titer was determined by p24 enzyme-linked immunosorbent assay (Perkin-Elmer, Waltham, MA). Cell-free viral stocks exceeding 50 ng/ml of p24 antigen were saved at −80°C.
Growth competition assay.
Stimulated PBMCs (5 × 105) were infected at a total multiplicity of infection of less than 0.001. Mutant virus was competed with the subtype-specific WT virus at various initial concentrations because the replicative differences should not rely on the initial viral concentration in five different competitions. After an overnight infection, the supernatant was removed and 500 μl of fresh PBMC maintenance medium was added. On day 4 after infection, the culture was transferred to a new plate with 5 × 105 freshly stimulated PBMCs. The cells were fed on day 8 with one-half volume of fresh medium, and the culture was discarded on day 12. Viral RNA was isolated from the cell supernatant on days 0, 4, 8, and 12 with the Qiaamp Viral RNA Mini Kit (Qiagen) with the addition of an RNase-free DNase I (Qiagen) digestion step. All competitions were carried out in PBMCs from the same blood donor in five different concentrations. The WT, 67N, and 70R viruses were competed in five additional wells in PBMCs from a different blood donor for a total of 10 replicates.
AZT IC50.
The AZT 50% inhibitory concentration (IC50) was determined by the protocol outlined previously (12) with some modification. PBMCs were placed in a 96-well plate at a concentration of 2 × 105 per well and stimulated with phytohemagglutinin A for 3 days. The cells were washed and resuspended in PBMC maintenance medium, and 50 μl of cells was added to each well. Fifty microliters of viral stock was also added to each well. After an overnight infection, 100 μl of AZT (Sigma-Aldrich) at concentrations from 0.002 to 200 μM was added to three wells for each dilution. The cells were fed on day 4 with fresh AZT solution, and on day 8, 50 μl of cell supernatant was saved at −80°C for p24 enzyme-linked immunosorbent assay determination. The IC50 was determined by nonlinear regression using SigmaPlot 2001 (Systat, San Jose, CA) by the Hill-Slope method. The expressed values are the percentages of growth at 100 μM AZT compared to growth with no AZT. This value was then normalized for each run with the WT, which was run with each set. Each mutant was tested in three different experiments in triplicate, except for the subtype C 67N70R215Y mutant, which was tested two times.
Quantitative PCR.
Competing viral concentrations were determined with a quantitative PCR using sequence tags in nef, as described by Boutwell et al. (2). Briefly, the concentration of each viral species was determined with a unique forward primer and a common reverse primer using the Quantifast SYBR green RT-PCR master mix (Qiagen). The WT forward primer sequence is 5′-CAACACAGCCGCCAATA-3′; the Mut forward primer is CAACACTCCGGCGAACA-3′. The common reverse primer is 5′-CCCACAAATCAAGGATCT-3′. Relative amounts of virus were determined by comparison to a standard curve generated by a linearized plasmid of approximately full viral length, 9,078 bp.
Relative replication capacity estimation and statistical analysis.
The relative replication capacity was determined with the formula described by Maree et al. (15). Briefly,
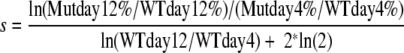 |
, where Mutday12% equals the percentage of total virus on day 12 that contains the mutation of interest, WTday12% equals the percentage of total virus on day 12 that is WT, Mutday4% equals the percentage of total virus on day 4 that contains the mutation of interest, WTday4% equals the percentage of total virus on day 4 that is WT, WTday12 equals the absolute number of copies of the WT virus on day 12, and WTday4 equals the absolute number of copies of WT virus on day 4. This formula is used to determine viral fitness, and relative viral fitness is s + 1 (15). Differences between two populations are determined with the Mann-Whitney rank sum test using SigmaStat 3.1 software (Systat).
RESULTS
Subtype B RT placement in the subtype C backbone.
