

Abstract
von Willebrand factor (VWF) performs its hemostatic functions through binding to various proteins. The A1 domain of VWF contains binding sites of not only physiologically important ligands, but also exogenous modulators that induce VWF-platelet aggregation. Sulfatides, 3-sulfated galactosyl ceramides, that are expressed on oligodendrocytes, renal tubular cells, certain tumor cells and platelets, have been suggested to interact with VWF under some pathological conditions. The binding of VWF to sulfatide requires the A1 domain, but its binding sites have not been precisely identified. Here, we report that alanine mutations at Arg1392, Arg1395, Arg1399 and Lys1423 led to decreased VWF–sulfatide binding. These sites have been reported to be the binding sites for platelet membrane glycoprotein (GP) Ib and/or snake venom botrocetin, and, interestingly, are identical to the monoclonal antibody (mAb) NMC4 epitope previously reported to inhibit the VWF-GPIb interaction. We observed that NMC4 also inhibited VWF interaction with sulfatides in a dose-dependent manner. Thus, we conclude that VWF binding sites of sulfatide overlap those of platelet GPIb and botrocetin.
Keywords: von Willebrand factor, A1 domain, Sulfatide, Alanine scanning mutagenesis
Introduction
von Willebrand factor (VWF) is a multimeric glycoprotein that plays a pivotal role in primary haemostasis. VWF circulates in the blood as a multimer with heterologous molecular weights, assembled from subunits of ∼250 kDa. The multimer sizes range from dimer of ∼500 to >10,000 kDa. Mature VWF consists of 12 domains, which are arranged in the following sequence: D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. The D′ and D3 domains adhere to and stabilize blood coagulation factor VIII [1]. Activated platelet integrin αIIbβ3 binds to VWF through a segment that includes the tri-peptide sequence Arg-Gly-Asp (RGD) in the VWF domain C1 [2]. Domain A3 has been shown to interact with collagen in a variety of different experimental models [3–5]. Particular interest has been focused on the A1 domain, since it contains binding sites for several molecules such as platelet surface glycoprotein (GP) Ibα [6, 7]. Exogenous materials such as snake venom protein botrocetin [7] and bitiscetin [8] also bind to the VWF A1 domain. The A1 domain is the major VWF binding site for heparin [9, 10] and unique sulfated glycolipid sulfatides [11–13].
Sulfatides are a group of cerebroside 3-sulfates, mainly comprising 3-sulfate esters of galactosylcerebrosides (galactosyl-3-sulfate esters) and found in mammalian tissues as the corresponding cerebroside group. Sulfatide is a major constituent of brain lipids and is found in trace amounts in other tissues. It is an essential glycosphingolipid in the peripheral as well as central nervous system, constituting 4–6 mole% of the total lipids in adult brain myelin. It is expressed on the surface of several types of cells, such as oligodendrocytes, renal tubular cells, certain tumor cells,and platelets. Diverse biological processes are mediated by sulfatide, including the regulation of cell growth protein trafficking, signal transduction, cell adhesion, neuronal plasticity, morphogenesis and thrombogenesis [14–17]. Sulfated glycolipids are also constituents of normal erythrocytes, but are not normally exposed; however, in sickle cell anemia, reversible sickling and oxidant damage to the membrane could modify and/or expose these molecules to the surface [18, 19]. The interaction between VWF and exposed sulfatide may cause periodic vaso-occlusion, which is the severest manifestation of sickle cell anemia [20].
Based on the study on the binding of dispase-cleaved VWF fragments, a sulfatide-binding site in the VWF A1 domain was localized between amino acids (AA) 1275 and 1436 [12]. Later, in 1995, it was reported that a synthetic peptide spanning AA1391–1409 inhibited VWF binding to bovine brain sulfatides [21]. To date, the precise binding sites have not been determined and are controversial to some measure. The aim of our study is to identify amino acids responsible for binding to sulfatides, by the alanine-scanning mutagenesis of VWF A1 domain, which has been previously adopted to identify those of GPIb, botrocetin, bitiscetin and heparin [7, 8, 10]. Here, we report that four amino acids in the A1 domain are important for sulfatide binding. Our findings are confirmed by monoclonal antibody (mAb) inhibition study that blocks the VWF A1- GPIb binding.
