

Abstract
The lysosomal compartment is essential for a variety of cellular functions, including the normal turnover of most long-lived proteins and all organelles. The compartment consists of numerous acidic vesicles (pH ∼4 to 5) that constantly fuse and divide. It receives a large number of hydrolases (∼50) from the trans-Golgi network, and substrates from both the cells’ outside (heterophagy) and inside (autophagy). Many macromolecules contain iron that gives rise to an iron-rich environment in lysosomes that recently have degraded such macromolecules. Iron-rich lysosomes are sensitive to oxidative stress, while ‘resting’ lysosomes, which have not recently participated in autophagic events, are not. The magnitude of oxidative stress determines the degree of lysosomal destabilization and, consequently, whether arrested growth, reparative autophagy, apoptosis, or necrosis will follow. Heterophagy is the first step in the process by which immunocompetent cells modify antigens and produce antibodies, while exocytosis of lysosomal enzymes may promote tumor invasion, angiogenesis, and metastasis. Apart from being an essential turnover process, autophagy is also a mechanism by which cells will be able to sustain temporary starvation and rid themselves of intracellular organisms that have invaded, although some pathogens have evolved mechanisms to prevent their destruction. Mutated lysosomal enzymes are the underlying cause of a number of lysosomal storage diseases involving the accumulation of materials that would be the substrate for the corresponding hydrolases, were they not defective. The normal, low-level diffusion of hydrogen peroxide into iron-rich lysosomes causes the slow formation of lipofuscin in long-lived postmitotic cells, where it occupies a substantial part of the lysosomal compartment at the end of the life span. This seems to result in the diversion of newly produced lysosomal enzymes away from autophagosomes, leading to the accumulation of malfunctioning mitochondria and proteins with consequent cellular dysfunction. If autophagy were a perfect turnover process, postmitotic ageing and several age-related neurodegenerative diseases would, perhaps, not take place.
Keywords: Ageing, Autophagy, Lipofuscin, Lysosomes, Mitochondria, Oxidative stress
Introduction
It was the discoveries made by Christian de Duve and his coworkers in the late 1950s that brought lysosomes to wider notice. The group, based at the Catholic University in Lovain, Belgium, was at the time conducting studies of glucose-6-phosphatase (reviewed in de Duve 2005). As a reference enzyme, they used unspecific acid phosphatase. One day, when they decided to reinvestigate a cell homogenate after keeping it for a day or two in a fridge, they found much higher activity of this enzyme compared to the initial measurement. In contrast, the activity of glucose-6-phosphatase had not changed. In this way enzymatic ‘latency’, a phenomenon that allows an enzyme to appear more active over time, was discovered. Using gradient sedimentation, a new compartment was found with a density close to that of mitochondria. Their findings strongly suggested that acid phosphatase was localized within cytosolic membrane-bound vesicles of limited stability, while glucose-6-phosphatase was not. With the help of cytochemists and electron microscopists, particularly Alex Novikoff and his colleagues, it was later found that a number of acidic hydrolases, capable of degrading most complex organic molecules, such as proteins, carbohydrates, lipids and nucleotides, exist together inside an organelle that was named ‘lysosome’ (lytic body). Lysosomes were observed to be a heterogeneous group of vacuoles of different sizes, form, and density. In the transmission electron microscope, some lysosomes were seen to contain still recognizable extra- and/or intracellular material under degradation, while others looked homogeneous or contained electron-dense whorls and clumps.
Soon after its recognition, the lysosomal compartment became thought of as kind of ‘stomach’ of the cell, and its importance in heterophagy (or endocytosis, which includes phagocytosis and pinocytosis) and autophagy (self-eating) was recognized. Some lysosomes were viewed as storage places for waste products, such as the age-pigment lipofuscin, and considered inactive without any remaining lytic capacity. Such structures were named ‘residual bodies’ or ‘telolysosomes’. Later the assumption of inactivity proved to be wrong, and such lysosomes are now recognized as integral parts of the lysosomal compartment and known to receive newly produced lysosomal enzymes by fusion with other lysosomes (Brunk and Ericsson 1972a).
Lysosomal enzymes are produced in the reticular network, matured in the cis-Golgi apparatus, and transported from the trans-Golgi network (TGN) within tiny vesicles, sometimes called primary lysosomes, although they are not acidic and thus not true lysosomes. The latter fuse with late endosomes and, maybe, also with autophagosomes (Dunn 1990). Mature lysosomes are part of a dynamic system. They fuse and divide, allowing their content to be propagated throughout the compartment (Brunk 1973; Luzio et al. 2007). Since lipofuscin, or age pigment, is non-degradable, the transfer of active hydrolases to lipofuscin-loaded lysosomes, in a futile attempt to degrade the pigment, means a waste of enzymes. In the event that lipofuscin-loaded lysosomes become plentiful, which certainly is the case in senescent long-lived postmitotic cells, the latter may run short of functional lysosomal enzymes. Although the total amount of lysosomal enzymes may be larger than in young cells, much of it is misdirected. In such circumstances, the result is a shortfall in the supply of hydrolytic enzymes for useful purposes and, above all, inadequate autophagy with an accumulation of malfunctioning, enlarged mitochondria and aggregates of aberrant proteins (see “Lipofuscin and postmitotic ageing”).