The subtype C HIV-1 molecular clone, MJ4, was the backbone for all viruses tested. The MJ4 RT was replaced with the HXB2 RT by adding an MfeI restriction site at nucleotide 2561 (HXB2 numbering). The new site was 3 amino acids, which are identical between the two clones, into RT. A BbsI site was added at 3603 in HXB2, which is 17 identical residues after RT ends. This created a DNA molecular clone with a subtype B RT in a subtype C backbone. This clone allows the comparison of RT mutations in the different subtypes without there being any other subtype-specific differences to complicate the analysis.
Quantification of competing viral species in the same culture.
Placing two distinct viruses in the same culture is the best way to determine small differences in replication capacities. A dual-infection culture allows the control of well-to-well variation that confounds single-culture methods by establishing a control or WT virus. The two viral isolates are distinguished from each other with a sequence tag in nef. Four nucleotides are changed in the wobble positions of consecutive codons, which allows differential primer detection in the real-time PCR assay.
The replication cost of the sequence tag is determined by competing two viruses that differ only in the tag (Fig. 1A and B). Each growth competition is carried out in five different initial concentrations in PBMCs from the same blood donor. The populations show some variation but essentially remain the same over 12 days of infection, indicating that there is no advantage or disadvantage to the tag in nef in this system.
FIG. 1.

Representative growth curves of virus with sequence tags in nef. (A) The infecting ratio of virus is 80:20, with the native species (WT) at 80% and the tagged sequence (tagWT) at 20%. (B) Reciprocal infecting ratio of 20:80, with the WT at 20% and tagWT at 80% in the initial culture. (C) The ratio of WT virus to 215Y virus is 90% to 10%. (D) Reciprocal ratio of 10% WT to 90% 215Y virus. (E) Closer initial inoculum ratio of 60% WT to 40% 215Y. (F) Reciprocal of panel E, with 40% WT and 60% 215Y.
To test the ability of the assay to detect small differences in replication capacities, the well-characterized AZT resistance mutation 215Y was added to the subtype B RT. Figure 1C and D shows the 215Y mutation in competition with the WT at initial concentrations of 10% to 90% and the reciprocal infection of 90% to 10%. There is a clear separation of the two viral species when the mutant starts at a low concentration and a nice crossing point when the mutant starts at the higher concentration. The differences between the two viruses’ abilities to replicate can be seen when they are infected with closer initial dilutions (Fig. 1E and F), demonstrating that the viral growth kinetics are not dependent on the starting culture conditions. The impact of the 215Y mutation on the ability of HIV-1 to replicate has been seen by other groups at a similar level (8, 10, 18).
Single mutants in the TAM pathway.
To investigate the replicative costs of TAMs, each mutation was added singly to both subtype B and C RTs. Each mutant infected PBMCs, along with its corresponding WT virus. Most single mutations behaved in the same manner regardless of subtype with two notable exceptions. In subtype B, the only mutations that were less fit than the WT were the 215Y and 215F mutations (P = 0.016 and P = 0.008, respectively); all other single mutations tested were similar to the WT. These values are similar to previously reported fitness indicators (18). In subtype C, the 215Y and 215F mutations were less fit than the WT (P = 0.008 for each), as well, and the 215Y results are shown in Fig. 2A and B. If the results for 215Y in subtype C are compared with the same graphs for subtype B (Fig. 1C and D), the results look similar, although the growth is somewhat delayed in subtype C.
FIG. 2.

(A) Competition with the subtype C WT (mjWT) and the 215Y mutant (mj215Y) with an initial inoculum of 90% WT to 10% mutant. (B) Reciprocal of panel A, with 10% WT to 90% 215Y. (C) Competition of the subtype C WT with the 67N mutant at a ratio of 90:10. (D) WT and 67N mutant at a ratio of 10:90. (E) 70R competition at a closer initial inoculum of 80% WT to 20% 70R. (F) Opposite of panel E, with 20% WT to 80% 70R. (G) Summary of all single-mutant competitions. Relative fitness is defined in Materials and Methods, and each mutant was competed with its subtype-specific WT. The bars represent the means, and the error bars are 1 standard deviation. The bars marked with asterisks are significantly different from the WT value according to the Mann-Whitney rank sum test, with a P value of less than 0.01.
