

Abstract
Viral infections often produce double-stranded RNA (dsRNA), which in turn triggers potent antiviral responses, including the global repression of protein synthesis mediated by protein kinase R (PKR) and 2′-5′ oligoadenylate synthetase (OAS). As a consequence, many viruses have evolved genes, such as those encoding dsRNA-binding proteins, which counteract these pathways. Human cytomegalovirus (HCMV) encodes two related proteins, pTRS1 and pIRS1, which bind dsRNA and can prevent activation of the PKR and OAS pathways. HCMV mutants lacking either IRS1 or TRS1 replicate at least moderately well in cell culture. However, as we demonstrate in the present study, an HCMV mutant lacking both IRS1 and TRS1 (HCMV[ΔI/ΔT]) has a severe replication defect. Infection with HCMV[ΔI/ΔT] results in a profound inhibition of overall and viral protein synthesis, as well as increased phosphorylation of eukaryotic initiation factor 2α (eIF2α). The vaccinia virus E3L gene can substitute for IRS1 or TRS1, enabling HCMV replication. Despite the accumulation of dsRNA in HCMV-infected cells, the OAS pathway remains inactive, even in HCMV[ΔI/ΔT]-infected cells. These results suggest that PKR-mediated phosphorylation of eIF2α is the dominant dsRNA-activated pathway responsible for inhibition of protein synthesis and HCMV replication in the absence of both IRS1 and TRS1 and that the requirement for evasion of the PKR pathway likely explains the necessity for IRS1 or TRS1 for productive infection.
Activation of double-stranded RNA (dsRNA)-mediated pathways contributes to the innate immune response to viral infection. Among the genes involved in this antiviral response are several interferon-induced genes, including those encoding protein kinase R (PKR) and 2′-5′ oligoadenylate synthetase (OAS). After binding to dsRNA, PKR dimerizes, autophosphorylates, and then phosphorylates the translation initiation factor eukaryotic initiation factor 2α (eIF2α). Phosphorylated eIF2α inhibits guanine nucleotide exchange factor eIF2B, preventing restoration of the eIF2α-tRNAMet-GTP ternary complex and thus halting protein synthesis at the level of initiation (reviewed in reference 20). OAS catalyzes the synthesis of 2′-5′ oligoadenylates, which activate latent RNase L, resulting in degradation of single-stranded mRNA and rRNA (reviewed in reference 44). Together, activation of the PKR and OAS pathways results in an antiviral environment by causing a shutdown of protein synthesis.
Since viral replication requires protein synthesis, many viruses have had to evolve strategies for evading these dsRNA-mediated antiviral response pathways (32, 36). For example, the vaccinia virus (VV) E3L protein binds to dsRNA and prevents activation of both PKR and OAS (14, 28, 50). VV lacking E3L (VVΔE3L) is sensitive to interferon, has a limited cellular host range, and is avirulent in mice (5, 9, 51). Replication of VVΔE3L in cell culture can be rescued by dsRNA-binding proteins (dsRBPs) from other viruses (30). For example, the dsRBPs pIRS1 and pTRS1 of human cytomegalovirus (HCMV) can inhibit eIF2α phosphorylation and RNase L activation, prevent the shutoff of protein synthesis, and rescue viral replication in VVΔE3L-infected human cells (17). In murine CMV (MCMV), two US22 family members, pm142 and pm143, have functions similar to those of pIRS1 and pTRS1 in that they rescue the replication of VVΔE3L in otherwise nonpermissive cells, and together they bind to dsRNA and block PKR activation (11, 16, 18, 48).
Deletions of individual dsRNA-binding genes have differing consequences in MCMV and HCMV systems. Deletion of either m142 or m143 from the MCMV genome eliminates viral replication and results in the activation of PKR and the inhibition of protein synthesis (48). Thus, both genes are essential and likely act together as a complex (16, 18). In contrast, neither TRS1 nor IRS1 is essential for HCMV replication. Several viruses with deletions of IRS1 have been reported, and each replicates as well as wild-type virus (6, 21, 31). Deletion of TRS1 inhibits viral replication, but only by approximately 2 log units and primarily after low multiplicity of infection (MOI) (6). TRS1 appears to have a role in viral assembly late in infection that accounts for the modest replication defect of the TRS1 deletion mutant (1). Notably, deletion of TRS1 does not appear to cause a defect in viral protein synthesis (6). Therefore, unlike MCMV, neither of the two PKR evasion genes of HCMV is essential.
To determine whether HCMV relies on having at least one of the two genes, TRS1 or IRS1, for replication and maintenance of protein synthesis, we constructed an HCMV mutant lacking both genes. This double-deletion virus (HCMV[ΔI/ΔT]) did not replicate in human foreskin fibroblasts (HF) and was unable to prevent the shutdown of protein synthesis during infection of wild-type HF. Infection with HCMV[ΔI/ΔT] resulted in increased phosphorylation of eIF2α but no activation of RNase L, suggesting that HCMV depends on expression of at least one of its two known dsRBPs in order to evade the PKR pathway.
MATERIALS AND METHODS
Cell lines.
All cells were maintained at 37°C in Dulbecco's modified Eagle medium supplemented with 10% Nu serum (BD Biosciences) and antibiotics as previously described (17). The pTRS1-expressing cell lines, HF-TRS1 and HF-2.7βTRS1, were derived by transduction of HF with retroviral vectors designed to express pTRS1 under the control of the simian virus 40 (SV40) promoter or the HCMV 2.7β (TRL4) promoter, respectively. The plasmid vector pEQ944, used to make HF-TRS1 cells, was constructed by inserting the HindIII fragment from pEQ876 (25) that contains the TRS1 reading frame into the HindIII site in LNSX (35). The 2.7β gene promoter from nucleotides −232 (relative to the first nucleotide of the 2.7β RNA) through +44 was PCR amplified from CMV Towne genomic DNA using primers 587 (5′-CCTTAGATCTTTCTTTTTTACATTATGAACGTGCCT-3′) and 588 (5′-CCTTGGATCCGGGCTTCTGGAGAACGCCGG-3′), followed by cloning in the TOPO vector pcDNA3.1/V5-His-TOPO (Invitrogen). The BamHI fragment containing the 2.7β promoter was then inserted into BamHI-digested pEQ944, replacing the SV40 promoter and generating pEQ1122. Retroviral vectors were produced by transfection of pEQ944 or pEQ1122, along with pLGPS (34) and pMD.G (37), into 293T cells. The supernatants were used to transduce HF, which were then selected in G418 (0.6 mg/ml, active weight).
Construction of mutant viruses.