Methods
Materials
mAb NMC-4, which recognizes the A1 domain of mature human VWF, was provided by Dr. Midori Shima (Nara Medical University, Japan). Anti-human VWF mAb B701, which has been reported to inhibit the VWF–sulfatide interaction [12, 22], was a kind gift from Dr. Dominique Meyer (INSERM University, France). Anti-human VWF mAb 33E12 was generously provided by Dr. Claudine Mazurier (CRTS, Lille, France). Anti-human VWF polyclonal antibody (pAb) D082 and peroxidase-conjugated anti-human VWF antibody P0226 were purchased from Dako (Glostrup, Denmark). We purchased sulfatide (cerebroside sulfate from the bovine brain), O-phenylene diamine (OPD), bovine serum albumin (BSA) and Tween-20 from Sigma (St Louis, MO, USA). U-bottomed ELISA plates were obtained from Nunc (Cambridge, MA, USA).
Plasmid constructs
The VWF segment targeted for mutagenesis consists of a section of 254 amino acid residues between His1226 and Gly1479 that contains the A1 domain and part of the D3 domain of mature human VWF. All 68 charged amino acids including arginine, lysine, aspartate, glutamate and histidine were changed singly or in small clusters to alanine. They were covered in a total of 49 constructs (Fig. 1). An expression plasmid, pSVvWF1.1, contains the full-length coding sequence of human VWF. Numbering of previously produced mutants started as +1 from the N-terminal Ser of the mature, processed VWF subunit after cleavage of the prepropeptide; thus, we renumbered all the mutants, adding 763 to the old name [23] used in the previous publications [7]. The names of clustered mutants are abbreviated with the range of residue numbers and the number of alanine substitutions [23].
Fig. 1.

Amino acid residues of human VWF targeted for charged-to-alanine mutagenesis. The amino acid sequence shown includes a part of domain D3 (1226–1259) and the entire A1 domain (1260–1479). The secondary structural elements of the A1 domain, as determined by Celikel et al. [26], are indicated below the sequence (α, α-helix; β, β-strand). Charged residues of His, Arg, Lys, Glu and Asp were targeted for mutagenesis and are shown by characters in bold. A total of 68 residues are covered by clustered or single mutants. A grey box surrounds each construct. Every clustered mutant is underlined. For example, construct 1226-HCD-1228 is named (1226–1228) 2A, containing alanine substitutions at two charged residues, His1226 and Asp1228, while E1290A represents the mutant Glu1290 that was changed to Ala
Expression of recombinant VWF
The preparation of recombinant VWF (rVWF) was described previously [24]. Briefly, human 293T cells, growing in Dulbecco’s modified Eagle’s medium (Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco-BRL), were transfected by the lipofection method using TransFast transfection kit (Promega, Madison, MI, USA) according to the manufacture’s instructions. After 24 h, the cells were washed with phosphate-buffered saline (PBS, Gibco), and then incubated with serum-free medium (Optimem-1, Gibco). After 48 h, rVWF secreted in the medium was collected and concentrated using Centriprep-30 and Centricon-100 devices (Millipore, Bedford, MA, USA). The amounts of rVWF were measured by ELISA, as described elsewhere [24]. VWF multimer analysis of the mutants was previously performed [7, 23]. Concentrated rVWF was diluted with PBS containing 5% BSA (PBSA) and PBS, adjusted to 1 μg/ml (the final BSA concentration was 1%). Diluted rVWF was stored at −20°C in aliquots until use.