We were surprised to find that in subtype C the 67N (Fig. 2C and D) and 70R (Fig. 2E and F) mutations were more fit than the WT (P = 0.008 for each). Figure 2E, in particular, shows a crossing point where the 70R mutant starts at a lower concentration than the WT (20% versus 80%) but by day 12 ends at a higher percentage. The 67N and 70R mutations in subtype B did not give a significant advantage. To test whether this was due to the larger amount of variability seen in these particular replicates, the growth competition was repeated in a second blood donor's PBMCs (data not shown but included in Fig. 2G). The enhanced replication phenotype is evident for 67N and 70R in subtype C even with the addition of five extra wells of competition data.
For the purposes of comparing many different mutants, it is best to use a summary statistic (23). The formula defined by Maree et al. (15) is a comparison of the difference in the growth rates of two viral species, which they call the coefficient of selectivity (s). Relative viral fitness is s + 1. For the comparison between different mutants, the s + 1 value is used and graphed (Fig. 2G). The relative fitness of the WT is defined as 1 because there is no advantage or disadvantage to the tag. More fit viruses will have values greater than 1, and less fit viruses will have values less than 1. All values are averages of five replicates, except for the WT, 67N, and 70R which have 10 replicates, and significance is determined by a P value of less than 0.01 as determined by the Mann-Whitney rank sum test.
Double and triple mutations.
Double mutants in the TAM pathway were tested as shown in Fig. 3. There are several interesting differences seen between the two subtypes in the relative costs of mutations along the TAM-1 pathway. The double mutant 41L215Y in subtype C does not significantly differ from WT levels of replication (Fig. 3A and B); however, in subtype B, it is less fit than the WT (P = 0.008). When the double mutant was compared to 215Y in subtype B, there was no significant change in fitness relative to 215Y alone (Fig. 3E).
FIG. 3.

(A) The subtype B WT competed with the 41L215Y double mutant at a ratio of 20:80, respectively. (B) The subtype C WT competed with the 41L215Y double mutant at the same initial ratio as in panel A. (C) The subtype B competition with the 70R215F mutant at a ratio of 40% WT to 60% mutant. (D) The 70R215F competition in subtype C at a ratio of 40% WT to 60% mutant. (E) Summary of all double-mutant competitions, with the 215Y and 215F single mutants included as a reference. The 41L215Y mutant is in the TAM-1 pathway. The 70R215F mutant is in the TAM-2 pathway, and the 70R215Y mutant is in the mixed TAM pathway. The bars are the means of five replicates, and the error bars are 1 standard deviation. All bars marked with asterisks have a P value of less than 0.01 according the Mann-Whitney rank sum test compared to the subtype-specific WT. The bars marked with a plus have a P value of less than 0.01 according to the Mann-Whitney rank sum test compared to each other.
The TAM-2 pathway mutations of 70R215F give values for relative fitness for subtype B and C (0.74 and 0.60, respectively) that are significantly different from that of the WT (P = 0.008 for each). However, only the subtype C double mutant was significantly less fit than the 215F mutation alone (P = 0.008). Figure 3C and D illustrates the small difference in impact on the two subtypes of the 70R215F mutant; the results for all double mutants are summarized in Fig. 3E.
The triple mutants show more differences between the two subtypes’ abilities to tolerate drug resistance mutations. The subtype C TAM-1 triple mutant, 41L210W215Y, has no replication capacity cost compared to the WT (Fig. 4B). In subtype B, the TAM-1 triple mutant is less fit than the WT (P = 0.032) (Fig. 4A) but has a relative fitness similar to that of the 215Y mutation alone. The TAM-2 triple mutant, 67N70R215F, confers a similar effect on each subtype. For both subtypes, the triple mutants are less fit than the WT and relatively less fit than the 215F mutation alone (P = 0.008 for each comparison). In fact, these mutations had the highest replicative cost of all mutants tested (Fig. 4E).