All mutant viruses were constructed on the basis of the AD169 bacterial artificial chromosome (BAC), which contains the full-length genome of the HCMV AD169 strain (27). Mutagenesis by homologous recombination was performed in Escherichia coli strain DY380 essentially as described previously (10). First, a kanamycin resistance gene (kan) flanked by FLP recombination target (FRT) sites was PCR amplified from plasmid pSLFRTkn (2) using oligonucleotide primers containing 50-nucleotide homologies immediately up- and downstream of TRS1 (5′-TGACGCGGGTTTGCTTCCTATATAGTGGACGTCGGAGGTGTCCGGCGCCCGTGGATAGCCTTCGAATTC-3′ and 5′-TTGTAAAACAAGTTTTCGAAACATAACGACAGCTGCAAAA GAAAACCAGTAGGACGACGACGACAAGTAA-3′) (homologies are shown in italics). The PCR product was then used to replace the TRS1 gene by homologous recombination. The kan gene was subsequently removed with FLP recombinase as described previously (10). To obtain HCMV[ΔI/ΔT] lacking IRS1 and TRS1, the IRS1 gene was replaced by a zeocin resistance gene in a fashion similar to that described above for TRS1. The following PCR primers were used: 5′-TGACGCGGGTTTGCTTCCTATATAGTGGACGTCGGAGGTGTCCGGCGCCCGAATTCAGTCCTGCTCCTCGGCCA-3′ and 5′-CAAGCGGAGAACGACAGCACGTCCTGACAACATATGGACTGGAGAGACTTGTTGACAATTAATCATCGGCAT-3′. To reinsert TRS1, the FRT-flanked kan gene was excised with EcoRI from pSLFRTkn and inserted into pcDNA-TRS1-HA (48) behind the TRS1 open reading frame. TRS1-hemagglutinin (HA) and the kan cassette were then PCR amplified with primers containing homologies up- and downstream of TRS1 as described above, and the linear PCR product was used for homologous recombination. The kan gene was subsequently removed with FLP recombinase. For construction of the HCMV[E3L] BAC, the VV E3L gene driven by an HCMV immediate-early (IE) promoter was excised with BglII and NotI from pCINeo-E3L (23) (kindly provided by Mariano Esteban, Centro Nacional de Biotecnología, Madrid, Spain) and inserted between the BamHI and NotI sites of pOri6k-Kan, a 1.6-kb plasmid consisting of a lambda π-dependent R6K origin of replication, a kan gene, a single FRT site, and a multiple cloning site (7) (kindly provided by Martin Messerle, Hannover Medical School, Hannover, Germany). The resulting plasmid, pOri6k-CMVE3L, was electroporated into E. coli carrying the HCMV[ΔI/ΔT] BAC, together with pCP20, a plasmid expressing FLP recombinase (15). This resulted in a FLP-mediated insertion of the entire pOri6K-CMVE3L plasmid into the HCMV[ΔI/ΔT] BAC.
Virus production.
BAC DNA was purified from E. coli using the NucleoBond BAC maxi kit (BD Biosciences) following the manufacturer's protocol. Virus was produced by electroporation into HF or HF-2.7βTRS1. Briefly, confluent 150-mm dishes of cells were trypsinized, pelleted, and resuspended in a 260-μl total volume of medium containing 100 μg of BAC DNA and 2 μg of a plasmid encoding Cre (pPGKCrebpA, obtained from J. Cooper, Fred Hutchinson Cancer Research Center [FHCRC]) and 2 μg of a pp71 expression plasmid (pBJ203, obtained from Bonita Biegalke, Ohio University). Electroporation was done in 0.4-cm cuvettes using a Bio-Rad Gene Pulser II at 260 V and 950 μF. The cells were then transferred to 100-mm dishes and split when confluent. Viruses were subjected to two rounds of plaque purification. HCMV[AD169] (ATCC), HCMV[TRS1-HA], and HCMV[E3L] were propagated and their titers were determined in HF. HCMV[ΔI/ΔT] was grown and its titer was determined in HF-TRS1. VVs (VC2) (47) and VVΔE3L (4) were propagated in BHK cells.
DNA analyses.
BAC DNA was digested with EcoRI, separated on 0.6% agarose gels, and stained with SYBR green II (Molecular Probes). HCMV DNA, purified from infected HF by lysis with 2% sodium dodecyl sulfate (SDS), followed by digestion with proteinase K and ethanol precipitation, was digested with KpnI, separated on 0.5% agarose gels, transferred to nitrocellulose, and analyzed by Southern blotting with a probe specific to the region downstream of TRS1. The probe was constructed by PCR of HCMV[AD169] DNA using primers 688 (GCTCCCGATGGAGAGCCC) and 689 (TCGGACCCATCGCCCC) to amplify the genomic region from nucleotides 225876 to 226112 (accession no. NC_001347).
Viral infections.
HF or HF-TRS1 were infected with HCMV at MOIs ranging from 0.1 to 5 PFU/cell, depending on the experiment. Except where otherwise indicated, infections were done using spin inoculation, in which virus was diluted in medium sufficient to cover the cells and the plates were centrifuged at 700 × g for 30 min at 7°C. The plates were then incubated at 37°C for an additional 30 min, after which the inoculum was aspirated and fresh medium was added. For viral production experiments, duplicate wells of HF or HF-TRS1 were infected without spin inoculation, virus in the medium was collected daily, and its titer was determined using HF-TRS1 cells. For VV infections, HF were infected at an MOI of 2 as previously described (19).
Immunoblot analyses.
At the indicated times postinfection (p.i.), cells were washed with phosphate-buffered saline (PBS) and lysed with 2% SDS. Cellular and viral DNAs were sheared in a Bransonic bath sonicator, and protein concentrations were determined by fluoraldehyde o-phthalaldehyde (Pierce) assay (24). Equivalent amounts of lysate were denatured, separated on SDS-polyacrylamide gel electrophoresis (PAGE) gels, and transferred to polyvinylidene difluoride membranes (GE Life Sciences) by electroblotting. Total and phospho-eIF2α were detected using phosphospecific or total eIF2α rabbit polyclonal antibodies (no. 9721 and 9722, both at 1:1,000; Cell Signaling Technology) and the Western Star chemiluminescence detection system (Tropix, Inc.) according to the manufacturers' recommendations. Viral protein levels were assessed using IE1 mouse monoclonal antibody (1:1,000; NEN), UL44 mouse monoclonal antibody (1:15,000; Virusys CA006-100), pp65 mouse monoclonal antibody (1:20,000; Virusys CA003-100), and pIRS1/pTRS1 rabbit polyclonal antiserum αp999 directed against amino acids 74 to 248 of TRS1 (1:1,000; see the supplemental material). Actin was detected using actin-specific antibody (1:1,000; Sigma A-2066).
Immunofluorescence.
HF were seeded into 12-well plates containing glass coverslips. The cells were infected at an MOI of 5 as described above. The coverslips were rinsed with PBS, fixed with 4% formaldehyde in PBS for 30 min at 4°C, and rinsed three times with PBS. The cells were permeabilized with 0.5% Triton X-100 (Sigma) in PBS, rinsed, and then blocked using 3% bovine serum albumin (BSA) in PBS for 30 min at room temperature (RT) and rinsed. The coverslips were incubated with J2 mouse monoclonal antibody (1:1,000 in 3% BSA in PBS; English and Scientific Consulting) or isotype-matched β-galactosidase mouse monoclonal antibody (Promega; Z378A) for 30 min at RT and rinsed three times in PBS before being stained with fluorescein isothiocyanate-labeled goat anti-mouse antibody (1:1,000 in 3% BSA in PBS) for 30 min at RT. Hoechst no. 33342 (5 μg/ml; Invitrogen) was used to stain nuclei. RNase treatments were completed after the blocking step using 8 U/ml ShortCut RNase III (New England Biolabs; M0245S) in the manufacturer-supplied buffer or 50 μg/ml RNase A (Sigma; R5503) in either 0.1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (low salt) or 2× SSC (high salt). After the addition of RNase, the coverslips were incubated at 37°C for 1 h before being washed with PBS and stained with J2 as described above. Ganciclovir (30 μM/ml; Syntex) was added at 1 h p.i. and remained on the cells throughout infection.