Sulfatide-binding assay
We performed this assay according to the method of Favaloro et al. [25] with some modifications. Briefly, ELISA plates were coated with 200 μl/well of sulfatide dissolved in methanol at a final concentration of 50 μg/ml and left uncovered for 2 days at 4°C to allow the methanol to evaporate. After washing three times with washing buffer (0.12 M NaCl, 0.02 M imidizole, 0.005 M citric acid, 0.1% BSA), the plates were incubated for 1 h with 200 μl of 5% PBSA to reduce non-specific background binding. The plates were re-washed twice and incubated with 35 μl of rVWF diluted with 3% PBSA (150 ng/ml, final) for 2 h at room temperature (RT). We used culture medium of 293T cells without transfection as a mock control. The plates were re-washed four times and incubated with the secondary antibody P0226 diluted in 3% PBSA (100 μl/well). After washing five times, 100 μl of OPD solution (400 μg/ml) was added followed by color development at 492 nm stopped by adding 2 N H2SO4 (50 μl/well). Independent duplicated experiments were performed three times. We confirmed the linear relationship between the optical density (OD) and concentration of added rVWF within the range of 0–200 ng/ml of rVWF (data not shown). We defined the relative percentage of sulfatide binding as follows:
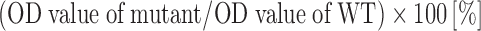 |
Epitope mapping of mAb B701
The binding assay of mAb to rVWF by ELISA has been described previously [23]. Briefly, round-bottomed ELISA plates were coated for 24 h at 4°C with 25 μl of B701, 7.5 μg/ml, in 0.1 M sodium carbonate, pH 9.6. The wells were washed with PBS containing 0.1% Tween 20 (PBS-T) and then incubated for 2 h at room temperature (RT) with 15 μl of various concentrations of wild-type or mutant rVWF diluted in 3% PBSA. The wells were washed three times in PBS-T and incubated for 1 h at RT with 20 μl of P0226 1:2000 diluted in 3% PBSA. The plates were washed three times, followed by color development of OPD solution, as described earlier. Binding of rVWF mutants was determined at a fixed concentration of rVWF (500 ng/ml) and normalized to the value obtained for wild-type rVWF. Control assays were performed with concentrated conditioned media from mock-transfected 293T cells that gave absorbance values of zero. We defined the relative binding percentage as follows.
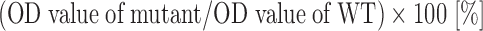 |
Effect of monoclonal antibodies on VWF binding to sulfatide
A volume of 20 μl of each mAb (NMC4, B701 and 33E12) diluted in 3% PBSA was mixed with 100 μl of rVWF (final concentrations of antibody and wild-type rVWF were 0.42–26.7 μg/ml and 182 ng/ml, respectively). After 15 min incubation at RT, 50 μl of the mixture was added to wells of sulfatide-coated ELISA plate, as described above. The wells were washed four times and incubated for 1 h at RT with P0226. The amounts of VWF bound to the ELISA plate were evaluated by measuring the OD value after adding the OPD buffer. This experiment was performed in triplicate and repeated at least twice. We defined the OD value without antibody as 100%. The inhibitory effect of antibody was expressed as bound VWF rate:
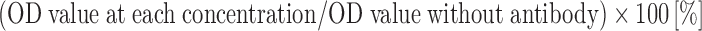 |
Statistical analysis
The significance of differences was evaluated by the Student’s t-test, and all statistical tests were two-sided.
Crystallographic structural representations
The PDB file of the VWF A1 domain [26] was downloaded from the RCSB PDB web site (http://www.rcsb.org/pdb/home/home.do), and its spherical surface plots were prepared with the program Swiss Pdb Viewer (downloadable at http://us.expasy.org/spdbv/).
Results
Sulfatide binding assay
At the start of the study of alanine scanning mutagenesis, several clustered mutants were generated, including (1290–1294)3A, (1332–1336)5A, (1359–1362)2A, 1376–1379)2A, (1392–1395)2A, (1405–1408)4A, (1419–1423)2A and (1426–1430)3A [23]; thereafter, these were replaced by the corresponding single mutants. The resulting charged-to-alanine rVWF mutant panel [7] was used for the study of VWF sulfatide binding. Mutants were expressed and secreted efficiently and the multimer distribution of all recombinant proteins was similar to that of wild-type rVWF or plasma VWF [7]. The folding of each mutant has been evaluated using a panel of conformation-dependent monoclonal antibodies [7]. Recognition of the A1 domain by these antibodies is impaired by reduction or denaturation, and mutants D1277A, D1283A, (1277–1283)2A, K1297A, (1312–1315)2A, R1315A, (1371–1374)3A and R1374A have reportedly shown markedly decreased binding to all 6 conformation-dependent mAbs, suggesting that these substitutions caused significant misfolding of the A1 domain [7]. These mutants were therefore excluded from further analysis [10].