It was soon recognized that many previously known storage diseases, often with a serious outcome, are due to mutated and inactive lysosomal hydrolases. As a consequence, substrates cannot be degraded by these faulty enzymes, but rather accumulate inside the lysosomal compartment. The analogy to lipofuscin accumulation in senescent long-lived postmitotic cells is obvious and, perhaps, we should look at ageing as a storage disorder where lipofuscin is the material that accumulates, although for other reasons than defective lysosomal enzymes (see “Lipofuscin and postmitotic ageing”). Recently it has been suggested that both lysosomal storage diseases and ageing are associated with disturbed autophagy that causes the accumulation of polyubiquitinated proteins and dysfunctional giant-type mitochondria (Kiselyov et al. 2007; Settembre et al. 2008, 2008; Terman 1995; Terman and Brunk 2005a). Consequently, senescence and lysosomal storage diseases might, possibly, be considered ‘autophagy disorders’.
Although de Duve made an early suggestion that lysosomes were ‘suicide bags’ (de Duve 1959), they were for a long time considered sturdy organelles, which would not release their potent mixture of hydrolases until cells were already dead. Lysosomal enzymes would then be responsible for dissolving dead cells only (necrosis). This notion was partly based on ultrastructural observations of seemingly intact lysosomes in devitalized cells. However, it later turned out that at least the lysosomal enzyme acid phosphatase could leak from lysosomes that look normal in the electron microscope (Brunk and Ericsson 1972b), and that lysosomes, contrary to general opinion at the time, are sensitive to stresses, especially to oxidants (Brunk and Svensson 1999; Kurz et al. 2004, 2006; Persson et al. 2003; Yu et al. 2003b). Many textbooks nevertheless still reflect the old opinion, and thus demonstrate the truth of Billings’ (1818–1885) statement: “The trouble with people is not that they don’t know, but that they know so much that ain’t so” (Billings 1874). The finding that the membranes of rat liver lysosomes contain a 37-fold higher specific content of α-tocopherol compared to other cellular membranes (Rupar et al. 1992) suggests that these membranes are in need of anti-oxidant protections, and is an indirect proof for the hypothesis that lysosomes normally are under the influence of substantial oxidative stress.
The first reports of lysosomal destabilization or LMP (lysosomal membrane permeability—compare with MMP, mitochondrial membrane permeability), as an early event in apoptosis appeared about fifteen years ago. The discovery was a result of experiments on the induction of apoptosis in cultured cells by moderate oxidative stress. LMP was found to be a response to such stress, which was then apparently followed by MMP and classical apoptosis (reviewed in Brunk et al. 2001). Even earlier, however, it was realized that a mitochondrial release of apoptogenic substances, such as cytochrome c, activates the caspase cascade and the internal pathway of apoptosis. Consequently, almost all researchers within the expanding field of programmed cell death/apoptosis focused on the roles of caspases and mitochondria in this process. The hypothesis that lysosomes might play an important role in, at least, some forms of apoptosis had few advocates among most cell death researchers. During the last two decades, a huge amount of knowledge has accumulated on apoptosis, but not until it was found that lysosomal calpain-like cysteine proteases truncate and activate the pro-apoptotic protein Bid (Cirman et al. 2004) did the role of lysosomes in apoptosis begin to become a somewhat more fashionable field of investigation. Additional evidence for the role of lysosomes in apoptosis was the finding that an endocytic uptake of the potent iron chelator desferrioxamine (DFO) gave rise to almost complete prevention (although only temporary – see further in “The stability of lysosomes under oxidative stress and their role in cellular iron metabolism”) of lysosomal rupture and ensuing apoptosis/necrosis when a number of different cell types were exposed to hydrogen peroxide (Antunes et al. 2001; Doulias et al. 2003; Yu et al. 2003b). This finding gave rise to two essential conclusions: (1) the importance of redox-active iron in lysosomal stability at oxidative stress, and (2) that hydrogen peroxide per se is not particularly harmful.
Even though the role of lysosomes in autophagy was recognized early (reviewed in Glaumann et al. 1981), this phenomenon was long considered a murky one. This, however, radically changed when it was recently found that autophagy is a much more regulated process than hitherto believed, and under the influence of more than a score of evolutionary well-preserved autophagy genes (ATG), which allow it to be selective and to take three different forms: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy (CMA) (reviewed in Cuervo et al. 2005; Shintani and Klionsky 2004; Suzuki and Ohsumi 2007; Yorimitsu and Klionsky 2005). More or less pronounced autophagy commonly occurs in apoptotic cells. Sometimes such autophagy is extensive, and apoptosis then deviates from the classical form. This form of apoptosis has been named autophagic cell death, or programmed cell death type II, and is often considered to be caspase-independent (see “Leaky lysosomes and apoptosis/necrosis”).
This review will focus on areas where lysosomes are important but not so often emphasized, and where the authors themselves have some experience. They include: how lysosomes participate in cellular iron metabolism; lysosomal behavior under oxidative stress; how lysosomes and mitochondria interact to induce apoptosis; and, how lysosomes are involved in postmitotic ageing.