FIG. 4.

Relative fitness of triple mutants. (A) Subtype B competition of the TAM-1 mutant, 41L210W215Y, with the WT at a ratio of 20% WT to 80% mutant. (B) Subtype C TAM-1 competition with the WT at the same ratio of 20% WT to 80% mutant. (C) Mixed TAM, 67N70R215Y, competition with WT at an inoculum of 40% WT to 60% mutant in subtype B RT. (D) Subtype C competition of WT to mixed TAM at a ratio of 40:60, respectively. (E) Summary of triple-mutant competitions, with the 215Y and 215F mutations included for reference. The bars are the means of five different competitions, and the error bars are 1 standard deviation. All bars marked with asterisks differ significantly from WT, with a P value of less than 0.05 as determined by the Mann-Whitney rank sum test. All bars marked with +, #, or ^ are comparisons between the two marked bars with a P value of less than 0.01.
The mixed TAM triple mutants show different results for each subtype, as well; Fig. 4C and D illustrates the different impacts these mutations had on the two RTs. The subtype C 67N70R215Y mutant grew less well than the WT (P = 0.008), but it replicated similarly to the 215Y single mutant (P = 0.421) and better than the TAM-2 triple mutant (P = 0.008). In subtype B, the mixed TAM triple mutant was not significantly less fit than the 215Y mutation alone (P = 0.095), and it had no advantage over the TAM-2 triple mutant (P = 0.421).
IC50.
The IC50s for the WT, 215Y, 215F, and all triple mutants were determined using the standard protocol of comparing viral growth after 8 days with AZT to growth without the drug (12). Due to the nature of the assay, the values tended to be variable because AZT resistance mutations did not always shift the inhibition curve to the right (data not shown). However, the inhibition curve was shifted up, implying that AZT could not restrict the growth of the mutants completely, even at concentrations as high as 100 μM. Figure 5 illustrates the amount of viral growth expressed as a percentage of the growth if no AZT was present and was normalized by the WT value to give the resistance at 100 μM. Most mutants were different from the WT, according to the Mann-Whitney rank sum test, but there was no difference between individual mutants. The 41L215Y double mutants were not different from the WT, but this may be explained by the variability of the assay.
FIG. 5.

AZT IC50 results expressed as the resistance over WT at a 100 μM concentration. The bars represent the means, and the error bars are 1 standard deviation. TAM-1 is the 41L210W215Y mutant, TAM-2 is the 67N70R215F mutant, and mTAM is the 67N70R215Y mutant. The asterisks signify a P value of less than or equal to 0.001 as determined by the Mann-Whitney rank sum test compared to the subtype-specific WT.
Competition of subtype B and C WT RTs.
To test if the underlying genetic differences between subtypes may be a cause of the tolerability of TAMs, the WT viruses were competed with each other in the same culture. It was found that the subtype B RT was significantly more fit than the subtype C RT (Fig. 6A and B). In fact, the relative fitness of subtype C compared to subtype B gave a value similar to that of the 215F mutation (1.20 and 1.22, respectively). If these RTs are representative of the subtypes as a whole, there may be a bigger biological explanation for the differences in the impacts of mutations on replication.
FIG. 6.

Competition of the subtype B and C WTs against each other. (A) Competition with an inoculum of 80% subtype B, hxWT, and 20% subtype C, mjWT. (B) Opposite of panel A, with an inoculum of 20% subtype B and 80% subtype C.
DISCUSSION
In order to better understand the evolution of drug resistance in HIV-1 subtype C, we compared AZT resistance mutations to the WT in subtype C and subtype B as a control. While our subtype B results confirmed what several other groups had seen (8, 10, 18), we found that mutations in the TAM pathways had different fitness costs in the two subtypes. The TAM-1 triple mutant in subtype B had a cost equal to that of the 215Y mutation alone, while the TAM-1 triple mutant was just as fast at replicating as the WT sequence in subtype C. If the TAM-1 mutants replicate as well as the WT virus, then there is no need for the virus to revert to the WT upon loss of drug pressure. This may change how transmitted resistance reverts to the WT or if it will revert at all (7). Even though there is no cost to the TAM-1 210W mutation in the subtype C background, it may occur rarely due to the fact that the genomic C consensus sequence requires 2 nucleotides to be changed (TTA→TGG), which if done in the wrong order can result in a stop codon.