RNA analysis.
Whole-cell RNA was harvested using TRIzol Reagent (Invitrogen), resolved on 1% formaldehyde agarose gels, and transferred to nitrocellulose. Northern blot hybridization was carried out using an 18S rRNA-specific probe as described previously (17).
Radiolabeling of virus-infected cells.
At 2, 24, 48, or 72 h post-HCMV infection, HF were pulse-labeled with Tran[35S]label (50 μCi/ml; Perkin-Elmer) in medium lacking methionine and cysteine for 1 h. The cells were then washed and lysed in 2% SDS. Equivalent amounts of each lysate were separated by SDS-PAGE, and the gels were dried and visualized by autoradiography.
RESULTS
Construction of viral mutants.
IRS1 and TRS1 encode proteins that bind to dsRNA and PKR, preventing the activation of PKR and subsequent phosphorylation of eIF2α (17, 26). In the context of VV infection, either gene can also substitute for E3L in blocking activation of the RNase L pathway and enable productive viral infection in human cells (17). Also, they each can rescue replication of HSVΔγ34.5 (12). However, these observations do not clarify which, if any, of the functions associated with these proteins are important during HCMV replication. The ability of viruses lacking either IRS1 or TRS1 to replicate in cell culture without any observed defects in viral-protein synthesis might be due to functions provided by the member of the pair that is still present in these single-deletion mutants. Thus, in order to evaluate the role of the shared activities of IRS1 and TRS1 during HCMV infection, we needed to construct an HCMV mutant virus in which both genes were deleted.
Using the AD169 BAC system (27), we first constructed a mutant virus genome lacking both IRS1 and TRS1 (HCMV[ΔI/ΔT]) and then a repair virus in which we inserted an HA-tagged TRS1 gene (HCMV[TRS1-HA]) back into the genome at the TRS1 locus as described in Materials and Methods (Fig. 1A). We also introduced the E3L gene, under the control of the HCMV major IE promoter, into the HCMV[ΔI/ΔT] BAC (HCMV[E3L]). To confirm the structures of the various BAC constructs, we first analyzed EcoRI-digested BAC DNA (Fig. 1B). Consistent with predictions, EcoRI digestion of HCMV[AD169] BAC DNA resulted in 8.8-kb and 11.3-kb fragments containing IRS1 and TRS1, respectively (Fig. 1B, lane 1). Replacement of IRS1 with the zeocin cassette containing an EcoRI site resulted in loss of the 8.8-kb band and generation of two bands at 4.3 kb and 2.5 kb. Deletion of TRS1 in HCMV[ΔI/ΔT] and reintroduction of the E3L gene in HCMV[E3L] were predicted to eliminate an 11.3-kb band and produce new bands at ∼4.0 kb and ∼4.5 kb, respectively. These bands were not clearly resolved from other bands in the same size range. Other than these expected alterations, the EcoRI fragments appeared the same among the various BAC DNAs, as expected.
FIG. 1.

Characterization of viral mutants. (A) Depiction of HCMV genomes used in this study. The HCMV genome contains unique long (UL) and unique short (US) segments flanked by inverted repeats. A zeocin gene cassette (Zeo), were inserted into the IRS1 locus. HCMV[ΔI/ΔT], constructed from an AD169 BAC, was used to construct repair viruses encoding TRS1 (HCMV[TRS1-HA]) and E3L (HCMV[E3L]), as described in Materials and Methods. Relevant KpnI sites are indicated, and the position of the probe (Pb; double line) used in panel C is shown. Also shown are FRT sites (arrowheads), the HCMV major IE promoter (Pr), and the R6K plasmid origin and kan sequences (rk). (B) BAC DNA was digested with EcoRI, electrophoretically separated, and detected using SYBR green to confirm the genomic structures. (C) Viral DNA was purified from infected HF or HF-TRS1 (HCMV[ΔI/ΔT]), digested with KpnI, separated by gel electrophoresis, and examined by Southern blotting using a 32P-labeled probe specific for the genomic region downstream of TRS1 (marked in panel A). (D) Lysates from mock-infected or HCMV-infected HF (MOI = 3) were prepared at 6 and 48 h p.i., separated by SDS-PAGE, and immunoblotted using αp999 antiserum specific for pIRS1 and pTRS1 (top) and for pp65 (bottom).
We transfected BAC DNA into HF to reconstitute the infectious viruses. The wild-type AD169 virus used in these experiments was obtained from the ATCC (VR-538) and was not BAC derived. For the mutants, we electroporated HF with BAC DNA, along with plasmids encoding Cre (to excise the bacterial sequences) and HCMV pp71 (pUL82, to enhance infectivity of viral DNA) (3). We were unsuccessful at producing infectious HCMV[ΔI/ΔT] following transfection into wild-type HF, so we constructed complementing cell lines by transducing HF with either of two retroviral vectors designed to express pTRS1 from the SV40 promoter or the HCMV 2.7β (TRL4) promoter. Using the latter cells, we succeeded in generating virus from the HCMV[ΔI/ΔT] BAC. However, since the HF-2.7βTRS1 cell line did not grow well, we used the SV40 promoter-driven pTRS1-expressing HF (HF-TRS1) for subsequent preparation of viral stocks and experiments.
We purified viral DNA from infected cells and performed Southern blotting to confirm the genomic structure (Fig. 1C). Probing for sequences just downstream from the TRS1 coding sequence yielded the expected KpnI 8.4-kb band from AD169 and HCMV[TRS1-HA] DNA. HCMV[ΔI/ΔT] virus produced a band of 6 kb, consistent with the removal of approximately 2.5 kb during the deletion of TRS1. HCMV[E3L] yielded the expected band of about 800 bp due to an additional KpnI site introduced with the E3L gene. We also examined the region around IRS1 by PCR to further confirm the viral structure (data not shown).
Using a rabbit antiserum directed against an identical region in the N terminus of pTRS1 and pIRS1, we tested for expression of these proteins in cells infected for 6 and 48 h with the various viruses. As expected, wild-type HCMV produced both pTRS1 and pIRS1, while HCMV[TRS1-HA] produced only pTRS1 (Fig. 1D). We detected no expression of pIRS1 or pTRS1 in cells infected with HCMV[ΔI/ΔT] or HCMV[E3L], although the antiserum detected a faint background band even in mock-infected HF. As a control to verify that each of the viruses infected the cells in this experiment, we also monitored pp65 (UL83) expression by immunoblot assay of the same lysates. pp65 was detected in each of the infected cell lysates at 6 h p.i., reflecting its delivery to cells by virions. In combination, these studies indicated that the viruses had the expected genomic structures.