The lysosomal compartment and its main functions
The lysosomal compartment is indispensable for cellular life and has several important functions. Lysosomes thus exist in all kinds of animal cells, except erythrocytes, which have a very specialized function and a minimal turnover of their constituents. The degradation of endocytosed or autophagocytosed materials takes place inside lysosomes that have an acidic (pH ∼4 to 5) environment, which is maintained by ATP-dependent proton pumps present in the lysosomal membrane. Such pumps are also present in the plasma membrane, especially in tumor cells with a pronounced anaerobic glycolysis (the Warburg effect) that results in a high production of lactic acid and a need to export protons to avoid acidification of their own cytosol (Bartrons and Caro 2007; Semenza et al. 2001).
Following synthesis in the endoplasmic reticulum, lysosomal hydrolases are tagged with mannose-6-phosphate (MP) in the cis-Golgi area and packaged into transport vesicles (sometimes named primary lysosomes although they have a neutral pH) in the trans-Golgi network (TGN) with the help of MP receptors. The newly produced hydrolases are transported to slightly acidic (pH about 6) late endosomes, which arise from early endosomes containing endocytosed material. Here, the lysosomal hydrolases are freed from MP receptors and activated, while the MP receptors are re-circulated to the Golgi apparatus. The late endosomes then mature to lysosomes that lack MP receptors, are rich in acid hydrolases, have a pH of 4–5 and contain endocytosed material to be degraded. Lysosomes fuse with autophagosomes/endosomes to form ‘hybrid’ organelles contaning material in the course of degradation originating both from outside and inside the cell. Following completed degradation of the enclosed material, lysosomes turn into ‘resting’ ones, which in the electron microscope look homogeneous and moderately electron-dense. They are then ready for new rounds of fusion (Luzio et al. 2007). Indeed, pronounced fusion and fission activity is a characteristic of the lysosomal compartment that allows lytic enzymes and other contents to spread through the compartment (see Fig. 1).
Fig. 1.

Autophagy can be subdivided into three forms: macro-, micro- and chaperone-mediated autophagy (CMA). In macroautophagy, a cup-formed phagophore surrounds cytosolic organelles thereby creating an autophagosome, which fuses with a lysosome or a late endosome. Small portions of cytoplasm enter lysosomes through invagination of the membrane (microautophagy). In CMA, proteins tagged with the amino acid sequence KFERQ are bound by molecular chaperones, e.g., Hsp73, and transported to lysosomes. The latter form from late endosomes from which they differ in that they do not have mannose-6-phosphate receptors (MPR). Lysosomal hydrolases are transported bound to MPRs in Golgi-derived vesicles to slightly acidified late endosomes and, possibly, autophagosomes. There, the enzymes are released and the MPRs are recycled back to the Golgi network
Following receptor-mediated endocytosis, the initially plasma membrane-bound receptors are often, but not always, returned to the plasma membrane, while the ligands are usually propagated further down the lysosomal compartment. An exception is the tranferrin receptor and its transferrin ligand, which are both returned to the plasma membrane, while the iron that is bound to transferrin is released by the acidity of the late endosomes (see “The stability of lysosomes under oxidative stress and their role in cellular iron metabolism”).
The processing and presentation of antigens in immuno-competent cells is dependent on a form of endocytotic–exocytotic activity, while autophagic degradation is of vital importance not only for the normal turnover of cellular constituents, but also for the removal of damaged structures and cytosolic microorganisms that have invaded the cell. Some cell types are able to exocytose lysosomal contents or even intact lysosomes (secretory lysosomes) (reviewed in Luzio et al. 2007). It has been recognized that tumor cells often secrete lysosomal proteases, which, in combination with acidification of their surroundings helps them to infiltrate and spread (de Milito and Fais 2005; Lorenzo et al. 2000; Martinez-Zaguilan et al. 1993; Montcourrier et al. 1997; Podgorski and Sloane 2003; Rochefort et al. 1990; Victor and Sloane 2007).
As pointed out above, lysosomes fuse with autophagosomes, or deliver part of their content (‘kiss-and-run’), to form autophagolysosomes where a variety of organelles and proteins are degraded into simple components, which in turn are reutilized by the anabolic machinery of the cell following transport to the cytosol (reviewed in Klionsky 2007; Terman et al. 2007).
From a physiological point of view, the lysosomal compartment can be looked upon as a box, built of vacuoles that constantly fuse and divide, that receives enzymes from the TGN and substrates from either the outside or the inside of the cell. Following substrate degradation inside individual lysosomes, the products diffuse or are actively transported to the cytosol for reutilization.
Most of the mechanisms involved in the formation of the autophagic double membrane (the phagophore), the inclusion of materials to be degraded, and the fusion of autophagosomes and lysosomes was, as mentioned above, recently worked out as a result of the discovery of a large family of phylogenetically well preserved autophagy-related genes (ATG) (Klionsky 2007; Shintani and Klionsky 2004; Suzuki and Ohsumi 2007; Yorimitsu and Klionsky 2005). Autophagy represents one of the main pathways for the turnover and reutilization of worn-out long-lived proteins and organelles and is a perfectly normal process. Interestingly, proteasomes, which also play an important role in the turnover of macromolecules, are themselves degraded by autophagy (Cuervo et al. 1995). The implication of this is that hampered autophagy might result in defective proteasomes since they, in common with mitochondria and other organelles, are then not properly renewed.