The genotype seen by our group in Botswana (17), which we call the mixed TAM pathway of 67N70R215Y, showed a less striking difference between the subtypes. The triple mutants in subtypes B and C were each just as fit as the 215Y mutant alone; however, only in subtype C was there a benefit for this mixed pathway compared to TAM-2. When the single mutations were considered alone, subtype C showed enhanced replication of the 67N and 70R mutations compared to the WT. This led to the hypothesis that these mutations may occur first, and adding a 215Y mutation to either of the other residues results in a more competent virus than if a 215F mutation occurs instead. Mutations occur randomly, but the more fit amino acids are more likely to persist and be seen clinically due to their ability to outcompete the rest of the viral reservoir. These data do not explain why this pathway was seen more than TAM-1 or TAM-2 in Botswana, but the published results (17) are based on a restricted number of patients.
The TAM-2 pathway was the least fit of the triple mutants in both subtypes. It is somewhat puzzling that this was the second most common set of mutations seen in the Tshepo study (17), but the observed genotypes may be driven by the replicative fitness of the 67N and 70R mutations. These mutations may be selected early, thereby negating the possibility of the TAM-1 pathway.
The Novitsky et al. study is based on virological-failure cases from a clinical trial in which all patients described in the paper were on the AZT/didanosine/efavirenz or nevirapine arm of the trial. Their unusual genotype of the mixed TAM pathway, 67N70R215Y, could be a result of the drug combination. However, when the results were compared with subtype B sequences from patients on a similar regimen, there was a significant difference in the frequency of the 41L and 210W mutations between the two subtypes (17). Whether there will be a similar pattern of resistance on the most common drug regimen of AZT/lamivudine remains to be seen.
A study done in India found that 50% of HIV-1 subtype C strains containing nonnucleoside RT inhibitor drug resistance mutations included the 41L mutation (19). In addition, there was only one instance of the mixed pathway as seen in the African study, but it came from an isolate that included viral clones with all of the accessory TAMs, including the 219Q mutation. The cohort in India was made up of individuals on diverse kinds and numbers of drug regimens for different amounts of time, whereas the Novitisky et al. report was from a drug treatment study in which each participant received only one drug regimen for a prolonged period. The results discussed here may reconcile these two studies. While the TAM-1 pathway is clearly the fittest pathway, early in treatment, the 67N and 70R mutations may evolve first due to their enhanced replication capacities in HIV-1 subtype C, and it may take continued drug pressure for the TAM-1 mutations to evolve. It will be interesting to see if there is an increase in frequency relative to subtype B of the TAM-1 and mixed- pathway genotypes in response to the most common drug regimen of AZT and lamivudine.
A growing concern is the transmissibility of drug resistance mutations. With an increased replicative capacity of 67N and 70R compared to the WT and the absence of a fitness cost to the TAM-1 mutations, one would expect to see these mutations be easily transmissible through all routes of infection. Several studies have been carried out to determine the amount of transmitted resistance in areas where subtype C is endemic (1, 4, 11, 13, 20). They found that transmitted resistance is rare, even though our results predict that some mutations would be transmitted more easily; however, the use of HAART in southern Africa has lagged for at least a decade with respect to developed countries. With the increasing roll-out of antiretroviral drugs worldwide, it will be interesting to determine if the 67N and 70R single mutations and the fully resistant TAM-1 mutants are transmitted more and persist longer in a subtype C background.
As treatment continues in the developing world, it is important to understand how subtypes may differ in their responses to antiviral therapy. We have shown here that HIV-1 subtypes B and C differ in their abilities to tolerate drug resistance mutations. In the future, it will be important to learn if the differences between subtypes can be seen in clinical samples and with clinical consequences.