Figure 2 indicates that 10 mutants, E1290A, R1308A, R1392A, R1395A, R1399A, Lys1405A, K1408A, H1419A, K1423A and K1430A, inhibited binding to sulfatide by more than 40% compared to wild-type rVWF. The four particular mutants, R1392A, R1395A, R1399A and K1423A, markedly inhibited binding by 68.2, 80.4, 65.3 and 66.4%, respectively, suggesting the importance of these four residues for binding to sulfatide.
Fig. 2.

Histogram of VWF binding to sulfatide. Wild- type or mutant rVWF, in comparison with the mock control (indicated on the left), was immobilized on ELISA plates coated with sulfatide. The relative sulfatide binding of each alanine mutant is shown as: (OD value of mutant/OD value of WT) × 100 (%). The results represent the means (±SD) of three independent experiments. The asterisk denotes a statistically significant difference compared to the control (P < 0.05). Relative binding of <60% is shown by the dashed line
Epitope mapping of mAb B701
mAb B701 did not bind to rVWF, lacking either the A1 or A1 + A3 domain. However, B701 bound to rVWF lacking only the A3 domain (data not shown). Therefore, B701 binds to the VWF-A1 domain, and Fig. 3 indicates that B701 lost the ability to bind to two particular mutants, K1348A and (1320–1326) 4A. The results suggest that Glu1320, His1322, Asp1323, His1326 and Lys1348 are candidates for the epitope of B701. B701 has been reported to disturb the VWF–sulfatide association [12, 22], but the epitope we identified was distinct from the candidate sulfatide binding site, Arg1392, Arg1395, Arg1399 and Lys1423, of which the mutation inhibited the binding to sulfatide (Fig. 2).
Fig. 3.

Mapping of mAb B701 epitope. The epitopes of mAb B701 were analyzed by binding each rVWF to B701-coated ELISA plates. Relative binding is shown as: (OD value of mutant/OD value of WT) × 100 [%]. The results represent the means (±SD) of three independent experiments. The asterisk denotes a statistically significant difference compared to the control (P < 0.05). Relative binding of <60% is shown by the dashed line
Effect of mAbs for sulfatide binding to VWF A1 domain
We studied the inhibitory effect of mAbs NMC4 and B701 that may interfere with VWF binding to GPIb or sulfatide. Figure 4 indicates that NMC4 blocked sulfatide binding in a dose-dependent manner, suggesting that NMC4 may interact with the sulfatide binding sites on the A1 domain. In contrast, Fig. 4 indicates that B701 did not affect the sulfatide binding. Control IgG 33E12, whose epitope is located in the VWF C-terminal region and has no effect on the interaction between the A1 domain and other proteins [23, 27], showed no effect on sulfatide binding (Fig. 4). Even higher concentrations of B701 (final: 30–120 μg/mL) did not inhibit sulfatide binding (data not shown). Taken together with the sulfatide-binding assay results of our mutants, the results of the epitope mapping experiment of B701 (Fig. 3) suggests that B701 may not interact with the VWF–sulfatide binding site. On the other hand, sulfatide-binding sites may overlap with the platelet GPIb-binding sites, and, thus, mAb NMC4 appears to interfere with not only VWF binding to GPIb, but also to sulfatide.
Fig. 4.

Inhibitory effects of mAb NMC4 and B701 on VWF–sulfatide binding. mAb NMC4, B701 and the control antibody 33E12 were tested for their inhibitiory effects on VWF–sulfatide binding. Serially diluted mAbs were mixed with a fixed concentration of WT rVWF (182 μg/ml). After incubation for 30 min, the mixture was applied to sulfatide-coated wells. The amounts of VWF bound were evaluated by measuring the OD value, as described in Sect. 2. OD values obtained in the absence of antibody were defined as 100%. The results represent the means (±SD) of two independent, triplicate experiments
Crystallographic structural representations
We addressed the locations of the putative binding sites of sulfatide and B701 in the three-dimensional structure of the VWF A1 domain as well as those of platelet GPIb and mAb NMC4. Our study indicated that four mutations at Arg1392, Arg1395, Arg1399 and Lys1423 markedly reduced the binding to sulfatide (Fig. 2). The first three are proposed GPIb-binding sites [7] located on the α4 helix (Fig. 1) and Fig. 5a shows that these residues (orange) are located close to each other on the surface of the molecule, although Lys1423 is located on the neighboring α5 helix. We then mapped several amino acid residues whose alanine mutants showed moderately decreased sulfatide binding (Fig. 2). Glu1290, Arg1308, Lys1405, Lys1408, His1419 and Lys1430 are distributed on the A1 surface, but are located in a distinct area from Arg1392, Arg1395 and Arg1399 (Fig. 5).