Growth characteristics of HCMV[ΔI/ΔT].
We next investigated the growth properties of the wild-type and mutant viruses in cell culture. We infected both HF and HF-TRS1 at a low MOI (0.1 PFU/cell) and analyzed virus production over time by determining the titer of the virus in the medium using HF-TRS1 (Fig. 2). We detected no infectious HCMV[ΔI/ΔT] at any time during 7 days of infection of HF. The repair virus, HCMV[TRS1-HA], grew to titers similar to those of wild-type AD169, indicating that pTRS1 expression is sufficient for wild-type replication properties, consistent with previous reports of wild-type growth of several IRS1-only deletion viruses (6, 21, 31). Interestingly, the VV E3L gene could substitute for deletion of IRS1 and TRS1. However, HCMV[E3L] production was slightly lower than that of wild-type AD169 at 5 to 7 days p.i., suggesting that pE3L may not be sufficient for full-scale virus production. The restoration of replication in HCMV[TRS1-HA] and HCMV[E3L] also argues that the replication defect of HCMV[ΔI/ΔT] is due to the deletion of the TRS1 and IRS1 genes and not to an undetected second-site mutation.
FIG. 2.
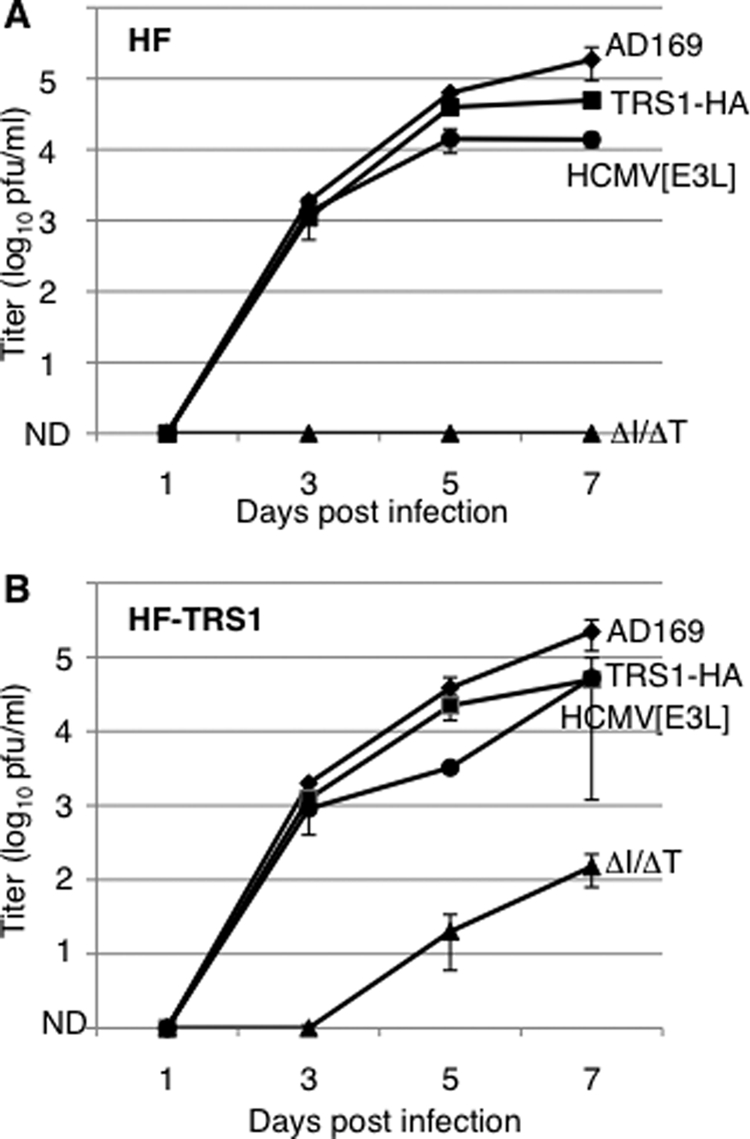
Replication of viral mutants. HF (A) or HF-TRS1 (B) were infected with wild-type AD169 HCMV or mutant viruses (MOI = 0.1). The mean (±SD) virus titer in the medium was determined on HF-TRS1. Infections were done in duplicate and are representative of three experiments. ND, none detected in 0.2 ml.
HCMV[ΔI/ΔT] did replicate in HF-TRS1 (Fig. 2B) but produced several log units less progeny than the other viruses. Among the possible explanations was that the pTRS1 expressed in these cells was not very active in blocking PKR-mediated translational repression. However, VVΔE3L replication was enhanced ∼30-fold in these cells compared to wild-type HF (data not shown), indicating that the pTRS1 expressed in HF-TRS1 was efficient at blocking the PKR response against VVΔE3L, at least. An alternative explanation for the low level of HCMV[ΔI/ΔT] replication was that the abundance of pTRS1 in the complementing cells may be lower than the levels produced during wild-type virus infections. Investigation of this possibility by immunoblot assay revealed that pTRS1 abundance in HF-TRS1 was generally less than that present late after wild-type HCMV infection of HF (data not shown). However, these results were somewhat variable, and we do not yet know whether the abundance of pTRS1 or another parameter, such as the timing of its expression or localization in the complementing cells, is suboptimal for full-scale viral production of HCMV[ΔI/ΔT] in HF-TRS1.
dsRNA accumulation during HCMV infection.
The observation that deletion of both known dsRBP genes of HCMV eliminated viral replication in wild-type HF suggests that either IRS1 or TRS1 is necessary to block dsRNA-activated antiviral pathways. To test the prediction of this hypothesis that dsRNA is in fact produced during HCMV infection, we infected HF with HCMV[AD169] and tested for dsRNA by immunofluorescence assays using a well-characterized dsRNA-specific monoclonal antibody, J2 (11, 46, 49), as described in Materials and Methods. As shown in Fig. 3A, we detected J2 reactivity in infected cells by 24 h p.i. In contrast, we observed only a small amount of background fluorescence in mock-infected cells or in infected cells stained with an isotype-matched control antibody. Consistent with the absence of detectable dsRNA at 7 h p.i. (Fig. 3A), we did not detect any dsRNA after infection in the presence of cycloheximide (data not shown), suggesting that it is not delivered with virions or produced by IE transcription. The intensity of the immunofluorescence was not substantially altered by ganciclovir treatment, indicating that late-gene expression is not required for dsRNA production. We used RNase III and RNase A digestions to evaluate the specificity of the J2 antibody for dsRNA in this setting (Fig. 3B). RNase III eliminated most of the immunofluorescence signal, consistent with its being primarily due to the presence of dsRNA. RNase A is generally considered to be a single-strand-specific RNase, but it can digest dsRNA under low-salt conditions (38). Thus, the loss of immunofluorescence signal under low-salt but not high-salt RNase A conditions further supports the conclusion that HCMV infection generates dsRNA. A similar pattern of accumulation of dsRNA was detected in cells infected with HCMV[ΔI/ΔT] (Fig. 3C). These data demonstrate that dsRNA is produced by 24 h post-HCMV infection, and its presence may explain the requirement for the dsRBPs pTRS1 and pIRS1 for HCMV replication.