During periods of starvation, enhanced autophagy serves as a way for cells to survive by degrading less important parts of themselves in order to produce ATP and building blocks for essential anabolic processes. Subsequent to cellular damage, reparative autophagy follows, in which altered and malfunctioning structures are replaced. Such reparative autophagy is commonly seen following, for example, ionizing irradiation, virus infection, and hypoxic or oxidative stress (Bergamini 2006; Kaushik and Cuervo 2006).
The stability of lysosomes under oxidative stress and their role in cellular iron metabolism
As indicated above, apart from the operations of professional scavengers with their high degree of endocytosis (heterophagy), autophagy is the main process that delivers substrates to the lysosomal compartment. Since many autophagocytosed macromolecules contain iron (e.g., ferritin and mitochondrial electron-transport complexes), the lysosomal compartment becomes rich in this essential, although potentially hazardous, transition metal (Brun and Brunk 1970; Miyawaki 1965; Persson et al. 2003; Radisky and Kaplan 1998; Roberts and Bomford 1988; Sakaida et al. 1990; Schraufstätter et al. 1988; Vaisman et al. 1997; Yu et al. 2003b; Zdolsek et al. 1993). In this respect, lysosomes are unique (reviewed in Kurz et al. 2007). It is important to recognize that lysosomes that have recently been engaged in an autophagic degradation of iron-rich compounds should contain high concentrations of iron, while others which have been inactive for a while with respect to such autophagy may contain only negligible amounts of it (Kurz et al. 2007; Nilsson et al. 1997). Similarly, for some limited period of time, certain cells in a population may have been inactive altogether with respect to autophagy. If so, inside a particular cell we expect to find lysosomes with a range of iron concentrations, and we also ought to find occasional cells with no, or almost no, lysosomal iron. This hypothesis is supported by a line of evidence: (1) recent findings indicate that the transport of released iron away from the lysosomal compartment is a rapid process (T. Kurz et al., in preparation); (2) a pronounced variation is found in the stability of individual lysosomes against oxidative stress in single cells, and also in the total lysosomal population amongst cells (Nilsson et al. 1997); (3) the cytochemical sulfide-silver method (SSM) for detection of loosely-bound iron shows substantial variability with respect to amounts of lysosomal iron within as well as between cells. Figure 2 shows the mainly lysosomal distribution of labile iron in J774 cells using the SSM. The heterogeneity with respect to lysosomal iron is demonstrated by the relation between the ‘development’ time and the number of positive lysosomes (for details see the legend to Fig. 2). The SSM demonstrates heavy metals, which normally means iron since it is the only such metal that normally occurs in any significant amount in most cells. Exceptions are pancreatic β-cells, prostatic epithelial cells and some neurons, all of which are rich in zinc (Zdolsek et al. 1993).
Fig. 2.

Cytochemical demonstration of lysosomal iron in J774 cells using the high pH, high S2− sulphide-silver method (SSM). After 30 min of development (a), no positive lysosomes are seen. However, after 40 min of development (b) a few dark-stained positive (iron-containing) lysosomes are visible (arrows), indicating particularly iron-rich lysosomes, while after 60 min of development (c) a strong and widespread lysosomal-type pattern is found, indicating at least some iron in most lysosomes
Taken together, these findings would satisfactorily explain the difference in the sensitivity to oxidative stress for lysosomes of individual cells as well as for a population of cells, where always some cells survive even substantial stress, while others undergo apoptosis at a rather low degree of such stress. Such an explanation has been pointed out as essential to find (Kroemer and Jäättelä 2005). The role of redox-active iron for the stability of lysosomes under oxidative stress is further substantiated by the finding that the simultaneous exposure to hydrogen peroxide and the potent lipophilic iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH) almost fully prevents both lysosomal rupture and apoptosis, as long as the stress is not overwhelmingly strong. Likewise, the endocytotic uptake of apo-ferritin and the autophagy of metallothioneins protect lysosomes against oxidative stress. Conversely, the endocytotic uptake of an iron–phosphate complex sensitizes to such stress (Fig. 3). The finding that hydrogen peroxide in concentrations that normally induce apoptosis or necrosis has no influence on cells in the presence of SIH (apart from briefly setting back their mitotic activity) tells us that hydrogen peroxide per se is not particularly toxic but rather must work in concert with iron in order to damage cells.
Fig. 3.

Labilization of lysosomes by hydrogen peroxide and its modulation by autophagocytosed metallothioneins and endocytosed apo-ferritin, desferrioxamine and a Fe phosphate complex. J774 cells were exposed to hydrogen peroxide (initially 100 μM in PBS) for 30 min (H2O2), to PBS only (Control), or to H2O2 in combination with 100 μM of the lipophilic iron-chelator SIH—and then returned to standard culture conditions. Some cells had previously been exposed to the hydrophilic iron chelator desferrioxamine (1 mM) for 3 h, to 1 μM apo-ferritin (Apo-Ft) for 4 h, or a Ferric-phosphate complex (30 μM) for 4 h, while other cells had been exposed to ZnSO4 for 12 h to induce the upregulation of metallothioneins and ensuing autophagy of this protein. Note that apo-ferritin, desferrioxamine, SIH, and metallothioneins stabilize lysosomes, while the Fe–phosphate complex has the opposite effect. The figure is compiled from a number of previously published ones
Increased amounts of iron can accumulate in some lysosomes due to the accretion of iron-rich non-degradable materials, such as lipofuscin and, especially, hemosiderin that, perhaps, could be considered an unusually iron-rich form of lipofuscin. Consequently, the sensitivity to oxidative stress of lysosomes loaded with lipofuscin or hemosiderin seems to be quite high (Seymour and Peters 1978; Terman et al. 1999a; Terman and Brunk 2004).