Fig. 5.

Location of amino acid residues whose mutants showed decreased binding to sulfatide, and epitopes of botrocetin, mAb NMC4 and B701 in VWF A1. A spherical surface model of the
VWF A1 domain is shown based on the coordinates reported by Emsley et al. [36]. Orange indicates residues whose alanine mutants showed markedly reduced sulfatide binding. Sky blue represents residues whose mutants showed moderately reduced binding to sulfatide. Mutation at Lys1362 (red) led to reduced GPIb binding, with normal binding to botrocetin, suggesting it to be the key residue in VWF-GPIb binding. Yellow indicates charged amino acid residues within residues Glu1320-His1326. Mutations at 1320–1326 and Lys1348 (purple) led to significant decreases in binding to mAb B701. a Surface representation of VWF A1 domain from the front. b Orientations from back. c Top view of the VWF A1 domain. The orientation of the front and behind is indicated. d The bottom view of the VWF A1 domain. Orientations from the front and back are indicated. The coloring of amino acids is the same in all four panels. In panel c, yellow indicates charged residues within Glu1320-His1326. Arrows show the binding sites of mAb NMC4, B724, B701, botrocetin and sulfatide
The epitopes of NMC4 are included in Arg1392, Arg1395 and Arg1399 [26], and are identical to the GPIb binding site [7]. It is thus highly likely that NMC4 binding to the A1 GPIb binding site results in interfering with the binding to sulfatide.
Lys1348 and four charged residues: Glu1320, His1322, Asp1323 and His1326 (yellow) between 1320 and 1326, which represent the B701 epitope, appear to be located near each other, but on the opposite site of the GPIb- and sulfatide-binding sites (Fig. 5).
Discussion
The current experiment indicated that ten alanine mutants showed decreased sulfatide-binding, and four novel mutations at Arg1392, Arg1395, Arg1399 and Lys1423 led to a marked reduction (Fig. 2). Our previous studies indicated that these mutants underwent no significant conformational changes [7]. It has been shown that Lys1362, Arg1392 and Arg1395 are important for binding to GPIb, among which Lys1362 is located at the center of the binding surfaces [7]. mAb NMC4 inhibits both ristocetin- and botrocetin-induced VWF binding to platelet GPIb [28], and the crystallographic structure has revealed that NMC4 interacts directly with the amino acid side chains of Arg1392, Arg1395 and Arg1399 in the VWF A1 domain [26], thus these sites are its epitope. Also, NMC4 effectively inhibited the sulfatide–VWF interaction (Fig. 4), suggesting the importance of these residues for binding not only to GPIb but also to sulfatide. This observation is supported by recent findings that sulfatide inhibits the binding of beads covalently coupled to the fragment of GPIbα to the VWF A1 domain [13].
A previous peptide inhibition study suggested that sulfatide binding sites were localized between amino acids, Lys1332 and Lys1348 [12]. However, our 7 alanine mutants between K1332A and K1348A showed normal sulfatide binding (Fig. 2). It is possible that the synthetic peptide interacted with other surface regions around the sulfatide binding sites.
It has been also indicated that mAb B724 effectively inhibited VWF–sulfatide interaction [12]. We previously showed that a mutation at Lys1423 did not decrease binding to GPIb, but inhibited binding to B724 [7]. In accordance with the previous observation that B724 inhibited VWF- binding to botrocetin [29], it is suggested that Lys1423 is important not only for binding to botrocetin, but also to sulfatide. The 3D structure of the VWF-A1 domain reveals that the three residues, Arg1392, Arg1395 and Arg1399, are localized as a cluster (Fig. 5), proposing their simultaneous accessibility to sulfatide. Lys1423, located in the α5 helix, is isolated from the former three residues, although a further study may be required to verify the cooperative role of this amino acid residue.