FIG. 3.

dsRNA production during HCMV infection. HF were mock infected or infected with HCMV at an MOI of 5 without (A and B) or with (C) spin inoculation and stained with the dsRNA-specific J2 antibody or an isotype control antibody, as described in Materials and Methods. Nuclei were stained using Hoechst no. 33342. (A) HCMV[AD169]-infected HF were stained using J2 at 7, 24, or 48 h p.i. (top rows) or an isotype control antibody (bottom row). Mock-infected and infected cells were also treated with ganciclovir, preventing viral late protein synthesis, and stained at 48 h p.i. (right). (B) HF infected with HCMV[AD169] for 48 h were digested with dsRNA-specific RNase III (left) or RNase A under low-salt (center) and high-salt (right) conditions before being stained with J2. RNase A is specific for single-stranded RNA under high-salt conditions (38). (C) HF were mock infected or infected with HCMV[AD169] (center) or HCMV[ΔI/ΔT] (right) and stained with J2 at 48 h p.i.
Protein synthesis shutdown during HCMV[ΔI/ΔT] infection.
Since dsRNA is produced during HCMV infection, we next investigated the hypothesis that the severe growth defect of HCMV[ΔI/ΔT] might be due to unopposed activation of dsRNA-activated pathways, resulting in inhibition of global protein synthesis. Therefore, we infected HF at an MOI of 3 and labeled newly made proteins with [35S]methionine at several time points (Fig. 4). We detected a nearly complete shutdown of protein synthesis between 24 and 48 h p.i. in HF infected with HCMV[ΔI/ΔT]. In contrast, protein synthesis was maintained in cells infected with HCMV[AD169], as well as HCMV[TRS1-HA], albeit at somewhat lower levels than in mock-infected cells. Additionally, protein synthesis continued in HF infected with HCMV[E3L] at levels similar to those infected with HCMV[AD169] (data not shown).
FIG. 4.

Protein synthesis in HF infected with HCMV mutants. HF were mock infected or infected with the indicated virus (MOI = 3) and labeled for 1 h with [35S]methionine at the indicated times p.i. Cell extracts were prepared as described in Materials and Methods and analyzed by SDS-PAGE and autoradiography. Molecular mass markers (kDa) are shown on the left.
Although we observed a generalized shutoff of protein synthesis in HCMV[ΔI/ΔT]-infected cells, it remained possible that viral protein synthesis was relatively preserved. Many other viruses repress overall protein synthesis but divert the remaining translational capacity toward making viral proteins at the expense of cellular proteins. Therefore, we analyzed expression of a subset of HCMV proteins of differing kinetic classes after infection with our wild-type and mutant viruses (Fig. 5). First, we monitored viral entry by assessing the intracellular abundance of the tegument protein pp65 by immunoblot assay. Similar to the results shown in Fig. 1D, all infected cells contained similar levels of pp65 at 2 h p.i., suggesting that each of the viruses, including HCMV[ΔI/ΔT], entered cells with comparable efficiencies (Fig. 5A). The abundance of the IE transcriptional activator IE1 (pUL123) was slightly lower in cells infected with HCMV[ΔI/ΔT] than in those infected with HCMV[AD169] and HCMV[TRS1-HA] at 24 h p.i. and remained lower at 48 h p.i. Consistent with the major reduction in overall protein synthesis between 24 and 48 h (Fig. 4), during which time early-late proteins accumulate, we detected substantially less of the early-late protein pUL44 in cells infected with HCMV[ΔI/ΔT] at 48 h but not at 24 h p.i. Also, note that pp65 accumulation was markedly reduced by 48 h p.i. with HCMV[ΔI/ΔT] (Fig. 1D). Thus, synthesis of both viral and cellular proteins is markedly inhibited when neither pIRS1 nor pTRS1 is present, consistent with there being a critical role for these dsRBPs in maintaining protein synthesis during HCMV infection.
FIG. 5.
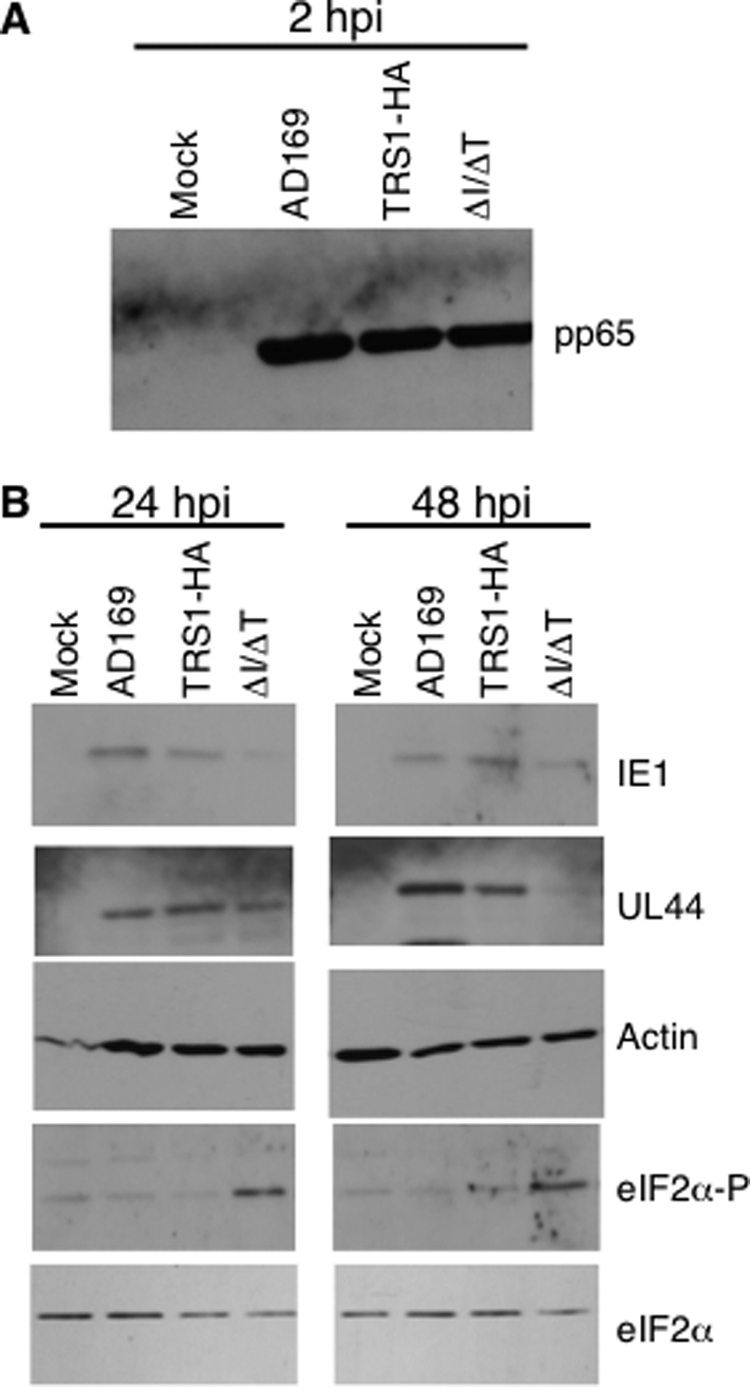
Specific protein synthesis is affected during HCMV[ΔI/ΔT] infection. HF were mock infected or infected (MOI = 3) with HCMV[AD169], HCMV[TRS1-HA], or HCMV[ΔI/ΔT], and lysates were prepared at 2, 24, and 48 h p.i., as shown, and analyzed by immunoblotting. (A) To confirm viral entry, lysates were analyzed at 2 h p.i. using pp65. (B) Levels of other viral and cellular proteins were analyzed at 24 and 48 h p.i. eIF2α-P, phosphorylated eIF2α.