Non-dividing cells mainly rely on the turnover and reutilization of iron, while growing populations of cells require its uptake from their surroundings. The uptake is tightly controlled and accomplished by a complex series of events. Iron is carried in the circulation by transferrin (Tf) in a non-redox-active form. The Tf–Fe complex binds to the transferrin receptor (TfR), and the Tf–Fe–TfR complex is endocytosed and propagated into late endosomes. As a consequence of the slightly acidic pH of late endosomes, iron is released and transported to the cytosol by the divalent metal transporter-1 (also known as Nramp 2 or SLC11A2). The Tf–TfR complex returns to the plasma membrane, where transferrin is freed from the TfR and released to the circulation to pick up more iron. At neutral pH, Fe–Tf binds to the TfR, while Tf does not. In the cytosol, low mass iron binds to specific iron regulating proteins (IRPs), which in turn up-regulate ferritin and down-regulate TfR, by interfering with the translation of their mRNA. By these mechanisms, cytosolic iron is kept at the lowest possible level that allows synthesis of necessary iron-containing macromolecules (Kruszewski 2003).
Even though it is not known exactly how low mass iron is transported from lysosomes to the cytosol following autophagic degradation of iron-containing macromolecules, it may be assumed that mechanisms akin to those that transport iron from late endosomes are involved. It is also not known whether iron that is bound to divalent metal transporters is redox-active. However, natural processes are usually operated in a benign manner and, thus, it may be anticipated that, first, cytosolic labile iron is in transit only briefly before being taken care of in a safe way and, second, that while in transit its reactivity with respect to electron transport is, at the least, restricted.
Recently, an alternative mechanism for the delivery of iron from late endosomes and lysosomes to mitochondria was suggested. It involves a direct transport between these two types of organelles following temporary close contact (kiss and run) (Zhang et al. 2005). For a schematic view of the involvement of lysosomes in iron metabolism, see Fig. 4.
Fig. 4.

The role of lysosomes in cellular iron metabolism. In serum, iron is bound in a transferrin-iron complex that attaches to transferrin receptors, which results in receptor-mediated endocytosis inside clathrin-coated vesicles. In the lowered pH of late endosomes, iron is released and transported to the cytoplasmic labile iron pool (LIP), while the transferrin and its receptor are recycled to the plasma membrane. LIP iron is probably temporarily bound by low affinity chelators before it is either used in the synthesis of iron-containing proteins or stored in ferritin. It is unknown if LIP iron is redox-active. The LIP also receives iron from lysosomes following degradation of iron-containing proteins, e.g., ferritin and mitochondrial electron transport complexes
Rapidly growing tumor cells have a high demand for iron and, therefore, iron chelators may be used to stop their proliferation and induce apoptosis. Encouraging therapeutic results have been obtained in clinical phase III studies using iron chelators, which seem to have a selective effect on tumor cells, while not harming normal ones that also need iron, e.g., erythroblasts (Richardson 2002).
Some investigators have found support for the hypothesis that when low mass iron is required, it is released directly from ferritin within the cytosol (de Domenico et al. 2006; Liu et al. 2003; Takagi et al. 1998). Others favor an alternative mechanism for iron release, namely autophagy followed by transport of released iron to the cytosol (Kidane et al. 2006; Radisky and Kaplan 1998; Roberts and Bomford 1988; Sakaida et al. 1990; Tenopoulou et al. 2005; Vaisman et al. 1997; T. Kurz et al., in preparation). The latter hypothesis is supported by the findings of many electron microscopists, who over the years have noticed ferritin inside autophagosomes (Koorts and Viljoen 2007). Other arguments for the autophagy hypothesis is that exposure of cells in culture to a panel of inhibitors to lysosomal enzymes prevents the release of iron from ferritin (Kidane et al. 2006), and the finding that exposure of cells to the iron chelator DFO, which is endocytosed and transported into the lysosomal compartment where it remains nondegraded and acts as a sink for iron, rapidly results in a major decrease of cytosolic labile (calcein-chelatable) iron (Tenopoulou et al. 2005). The latter finding supposedly reflects a rapid incorporation of cytosolic iron into ferritin or iron-containing macromolecules under construction. Such a rapid disappearance of calcein-chelatable iron from the cytosol is in agreement with the above notion that cytosolic labile iron is in rapid transit and kept at the lowest possible concentration in order to avoid Fenton-type reactions (Kruszewski 2003). If iron were liberated from ferritin directly in the cytosol, and its concentration regulated by a feedback mechanism (de Domenico et al. 2006), one would expect a preserved cytosolic concentration of chelatable iron for quite a while. However, whether autophagy and a mechanism that involves direct iron-release from ferritin act in concert, or if one of them is dominant—or even the only one—remains to be established.