Acknowledgments
These studies were supported in part by a grant from the Secure the Future Foundation.
We thank C. Boutwell and M. F. McLane for scientific advice, R. Bosch for statistical advice, and L. Melton for editorial assistance.
Footnotes
Published ahead of print on 18 February 2009.
REFERENCES
- 1.Arora, S. K., S. Gupta, J. S. Toor, and A. Singla. 2008. Drug resistance-associated genotypic alterations in the pol gene of HIV type 1 isolates in ART-naive individuals in North India. AIDS Res. Hum. Retrovir. 24125-130. [DOI] [PubMed] [Google Scholar]
- 2.Boutwell, C. L., C. F. Rowley, and M. Essex. 2009. Reduced viral replication capacity of HIV-1 subtype C caused by CTL escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 832460-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner, B. G., M. Oliveira, F. Doualla-Bell, D. D. Moisi, M. Ntemgwa, F. Frankel, M. Essex, and M. A. Wainberg. 2006. HIV-1 subtype C viruses rapidly develop K65R resistance to tenofovir in cell culture. AIDS 20F9-F13. [DOI] [PubMed] [Google Scholar]
- 4.Bussmann, H., V. Novitsky, W. Wester, T. Peter, K. Masupu, L. Gabaitiri, S. Kim, S. Gaseitsiwe, T. Ndungu, R. Marlink, I. Thior, and M. Essex. 2005. HIV-1 subtype C drug-resistance background among ARV-naive adults in Botswana. Antivir. Chem. Chemother. 16103-115. [DOI] [PubMed] [Google Scholar]
- 5.Collins, J. A., M. G. Thompson, E. Paintsil, M. Ricketts, J. Gedzior, and L. Alexander. 2004. Competitive fitness of nevirapine-resistant human immunodeficiency virus type 1 mutants. J. Virol. 78603-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conway, B., M. A. Wainberg, D. Hall, M. Harris, P. Reiss, D. Cooper, S. Vella, R. Curry, P. Robinson, J. M. Lange, and J. S. Montaner. 2001. Development of drug resistance in patients receiving combinations of zidovudine, didanosine and nevirapine. AIDS 151269-1274. [DOI] [PubMed] [Google Scholar]
- 7.Gandhi, R. T., A. Wurcel, E. S. Rosenberg, M. N. Johnston, N. Hellmann, M. Bates, M. S. Hirsch, and B. D. Walker. 2003. Progressive reversion of human immunodeficiency virus type 1 resistance mutations in vivo after transmission of a multiply drug-resistant virus. Clin. Infect. Dis. 371693-1698. [DOI] [PubMed] [Google Scholar]
- 8.Harrigan, P. R., S. Bloor, and B. A. Larder. 1998. Relative replicative fitness of zidovudine-resistant human immunodeficiency virus type 1 isolates in vitro. J. Virol. 723773-3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holguin, A., E. Ramirez de Arellano, P. Rivas, and V. Soriano. 2006. Efficacy of antiretroviral therapy in individuals infected with HIV-1 non-B subtypes. AIDS Rev. 898-107. [PubMed] [Google Scholar]
- 10.Hu, Z., F. Giguel, H. Hatano, P. Reid, J. Lu, and D. R. Kuritzkes. 2006. Fitness comparison of thymidine analog resistance pathways in human immunodeficiency virus type 1. J. Virol. 807020-7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobs, G. B., A. Laten, E. J. van Rensburg, J. Bodem, B. Weissbrich, A. Rethwilm, W. Preiser, and S. Engelbrecht. 2008. Phylogenetic diversity and low level antiretroviral resistance mutations in HIV type 1 treatment-naive patients from Cape Town, South Africa. AIDS Res. Hum. Retrovir. 241009-1012. [DOI] [PubMed] [Google Scholar]