Phosphorylation of eIF2α during HCMV[ΔI/ΔT] infection.
The shutoff of protein synthesis after HCMV[ΔIΔT] infection could be the result of dsRNA activation of the PKR pathway, the OAS pathway, or both. In the context of VV infection, pTRS1 and pIRS1 are capable of blocking both pathways (17), but those observations do not reveal whether both, one, or neither of the pathways is targeted in the context of HCMV infection.
We first investigated whether HCMV[ΔI/ΔT] infection activates the PKR pathway by monitoring the level of eIF2α phosphorylation. By immunoblot assay, we observed increased levels of eIF2α phosphorylation in cells infected with HCMV[ΔI/ΔT] by 24 h p.i. and continuing until at least 48 h p.i. (Fig. 5). The levels of phospho-eIF2α in HCMV[AD169]- and HCMV[TRS1-HA]-infected cells were similar to those in mock-infected cells, and total eIF2α levels were similar in all lysates. The phosphorylation of eIF2α by 24 h p.i. in HCMV[ΔI/ΔT]-infected cells is consistent with the shutoff of protein synthesis we observed beginning between 24 and 48 h p.i. Thus, is seems likely that eIF2α phosphorylation contributes to the shutdown of translation after infection with HCMV[ΔI/ΔT] and that pTRS1 is able to prevent this outcome.
Prevention of RNase L-mediated RNA degradation does not require pTRS1 expression.
The detection of elevated levels of eIF2α phosphorylation does not exclude a possible contributory role for the OAS/RNase L pathway in the shutoff of protein synthesis in cells infected with HCMV[ΔI/ΔT]. Infection with VVΔE3L results in activation of RNase L, and coinfection with HCMV or expression of pE3L, pTRS1, or pIRS1 eliminates this phenotype (17, 19), thus revealing that these HCMV dsRBPs are able to block activation of the OAS/RNase L pathway.
To evaluate the role of RNase L in the protein-synthetic defect in cells infected with HCMV[ΔI/ΔT], we assessed the integrity of the 18S rRNA by Northern blot assay of RNA isolated from mock-infected and virus-infected cells (Fig. 6). Consistent with previous results using HCMV Towne (19), 18S rRNA remained intact in mock-infected cells and cells infected for 48 h with HCMV[AD169], HCMV[TRS1-HA], and HCMV[E3L]. Interestingly, the 18S rRNA also remained intact in HCMV[ΔI/ΔT]-infected cells. Thus, at a time when overall protein synthesis is markedly inhibited due to lack of TRS1 and IRS1 and when dsRNA has been produced, RNase L remains inactive.
FIG. 6.
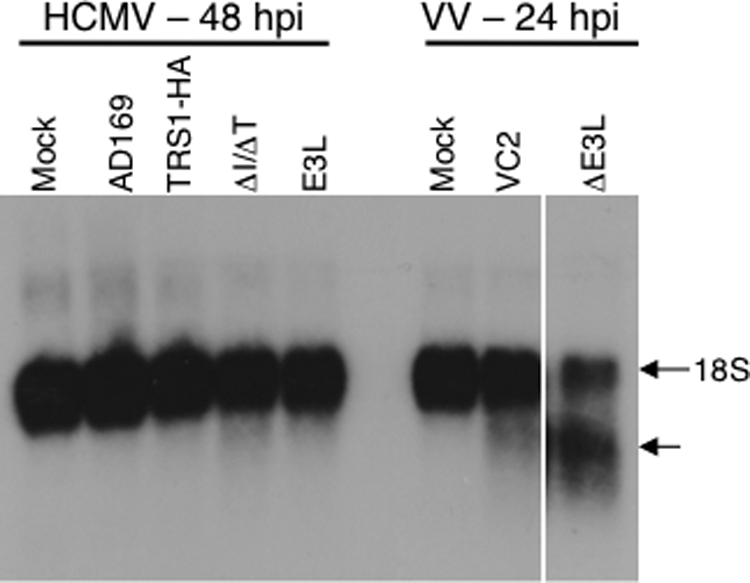
RNase L is not activated during HCMV infection. HF were mock infected or infected with HCMV (MOI = 5) or VV (MOI = 2). At 24 h p.i. (VV) or 48 h p.i. (HCMV), RNA was harvested, and 1 μg of each sample was analyzed by Northern blot hybridization using a 32P-labeled 18S rRNA-specific probe.
DISCUSSION
To counteract viral infection, cells activate a variety of antiviral responses. One of the major arms of the innate immune response is the interferon system, induction of which results in the expression of multiple antiviral genes and creation of an environment hostile to viral replication. Two of these antiviral genes, PKR and OAS, produce proteins that are activated by binding to dsRNA and trigger pathways that shut down general protein synthesis and viral replication. Since viral replication depends on continued protein synthesis, many viruses have evolved genes that inhibit the PKR and OAS pathways. In fact, these viral evasion factors appear to have exerted intense evolutionary pressure on the host antiviral genes, as illustrated by a remarkably strong signature of positive selection in the PKR gene among vertebrates (22, 42).
Viral genes involved in evading innate immune responses are known to target a variety of steps in these pathways. In the case of the PKR pathway, one common viral strategy is the expression of proteins that bind to dsRNA and to PKR and thereby interfere with PKR homodimerization or kinase activity. Among herpesviruses, the Us11 gene of herpes simplex virus type 1 (HSV-1) (39, 40), TRS1 and IRS1 of HCMV (25, 26), and the combination of m142 and m143 in MCMV (11, 16, 18) act in this fashion. Less is known about viral inhibitors of the OAS pathway, but direct binding to dsRNA and OAS may be important, at least for HSV Us11 function (43).
That HCMV IRS1 and TRS1 share several functions is not surprising, since they are found in the inverted repeats flanking the unique short region of the genome and are identical over their N-terminal 549 codons. The C-terminal regions diverge, but even these regions are ∼35% identical. The first reported activity of these genes was the ability of each to act as a coactivator of reporter gene expression in transfection assays when combined with transcriptional activators IE1 and IE2 (41, 45). We showed that their dsRNA-binding activities map to a region within the identical N-termini (25), with PKR binding dependent on divergent C-terminal sequences (26). Each protein binds to PKR, blocks activation of the PKR and OAS/RNase L pathways resulting from infection by VVΔE3L, and causes relocalization of PKR to the nucleus (17, 26). Both genes can also rescue replication of VVΔE3L and of HSV-1 lacking γ34.5 (13).