As mentioned above, excessive iron may accumulate within lysosomes as a component of lipofuscin (reviewed in Brunk and Terman 2002; Jolly et al. 1995) or in the form of hemosiderin. The latter substance seems to represent a particularly iron-rich form of lipofuscin (or ceroid, see below) composed of non-degradable polymerized aldehyde-bridged ferritin residues. Because cells do not have the capacity to rid themselves of intralysosomal iron that is bound to lipofuscin, it slowly accumulates over time, even if the uptake of iron is effectively regulated. This accumulation is particularly evident in the reticulo-endothelial system, in hepatocytes, and in long-lived postmitotic cells, such as neurons and cardiac myocytes. It seems most probable that the sensitivity to oxidative stress is enhanced in such cells at the end of their lifespan.
As long as Fe(II) has at least one of its six coordinates free, it is capable of catalyzing a homolytic splitting of hydrogen peroxide resulting in the formation of the extremely reactive hydroxyl radical (Fenton reaction).
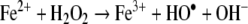 |
The favourable conditions for intralysomal Fenton reactions are a low internal pH (∼4 to 5) and the presence of reducing equivalents, such as cysteine, ascorbic acid or glutathione (Baird et al. 2006; Pisoni et al. 1990; Schafer and Buettner 2000). At this pH, cysteine, as well as the other reducing agents, easily reduces iron(III) to iron(II). An additional reducing agent inside autophagolysosomes may be the superoxide anion radical (O2•−). Mitochondria undergoing autophagic degradation probably continue to produce these radicals for a while, perhaps even in increased amounts, following disabling of the mitochondrial electron transport complexes secondary to the proteolytic attack.
Hydrogen peroxide forms continuously in cells, mainly by loss of electrons from the mitochondrial electron transporting chain, but also from a variety of oxido-reductases that operate by one-electron transfers. Some hydrogen peroxide may escape the effective cytosolic degradation by catalase and glutathione peroxidase and diffuses into lysosomes (Walton and Lewis 1916), which do not contain these enzymes (those that are autophagocytosed are rapidly degraded), to react with labile iron and form hydroxyl radicals, or similarly reactive ferryl or perferryl compounds (reviewed in Eaton and Qian 2002) (see Fig. 5).
Fig. 5.

Lysosomal destabilization by oxidative stress. The cell’s antioxidant defense protects against moderate oxidative events. However, if the protective shield is overwhelmed, hydrogen peroxide, which is mainly produced by mitochondria, diffuses into lysosomes in abnormal amounts. Since many lysosomes are rich in redox-active iron due to degradation of iron-containing macromolecules, Fenton-type reactions then take place resulting in lysosomal rupture with release of powerful lytic enzymes
In normal conditions, with a low and steady state rate of autophagy, the result of the inevitable Fenton-type reactions inside lysosomes is a slow formation of lipofuscin (age-pigment) over time. Following enhanced autophagy, for example because of reparative efforts after injury, when often oxidative stress is present as well, a much more rapid formation of the pigment occurs. In such cases that are unrelated to normal ageing, the pigment is called ceroid, although both lipofuscin and ceroid basically have the same composition and mode of origin, i.e., intralysosomal iron-catalyzed peroxidation of material under degradation (Brunk and Terman 2002; Terman and Brunk 1998).
When enhanced oxidative stress results in peroxidation not only of the lysosomal contents but also of its membrane, lysosomes become leaky after a delay that is dependent on the magnitude of oxidative stress. This happens also if cells are exposed only briefly to oxidative stress before being returned to standard culture conditions without further stress. Consequently, any satisfactory hypothesis on the mechanisms behind oxidation-induced lysosomal rupture must provide firm and distinct links between the triggering events, which occur within minutes, and the ultimate leak that may be delayed by hours. This link seems to be lysosomal membrane peroxidation that takes place as a chain-reaction that results in the formation of peroxides that slowly decompose spontaneously or, much more rapidly, in the presence of catalyzing iron. Therefore, initially, only the most iron-rich lysosomes start to leak and the damage done by released proteolytic enzymes, many of which are partly active (although less stable) also at neutral pH (Turk et al. 2002), is followed by reparative autophagy. If the leak is moderate, the cell may survive, while the apoptotic machinery is activated if the leak is more substantial (Antunes et al. 2001).
A protective side-effect of ferritin autophagy, in relation to lysosomal and cellular resistance to oxidative stress, is that ferritin, which is only partly iron-saturated, seems to be able to temporarily chelate intralysosomal labile iron before being degraded itself, thereby reducing its concentration of redox-active iron and, therefore, the sensitivity of lysosomes to oxidative stress (Garner et al. 1997, 1998). Each molecule of ferritin can store around 4,500 atoms of iron. Consequently, we have postulated that even a limited number of autophagocytosed ferritin molecules are able to substantially reduce lysosomal redox-active iron (although not the total amount of lysosomal iron). Since autophagy is a continuous process, degraded lysosomal ferritin is constantly replaced—and a state of equilibrium created between cytosolic and lysosomal ferritin. If this is correct, the concentration of lysosomal labile iron is determined by the rate of autophagic turnover of ferruginous materials and cytosolic iron-binding proteins, such as ferritin and metallothioneins, which are a family of iron-binding proteins (Baird et al. 2006; Garner et al. 1997, 1998; Persson et al. 2001; T. Kurz et al., in preparation). This proposal is compatible with the fact that cells rich in ferritin, metallothioneins and some other stress-induced phase II proteins are resistant to oxidative stress (Arosio and Levi 2002; Baird et al. 2006; Garner et al. 1997; Viarengo et al. 2000). If this hypothesis is correct, there is another side to the coin. In the case of diseases involving iron-overload, such as advanced conditions of hemochromatosis, in which ferritin may be iron-saturated, then, rather than acting as a binder of lysosomal redox-active iron, iron-saturated ferritin might just increase it and, therefore, enhance lysosomal sensitivity to oxidative stress. These hypotheses are supported by findings that endocytotic uptake of apo-ferritin, or the autophagy of metallothioneins protects lysosomes against oxidative stress, while the endocytosis of a Fe–phosphate complex makes them much more vulnerable to such stress (Fig. 3).