- 12.Japour, A. J., D. L. Mayers, V. A. Johnson, D. R. Kuritzkes, L. A. Beckett, J. M. Arduino, J. Lane, R. J. Black, P. S. Reichelderfer, R. T. D'Aquila, et al. 1993. Standardized peripheral blood mononuclear cell culture assay for determination of drug susceptibilities of clinical human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 371095-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lall, M., R. M. Gupta, S. Sen, K. Kapila, S. P. Tripathy, and R. S. Paranjape. 2008. Profile of primary resistance in HIV-1-infected treatment-naive individuals from Western India. AIDS Res. Hum. Retrovir. 24987-990. [DOI] [PubMed] [Google Scholar]
- 14.Marcelin, A. G., C. Delaugerre, M. Wirden, P. Viegas, A. Simon, C. Katlama, and V. Calvez. 2004. Thymidine analogue reverse transcriptase inhibitors resistance mutations profiles and association to other nucleoside reverse transcriptase inhibitors resistance mutations observed in the context of virological failure. J. Med. Virol. 72162-165. [DOI] [PubMed] [Google Scholar]
- 15.Maree, A. F., W. Keulen, C. A. Boucher, and R. J. De Boer. 2000. Estimating relative fitness in viral competition experiments. J. Virol. 7411067-11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ndung'u, T., B. Renjifo, and M. Essex. 2001. Construction and analysis of an infectious human immunodeficiency virus type 1 subtype C molecular clone. J. Virol. 754964-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novitsky, V., C. W. Wester, V. DeGruttola, H. Bussmann, S. Gaseitsiwe, A. Thomas, S. Moyo, R. Musonda, E. Van Widenfelt, R. G. Marlink, and M. Essex. 2007. The reverse transcriptase 67N 70R 215Y genotype is the predominant TAM pathway associated with virologic failure among HIV type 1C-infected adults treated with ZDV/ddI-containing HAART in southern Africa. AIDS Res. Hum. Retrovir. 23868-878. [DOI] [PubMed] [Google Scholar]
- 18.Paintsil, E., A. Margolis, J. A. Collins, and L. Alexander. 2006. The contribution of HIV fitness to the evolution pattern of reverse transcriptase inhibitor resistance. J. Med. Virol. 78425-430. [DOI] [PubMed] [Google Scholar]
- 19.Sen, S., S. P. Tripathy, A. A. Patil, V. M. Chimanpure, and R. S. Paranjape. 2007. High prevalence of human immunodeficiency virus type 1 drug resistance mutations in antiretroviral treatment-experienced patients from Pune, India. AIDS Res. Hum. Retrovir. 231303-1308. [DOI] [PubMed] [Google Scholar]
- 20.Soares, E. A., A. M. Martinez, T. M. Souza, A. F. Santos, V. Da Hora, J. Silveira, F. I. Bastos, A. Tanuri, and M. A. Soares. 2005. HIV-1 subtype C dissemination in southern Brazil. AIDS 19(Suppl. 4)S81-S86. [DOI] [PubMed] [Google Scholar]
- 21.Soares, E. A., A. F. Santos, T. M. Sousa, E. Sprinz, A. M. Martinez, J. Silveira, A. Tanuri, and M. A. Soares. 2007. Differential drug resistance acquisition in HIV-1 of subtypes B and C. PLoS ONE 2e730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UNAIDS. 2007. 2007 AIDS epidemic update. United Nations Programme on HIV/AIDS and World Health Organization, Geneva, Switzerland.
- 23.Wang, G. P., and F. D. Bushman. 2006. A statistical method for comparing viral growth curves. J. Virol. Methods 135118-123. [DOI] [PubMed] [Google Scholar]
- 24.Yahi, N., C. Tamalet, C. Tourres, N. Tivoli, F. Ariasi, F. Volot, J. A. Gastaut, H. Gallais, J. Moreau, and J. Fantini. 1999. Mutation patterns of the reverse transcriptase and protease genes in human immunodeficiency virus type 1-infected patients undergoing combination therapy: survey of 787 sequences. J. Clin. Microbiol. 374099-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]