In contrast to these shared activities, TRS1 and IRS1 have been reported to have properties distinct from each other. Romanowski and Shenk described a carboxy-terminal 263-amino-acid product of IRS1 that represses the reporter gene-activating function (41). Deletion of TRS1 attenuates viral growth, particularly during low-MOI infection, a phenotype associated with a late assembly defect (1, 6); this phenotype is not detected with IRS1 deletion viruses. Other than these differences, the significance of HCMV containing two highly related genes with many redundant functions is not yet known.
We hypothesized that the ability of HCMV containing single deletions of either IRS1 or TRS1 to replicate without any apparent defects in protein-synthetic capacity might be due to the ability of the remaining member of the pair to block one or more of the dsRNA-activated pathways. To test this hypothesis, we constructed a virus lacking both proteins. After our initial attempts to produce HCMV[ΔI/ΔT] from the corresponding BAC by transfection into normal HF or complementing HF transduced with a retrovirus expressing TRS1 under the control of the SV40 promoter were unsuccessful, we generated an HF line transduced with a retroviral vector in which the strong HCMV 2.7β promoter regulated TRS1 expression, hoping that we might avoid potential toxicity from constitutive expression of pTRS1 but achieve a high level of induction of TRS1 in infected cells. Although this strategy proved to be successful for reconstituting HCMV[ΔI/ΔT], immunoblot assays suggested that the level of pTRS1 expression in the 2.7β-TRS1 cells was not substantially different from that in the SV40-TRS1 cells (data not shown). Thus, our data are inconclusive about whether use of the 2.7β promoter might be a generally applicable tool for complementing HCMV mutants in trans.
The complementing cells, HF-TRS1, supported the replication of HCMV[ΔI/ΔT] but did not completely rescue its replication to wild-type levels. HF-TRS1 cells infected with HCMV[ΔI/ΔT] synthesized proteins at a rate similar to those infected with HCVM[AD169] at least as late as 72 h p.i. (data not shown), suggesting that the pTRS1 produced in the HF-TRS1 was sufficient to prevent the shutdown of protein synthesis at least up to that time point. We suspect that altered abundance, kinetics of expression, and/or subcellular localization of pTRS1 in the infected complementing cells is not optimal for the virion assembly function of pTRS1.
HCMV[ΔI/ΔT] does not replicate at all in wild-type HF, likely as a result of the profound shutoff of protein synthesis that is most evident in comparisons of metabolically labeled cells at 24 and 48 h p.i. (Fig. 4). This timing is quite consistent with our observation that dsRNA accumulates and eIF2α becomes phosphorylated in HCMV-infected cells by 24 h p.i. (Fig. 3 and 5). A similar pattern of dsRNA accumulation and eIF2α phosphorylation occurs during infection with MCMV lacking either m142 or m143 or both (11, 48). Although the nature of the dsRNAs expressed during infections by these and other large DNA viruses is not yet known, it has been postulated that polyadenylation at the normal sites becomes inefficient at late times p.i. and results in long transcripts from both strands of the genome that can anneal and trigger dsRNA-activated pathways (reviewed in reference 29). Our data are consistent with this idea, except that we found that the accumulation of dsRNA occurs even in the presence of ganciclovir, suggesting that late-gene expression is not essential. Alternatively, in the case of viruses like HCMV that have GC-rich genomes, normal viral transcripts may assume enough secondary structure to mimic dsRNA and activate the antiviral response pathways.
Our analyses of HCMV[E3L] bolster the conclusion that the requirement that HCMV contain either TRS1 or IRS1 is explained by the need to block cytoplasmic dsRNA-activated antiviral response pathways. The observation that HCMV lacking TRS1 but containing IRS1 has only a modest replication defect (6) might be explained by pIRS1 being able to complement in part an essential role for pTRS1 in virion assembly. However, it seems quite unlikely that pE3L functions directly in HCMV assembly. Rather, the well-established role of pE3L in blocking dsRNA-activated pathways is the more plausible explanation for its rescuing replication of HCMV[ΔI/ΔT]. In previous studies, we showed that pIRS1 and pTRS1, but not pE3L, cause PKR accumulation in the nucleus (26). The ability of HCMV[E3L] to replicate quite efficiently therefore suggests that nuclear accumulation of PKR is not critical for HCMV replication, although additional experiments need to be done to evaluate the localization of PKR after infection with HCMV[E3L].
Beside the PKR-mediated response, another major arm of the dsRNA-mediated antiviral response is the activation of OAS, leading to activation of RNase L and subsequent cleavage of mRNA and rRNA. OAS is strongly induced immediately upon HCMV infection; in fact, binding of the viral envelope protein glycoprotein B is sufficient to induce OAS expression (8). Since wild-type HCMV infection did not result in RNase L activation and pTRS1 or pIRS1 is able to block RNase L activation by VVΔE3L infection, we expected that blocking the OAS pathway might be another important function of pTRS1 and pIRS1. However, our data demonstrate that even after infection with HCMV[ΔI/ΔT], RNase L is not activated. This result might be due to expression of another inhibitor of the pathway. Other members of the US22 gene family bind dsRNA (reference 18 and unpublished data), and one of them might block OAS/RNase L activation during HCMV[ΔI/ΔT] infection. Similarly, MCMV infection does not result in RNase L activation, regardless of whether it contains its dsRBP genes, m142 and m143, suggesting that the dsRNAs produced during infection by CMVs may not be sufficiently abundant or correctly localized to activate this pathway (11). Although TRS1 and IRS1 appear not to be needed to block RNase L under the conditions we used here, it remains possible that their ability to block the pathway plays a role in other cell types or under other conditions.
Many viruses encode multiple proteins for evading the dsRNA-mediated antiviral response, but HCMV is unusual in that the genes appear to be redundant in this respect. In the most closely related virus studied thus far, MCMV, two genes are also present, but they act together, and deletion of either one blocks viral replication (18, 48). VV and HSV-1 each also have at least two genes involved in evading the dsRNA responses, but in both cases, the genes act at different steps in the PKR pathway (36). In the case of VV, the two genes may be functionally important in alternative cell types (33). Regardless, our research has now established that HCMV depends on having at least one of two dsRNA-binding, PKR-binding proteins that can prevent eIF2α phosphorylation and maintain protein synthesis. By analogy to genes with similar functions in other viruses, it is very likely that these proteins play a vital role in pathogenesis during infection of humans, and understanding the mechanism may have implications for new antiviral strategies and vaccine design.
Supplementary Material
Acknowledgments
We thank Katherine DeNiro, Morgan Hakki, and the Genomics Core Facility of the Fred Hutchinson Cancer Research Center for technical assistance. We are grateful to Bonita Biegalke (Ohio University), Jon Cooper (FHCRC), Dusty Miller (FHCRC), Martin Messerle (MHH Hannover), and Mariano Esteban (CNB Madrid) for plasmids and to Bertram Jacobs (Arizona State University) for vaccinia viruses.
This work was supported by NIH grant AI26672 (to A.P.G.) and DFG grant BR 1730/3-1 (to W.B.). E.E.M. was supported by NIH grant T32 CA09929.