Our study also identified the epitope of mAb 701. Two mutants, (1320–1326) 4A and K1348A, showed significant decrease in binding to B701 (Fig. 3), and residues Glu1320, His1322 Asp1323, His1326 and Lys1348 are localized in close proximity (Fig. 5c), suggesting that they include the B701 epitope. The epitope is, however, located on the opposite site of the GPIb- and sulfatide- binding site (Fig. 5c), which is consistent with our observation that B701 showed no effects on the sulfatide–VWF interaction (Fig. 4).
The effects of sulfatides on thrombogenesis have been evaluated for many years, but still remain controversial [30]. The recent findings of Borthaker et al. showed that sulfatide inhibits the binding of beads covalently coupled to the fragment of GPIbα to the VWF-A1 domain [13]. The binding of VWF to GPIb requires conformational change of VWF, induced by shear stress under irregular blood flow [24, 31]. Therefore, it is likely that VWF–sulfatide binding competes with VWF–GPIb binding, and that sulfatides possess antithrombotic properties under some pathological conditions, such as sites of arteriosclerosis or vascular injury where the VWF–GPIb interaction initiates thrombus formation [32]. However, interestingly, administration of sulfatides into a rat deep vein thrombosis model in which the inferior vena cava was ligated led to enhanced thrombus formation compared to the control, which received an injection of the vehicle alone [33]. Our result and those of other studies showed that sulfatides could bind to intact VWF (with no conformational change) [11, 25]. Thus, sulfatides may induce thrombus formation through binding to intact VWF under conditions whereby blood flow is congestive, in which VWF–GPIb does not play a pivotal role in thrombus formation [34, 35]. Our in vitro experiments revealed that mAb NMC4 inhibited the VWF–sulfatide association in a dose-dependent manner (Fig. 4), suggesting the possibility that the in vivo neutralization of VWF-dependent adhesion could lead to a possible intervention treatment to prevent vaso-occlusion under such condition.
Acknowledgments
We are grateful to Ms. Chika Wakamatsu for performing excellent, skilled techniques in molecular biology. Particular gratitude is expressed to Dr. Dominique Meyer for providing information on mAb 701 and B724. We also thank Dr. Wayne Albers for his technical advice regarding the 3D structure, and are grateful to Dr. Sashi Kesavapany for the helpful suggestions.
References
- 1.Foster PA, Fulcher CA, Marti T, Titani K, Zimmerman TS. A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J Biol Chem. 1987;262:8443–6. [PubMed] [Google Scholar]
- 2.Fujimoto T, Ohara S, Hawiger J. Thrombin-induced exposure and prostacyclin inhibition of the receptor for factor VIII/von Willebrand factor on human platelets. J Clin Invest. 1982;69:1212–22. doi: 10.1172/JCI110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth GJ, Titani K, Hoyer LW, Hickey MJ. Localization of binding sites within human von Willebrand factor for monomeric type III collagen. Biochemistry. 1986;25:8357–61. doi: 10.1021/bi00374a004. [DOI] [PubMed] [Google Scholar]
- 4.Kalafatis M, Takahashi Y, Girma JP, Meyer D. Localization of a collagen-interactive domain of human von Willebrand factor between amino acid residues Gly 911 and Glu 1,365. Blood. 1987;70:1577–83. [PubMed] [Google Scholar]
- 5.Lankhof H, van Hoeij M, Schiphorst ME, et al. A3 domain is essential for interaction of von Willebrand factor with collagen type III. Thromb haemost. 1996;75:950–8. [PubMed] [Google Scholar]