Besides hemochromatosis, an increasing number of diseases have been shown to be associated with the intracellular accumulation of iron (in particular within mitochondria and lysosomes) with consequently enhanced sensitivity to oxidative stress. As pointed out earlier in this review, there is a close relation between lysosomes and mitochondria, which is the basis of the lysosomal–mitochondrial axis theories of ageing and apoptosis (Terman et al. 2006). Because mitochondria are degraded by lysosomes, accumulation of iron inside the former will also result in lysosomal iron loading. Moreover, oxidative stress due to normal production of hydrogen peroxide by mitochondria will labilize lysosomes, while released lysosomal enzymes will damage mitochondria and induce further oxidative stress.
Friedreich’s ataxia is associated with a deficiency of frataxin protein, a mitochondrial iron chaperone that chelates iron in a non-redox-active form and thus protects from Fenton reactions. Frataxin participates in the formation of iron–sulphur clusters, which are cofactors of several enzymes. Lack of frataxin results in the accumulation of mitochondrial redox-active iron with increased mitochondrial vulnerability to oxidative stress and ensuing neuronal damage (Pandolfo 2006). Aceruloplasminemia patients lack ferroxidase ceruloplasmin activity, which results in iron accumulation within a variety of tissues. Neurological symptoms, retinal degeneration, and diabetes follow (Berg and Youdim 2006; Miyajima 2003). Sideroblastic anemia is characterized by iron-laden perinuclear mitochondria in developing red blood cells and, sometimes, also in neurons and other cell types, due to an enzymatic defect of heme synthesis (Fontenay et al. 2006). There is also accumulating evidence that iron accumulation within neurons contributes to the development of several common neurodegenerative diseases, such as the Alzheimer’s, Parkinson’s and Huntington’s diseases (Berg and Youdim 2006; Sipe et al. 2002).
Leaky lysosomes and apoptosis/necrosis
An active role for lysosomes in the induction of cell death was an early suggestion (reviewed in de Duve and Wattiaux 1966). Apoptosis or programmed cell death (PCD) was not yet identified in those days, although cells with characteristics typical for apoptosis had long been described by morphologists. As pointed out in the introduction to this review, the role lysosomes play in apoptosis did not become recognized until recently—and is still being illuminated. Major reasons for this considerable delay were the facts that (1) lysosomal membrane permeabilization may occur without any apparent ultrastructural alterations, creating an impression, still reflected in textbooks, that lysosomes are sturdy organelles that only break when cells are dead or almost dead, and that (2) many commonly used caspase inhibitors also inhibit cysteine proteases in general, including lysosomal cathepsins (Kroemer and Jäättelä 2005).
The finding a few years ago that some apoptogenic drugs, which are also lysosomotropic detergents or aldehydes—such as MSDH (o-methylserine dodecylamide hydrochloride), sphingosine, Leu-Leu-Ome, and 3-aminopropanal—specifically destabilize lysosomes led to a further focus on the possibility that lysosomal rupture may be an upstream event in some forms of apoptosis (Boya et al. 2003; Brunk et al. 1997; Cirman et al. 2004; Kågedal et al. 2001). A strong argument for the idea that moderate lysosomal rupture causes apoptosis was the finding that the apoptogenic effects of these lysosomotropic drugs are prevented by an initial exposure to pH-raising lysosomotropic substances, for example ammonia or chloroquine (Ohkuma and Poole 1978). The raised lysosomal pH prevents the intralysosomal accumulation of the apoptogenic lysosomotropic agents (Kågedal et al. 2001; Li et al. 2003; Yu et al. 2003a). The resulting preservation of lysosomal stability and lack of apoptosis certainly links lysosomal leak to apoptosis, although the causal molecular mechanisms were not explained by theses experiments.
Oxidative stress during apoptosis, and lysosomal involvement in apoptosis due to such stress, is increasingly recognized (Brunk et al. 2001; Cirman et al. 2004; Guicciardi et al. 2004; Nylandsted et al. 2004). The influence of such stress on cells depends on the dose.
Figure 6 illustrates the heterogeneity between cells and lysosomes with respect to lysosomal stability following oxidative stress. It shows J774 cells following photo-oxidative stress (AO-loaded cells were exposed to multiple pulses of blue light). Some cells show apoptotic morphology and the number of intact lysosomes is markedly reduced, while other cells look almost normal.
Fig. 6.