Footnotes
Published ahead of print on 11 February 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adamo, J. E., J. Schroer, and T. Shenk. 2004. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J. Virol. 7810221-10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atalay, R., A. Zimmermann, M. Wagner, E. Borst, C. Benz, M. Messerle, and H. Hengel. 2002. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcγ receptor homologs. J. Virol. 768596-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldick, C. J., Jr., A. Marchini, C. E. Patterson, and T. Shenk. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 714400-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beattie, E., K. L. Denzler, J. Tartaglia, M. E. Perkus, E. Paoletti, and B. L. Jacobs. 1995. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 69499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beattie, E., E. B. Kauffman, H. Martinez, M. E. Perkus, B. L. Jacobs, E. Paoletti, and J. Tartaglia. 1996. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 1289-94. [DOI] [PubMed] [Google Scholar]
- 6.Blankenship, C. A., and T. Shenk. 2002. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J. Virol. 7612290-12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borst, E. M., and M. Messerle. 2005. Analysis of human cytomegalovirus oriLyt sequence requirements in the context of the viral genome. J. Virol. 793615-3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle, K. A., R. L. Pietropaolo, and T. Compton. 1999. Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol. Cell. Biol. 193607-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandt, T. A., and B. L. Jacobs. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 75850-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brune, W., M. Nevels, and T. Shenk. 2003. Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J. Virol. 7711633-11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budt, M., L. Niederstadt, R. S. Valchanova, S. Jonjic, and W. Brune. 2009. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J. Virol. 831260-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cassady, K. A. 2005. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 798707-8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cassady, K. A., M. Gross, and B. Roizman. 1998. The herpes simplex virus US11 protein effectively compensates for the γ1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 728620-8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang, H. W., J. C. Watson, and B. L. Jacobs. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 894825-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1589-14. [DOI] [PubMed] [Google Scholar]
- 16.Child, S. J., and A. P. Geballe. 2009. Binding and relocalization of PKR by murine cytomegalovirus. J. Virol. 831790-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Child, S. J., M. Hakki, K. L. De Niro, and A. P. Geballe. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78197-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Child, S. J., L. K. Hanson, C. E. Brown, D. M. Janzen, and A. P. Geballe. 2006. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J. Virol. 8010173-10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Child, S. J., S. Jarrahian, V. M. Harper, and A. P. Geballe. 2002. Complementation of vaccinia virus lacking the double-stranded RNA-binding protein gene E3L by human cytomegalovirus. J. Virol. 764912-4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dever, T. E., A. C. Dar, and F. Sicheri. 2007. The eIF2α kinases, p. 319-344. In M. B. Matthews, N. Sonenberg, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 21.Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 10014223-14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elde, N. C., S. J. Child, A. P. Geballe, and H. Malik. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457485-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia, M. A., S. Guerra, J. Gil, V. Jimenez, and M. Esteban. 2002. Anti-apoptotic and oncogenic properties of the dsRNA-binding protein of vaccinia virus, E3L. Oncogene 218379-8387. [DOI] [PubMed] [Google Scholar]
- 24.Geballe, A. P., and E. S. Mocarski. 1988. Translational control of cytomegalovirus gene expression is mediated by upstream AUG codons. J. Virol. 623334-3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakki, M., and A. P. Geballe. 2005. Double-stranded RNA binding by human cytomegalovirus pTRS1. J. Virol. 797311-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hakki, M., E. E. Marshall, K. L. De Niro, and A. P. Geballe. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 8011817-11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobom, U., W. Brune, M. Messerle, G. Hahn, and U. H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 747720-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobs, B. L. 2000. Translational control in poxvirus-infected cells, p. 951-971. In W. B. Hershey, M. B. Matthews, and N. Sonenberg (ed.), Translational control. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 29.Jacobs, B. L., and J. O. Langland. 1996. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 219339-349. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs, B. L., J. O. Langland, and T. Brandt. 1998. Characterization of viral double-stranded RNA-binding proteins. Methods 15225-232. [DOI] [PubMed] [Google Scholar]
- 31.Jones, T. R., and V. P. Muzithras. 1992. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J. Virol. 662541-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langland, J. O., J. M. Cameron, M. C. Heck, J. K. Jancovich, and B. L. Jacobs. 2006. Inhibition of PKR by RNA and DNA viruses. Virus Res. 119100-110. [DOI] [PubMed] [Google Scholar]
- 33.Langland, J. O., and B. L. Jacobs. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299133-141. [DOI] [PubMed] [Google Scholar]
- 34.Miller, A. D., J. V. Garcia, N. von Suhr, C. M. Lynch, C. Wilson, and M. V. Eiden. 1991. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J. Virol. 652220-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller, A. D., and G. J. Rosman. 1989. Improved retroviral vectors for gene transfer and expression. BioTechniques 7980-982, 984-986, 989-990. [PMC free article] [PubMed] [Google Scholar]
- 36.Mohr, I. J., T. Pe'ery, and M. B. Mathews. 2007. Protein synthesis and translational control during viral infection, p. 545-599. In M. B. Matthews, N. Sonenberg, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 37.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272263-267. [DOI] [PubMed] [Google Scholar]
- 38.Nichols, N. M., and D. Yue. 2008. Ribonucleases, p. 3.13.1-3.13.8. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current topics in molecular biology. John Wiley and Sons, New York, NY.
- 39.Peters, G. A., D. Khoo, I. Mohr, and G. C. Sen. 2002. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 7611054-11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poppers, J., M. Mulvey, D. Khoo, and I. Mohr. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 7411215-11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romanowski, M. J., and T. Shenk. 1997. Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J. Virol. 711485-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rothenburg, S., E. J. Seo, J. S. Gil, T. E. Dever, and K. Dittmar. 2009. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 1663-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanchez, R., and I. Mohr. 2007. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 813455-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverman, R. H. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 8112720-12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stasiak, P. C., and E. S. Mocarski. 1992. Transactivation of the cytomegalovirus ICP36 gene promoter requires the alpha gene product TRS1 in addition to IE1 and IE2. J. Virol. 661050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Targett-Adams, P., S. Boulant, and J. McLauchlan. 2008. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 822182-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tartaglia, J., M. E. Perkus, J. Taylor, E. K. Norton, J. C. Audonnet, W. I. Cox, S. W. Davis, J. van der Hoeven, B. Meignier, M. Riviere, et al. 1992. NYVAC: a highly attenuated strain of vaccinia virus. Virology 188217-232. [DOI] [PubMed] [Google Scholar]
- 48.Valchanova, R. S., M. Picard-Maureau, M. Budt, and W. Brune. 2006. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 8010181-10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber, F., V. Wagner, S. B. Rasmussen, R. Hartmann, and S. R. Paludan. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 805059-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitaker-Dowling, P., and J. S. Youngner. 1984. Characterization of a specific kinase inhibitory factor produced by vaccinia virus which inhibits the interferon-induced protein kinase. Virology 137171-181. [DOI] [PubMed] [Google Scholar]
- 51.Xiang, Y., R. C. Condit, S. Vijaysri, B. Jacobs, B. R. Williams, and R. H. Silverman. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 765251-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.