- 6.Fujimura Y, Titani K, Holland LZ, et al. von Willebrand factor. A reduced and alkylated 52/48-kDa fragment beginning at amino acid residue 449 contains the domain interacting with platelet glycoprotein Ib. J Biol Chem. 1986;261:381–5. [PubMed] [Google Scholar]
- 7.Matsushita T, Meyer D, Sadler JE. Localization of von Willebrand factor-binding sites for platelet glycoprotein Ib, botrocetin by charged-to-alanine scanning mutagenesis. J Biol Chem. 2000;275:11044–9. doi: 10.1074/jbc.275.15.11044. [DOI] [PubMed] [Google Scholar]
- 8.Matsui T, Hamako J, Matsushita T, Nakayama T, Fujimura Y, Titani K. Binding site on human von Willebrand factor of bitiscetin, a snake venom-derived platelet aggregation inducer. Biochemistry. 2002;41:7939–46. doi: 10.1021/bi020004b. [DOI] [PubMed] [Google Scholar]
- 9.Fujimura Y, Titani K, Holland LZ, et al. A heparin-binding domain of human von Willebrand factor. Characterization and localization to a tryptic fragment extending from amino acid residue Val-449 to Lys-728. J Biol Chem. 1987;262:1734–9. [PubMed] [Google Scholar]
- 10.Adachi T, Matsushita T, Dong Z, et al. Identification of amino acid residues essential for heparin binding by the A1 domain of human von Willebrand factor. Biochem Biophys Res Commun. 2006;339:1178–83. doi: 10.1016/j.bbrc.2005.11.126. [DOI] [PubMed] [Google Scholar]
- 11.Roberts DD, Williams SB, Gralnick HR, Ginsburg V. von Willebrand factor binds specifically to sulfated glycolipids. J Biol Chem. 1986;261:3306–9. [PubMed] [Google Scholar]
- 12.Christophe O, Obert B, Meyer D, Girma JP. The binding domain of von Willebrand factor to sulfatides is distinct from those interacting with glycoprotein Ib, heparin, collagen and resides between amino acid residues Leu 512 and Lys 673. Blood. 1991;78:2310–7. [PubMed] [Google Scholar]
- 13.Borthakur G, Cruz MA, Dong JF, et al. Sulfatides inhibit platelet adhesion to von Willebrand factor in flowing blood. J Thromb Hemost. 2003;1:1288–95. doi: 10.1046/j.1538-7836.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- 14.Roberts DD, Ginsburg V. Sulfated glycolipids and cell adhesion. Arch Biochem Biophys. 1988;267:405–15. doi: 10.1016/0003-9861(88)90046-X. [DOI] [PubMed] [Google Scholar]
- 15.Aruffo A, Kolanus W, Walz G, Fredman P, Seed B. CD62/P-selectin recognition of myeloid and tumor cell sulfatides. Cell. 1991;67:35–44. doi: 10.1016/0092-8674(91)90570-O. [DOI] [PubMed] [Google Scholar]
- 16.Merten M, Thiagarajan P. Role for sulfatides in platelet aggregation. Circulation. 2001;104:2955–60. doi: 10.1161/hc4901.100383. [DOI] [PubMed] [Google Scholar]
- 17.Ishizuka I. Chemistry and functional distribution of sulfoglycolipids. Prog Lipid Res. 1997;36:245–319. doi: 10.1016/S0163-7827(97)00011-8. [DOI] [PubMed] [Google Scholar]
- 18.Barabino GA, Liu XD, Ewenstein BM, Kaul DK. Anionic polysaccharides inhibit adhesion of sickle erythrocytes to the vascular endothelium and result in improved hemodynamic behavior. Blood. 1999;93:1422–9. [PubMed] [Google Scholar]
- 19.Hillery CA, Du MC, Montgomery RR, Scott JP. Increased adhesion of erythrocytes to components of the extracellular matrix: isolation and characterization of a red blood cell lipid that binds thrombospondin and laminin. Blood. 1996;87:4879–86. [PubMed] [Google Scholar]
- 20.Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61. [PubMed] [Google Scholar]
- 21.Andrews RK, Booth WJ, Bendall LJ, Brendt MC. The amino acid sequence glutamine-628 to valine-646 within the A1 repeat domain mediates binding of von Willebrand factor to bovine brain sulfatides and equine tendon collagen. Platelets. 1995;6:245–51. doi: 10.3109/09537109509023562. [DOI] [PubMed] [Google Scholar]
- 22.Pietu G, Ribba AS, Cherel G, et al. Epitope mapping of inhibitory monoclonal antibodies to human von Willebrand factor by using recombinant cDNA libraries. Thromb Hemost. 1994;71:788–92. [PubMed] [Google Scholar]
- 23.Matsushita T, Sadler JE. Identification of amino acid residues essential for von Willebrand factor binding to platelet glycoprotein Ib. Charged-to-alanine scanning mutagenesis of the A1 domain of human von Willebrand factor. J Biol Chem. 1995;270:13406–14. doi: 10.1074/jbc.270.22.13406. [DOI] [PubMed] [Google Scholar]
- 24.Nakayama T, Matsushita T, Dong Z, et al. Identification of the regulatory elements of the human von Willebrand factor for binding to platelet GPIb. Importance of structural integrity of the regions flanked by the CYS1272-CYS1458 disulfide bond. J Biol Chem. 2002;277:22063–72. doi: 10.1074/jbc.M201327200. [DOI] [PubMed] [Google Scholar]
- 25.Favaloro EJ. Sulfatide-binding assay for von Willebrand factor. Detection of von Willebrand’s disease without discrimination of vWD subtypes. Thromb Res. 2000;98:213–9. doi: 10.1016/S0049-3848(99)00232-7. [DOI] [PubMed] [Google Scholar]
- 26.Celikel R, Varughese KI, Madhusudan, Yoshioka A, Ware J, Ruggeri ZM. Crystal structure of the von Willebrand factor A1 domain in complex with the function blocking NMC-4 Fab. Nat Struct Biol. 1998;5:189–94. doi: 10.1038/nsb0398-189. [DOI] [PubMed] [Google Scholar]
- 27.Fujimura Y, Miyata S, Nishida S, et al. The interaction of botrocetin with normal or variant von Willebrand factor (types IIA and IIB) and its inhibition by monoclonal antibodies that block receptor binding. Thromb Hemost. 1992;68:464–9. [PubMed] [Google Scholar]
- 28.Fujimura Y, Usami Y, Titani K, et al. Studies on anti-von Willebrand factor (vWF) monoclonal antibody NMC-4, which inhibits both ristocetin- and botrocetin-induced vWF binding to platelet glycoprotein Ib. Blood. 1991;77:113–20. [PubMed] [Google Scholar]
- 29.Christophe O, Rouault C, Obert B, Pietu G, Meyer D, Girma JP. A monoclonal antibody (B724) to von Willebrand factor recognizing an epitope within the A1 disulphide loop (Cys509-Cys695) discriminates between type 2A and type 2B von Willebrand disease. Br J Haematol. 1995;90:195–203. doi: 10.1111/j.1365-2141.1995.tb03400.x. [DOI] [PubMed] [Google Scholar]
- 30.Kyogashima M. The role of sulfatide in thrombogenesis and haemostasis. Arch Biochem Biophys. 2004;426:157–62. doi: 10.1016/j.abb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Siedlecki CA, Lestini BJ, Kottke-Marchant KK, Eppell SJ, Wilson DL, Marchant RE. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 1996;88:2939–50. [PubMed] [Google Scholar]
- 32.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 33.Kyogashima M, Onaya J, Hara A, Taketomi T. Sulfatide can markedly enhance thrombogenesis in rat deep vein thrombosis model. Glycoconj J. 1998;15:915–22. doi: 10.1023/A:1006967217828. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto H, Vreys I, Stassen JM, Yoshimoto R, Vermylen J, Hoylaerts MF. Antagonism of vWF inhibits both injury induced arterial and venous thrombosis in the hamster. Thromb Hemost. 1998;79:202–10. [PubMed] [Google Scholar]
- 35.Wu YP, Vink T, Schiphorst M, et al. Platelet thrombus formation on collagen at high shear rates is mediated by von Willebrand factor–glycoprotein Ib interaction and inhibited by von Willebrand factor–glycoprotein IIb/IIIa interaction. Arterioscler Thromb Vasc Biol. 2000;20:1661–7. doi: 10.1161/01.ATV.20.6.1661. [DOI] [PubMed] [Google Scholar]
- 36.Emsley J, Cruz M, Handin R, Liddington R. Crystal structure of the von Willebrand factor A1 domain and implications for the binding of platelet glycoprotein Ib. J Biol Chem. 1998;273:10396–401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]