Lysosomes are differently sensitive to photooxidation. Using a 488 nm argon laser, AO-stained J774 cells were depicted by confocal laser scanning microscopy. Pictures were taken in intervals of a few minutes during which the samples were subjected to pulses of blue light. Panel a shows the first of these ones presenting distinct red fluorescence of lysosomes and a weak green fluorescence of the nuclei and cytoplasm. In panel b a few cells (some are arrowed) show enhanced green fluorescence that indicates early lysosomal burst, while in panel c more advanced lysosomal rupture has resulted in a maximally increased green fluorescence. Also note the disappearance of many lysosomes from the cells. In panels d–f, the green fluorescence is successively diminishing due to increased plasma membrane permeability, secondary to the attack of lytic lysosomal enzymes, with escape of cytosolic AO to the surrounding medium. Note some remaining intact lysosomes in a few cells in f
Moderate lysosomal rupture induces apoptosis, while pronounced lysosomal leakage results in necrosis without caspase activation (Zhao et al. 2003). The puzzling lack of apoptotic response following major lysosomal damage was difficult to explain. It was hypothesized to be a result of a violent degradation of pro-caspases that, however, could not be verified. Very recently, Galaris and coworkers pointed out that a pronounced lysosomal breakup results in a major release to the cytosol of low mass lysosomal iron that seemed to prevent the activation of caspase-9 within the apoptosome that is a complex of pro-caspase-9, cytochrome c and ATP. This lack of activation was suggested to be an effect of iron binding to thiol groups in the active center of the pro-caspase (Barbouti et al. 2007; Brunk and Eaton 2007).
As pointed out above, the amount of redox-active lysosomal iron seems to be crucial for lysosomal stability during oxidative stress, probably explaining the variability in lysosomal sensitivity to such stress (Nilsson et al. 1997). The fact that cells with upregulated metallothioneins, heat shock proteins, or apo-ferritin are relatively resistant to oxidative stress may, at least partially, be explained by autophagic transfer of such macromolecules into the lysosomal compartment, where they may temporarily bind redox-active iron before being degraded (see above). In the event that autophagy of such proteins continues, as would be expected as a part of normal turnover, a steady state between lysosomal redox-active and non-redox active iron would be established that is dependent on the cytosolic amounts of these proteins.
Lysosomal enzymes have been shown to induce MMP as well as MPT (mitochondrial permeability transition), through proteolytic activation of phospholipases or the Bcl-2 family members Bid, Bax and Bak. Calpain-like lysosomal cysteine proteases were found to induce cleavage of Bid (Cirman et al. 2004), which in its truncated form relocates to mitochondria resulting in Bax/Bak activation with ensuing formation of pores in the outer membrane and resulting escape of cytochrome c. Moreover, cathepsin D, although not cathepsin B, has been reported to modify/activate Bax to a pore-forming structure (Castino et al. 2007). Consequently, the release into the cytosol of pro-apoptotic mitochondrial factors such as cytochrome c, apoptosis inducing factor (AIF) and Smac/DIABLO will trigger cell death (Bidere et al. 2003; Brunk et al. 2001; Cirman et al. 2004; Guicciardi et al. 2004; Heinrich et al. 2004). Moreover, the direct activation of caspases-2 and -8 by lysosomal cathepsins has been described for at least certain cell types (Baumgartner et al. 2007; Yeung et al. 2006), although further studies are needed in order to confirm this.
The mechanisms described above all work through activation of the internal (mitochondrial) pathway of apoptosis. There are, however, also indications that lysosomal destabilization is involved in apoptosis following ligation of death receptors on the plasma membrane. Ligation of the FAS receptor was found to give rise to early LMP, although neither the mechanisms nor the possible relation to activation of caspase-8 were initially understood (Brunk and Svensson 1999). Recently; however, it has been found that ligation of the receptors for tumor necrosis factor and TRAIL, at least in certain types of cells, induces not only formation of active caspase-8 from its pro-form, but also caspase-8-mediated activation of the pro-apoptotic protein Bax. This pro-apoptotic Bcl-2 family member, which was formerly believed to be involved only in MMP, was found also to participate in LMP, perhaps by forming pores similar to those of the mitochondria (Werneburg et al. 2007).
Moreover, the multifunctional cytokine tumor necrosis factor (TNF) that induces apoptosis has been found to induce cellular stress, probably by stimulating the production of superoxide by NADPH oxidase (Autelli et al. 2005; Frey et al. 2002; Kroemer and Jäättelä 2005; Tang et al. 1999). It seems reasonable to believe that the resulting oxidative stress may induce lysosomal iron-mediated rupture with ensuing apoptosis or necrosis.
All these new findings suggest that lysosomal destabilization might play an integral part in apoptosis of both the internal and external pathways, and suggest that the combined action of lysosomal hydrolases and caspases is necessary for apoptosis. Caspases may be needed for the degradation of a limited number of specialized proteins relevant to apoptosis, such as poly(ADP-ribose)polymerase and laminin, while lysosomal hydrolases may be needed for bulk degradation.
A form of apoptosis that is associated with pronounced autophagy is programmed cell death type II, or autophagic cell death. It is often considered caspase-independent, though whether this is completely correct still remains to be clarified. An alternative interpretation of this phenomenon, at least in some cases, is that damaged cells try to heal themselves by reparative autophagy, which, in the event that it fails, is followed by apoptosis. If a majority of cells succeed in their repair efforts, the activation of caspases in only a few cells may be difficult to detect using biochemical methods. There are indications of a mutually reinforcing link between autophagy and apoptosis, although mutual inhibition under certain conditions has also been suggested (Kroemer and Jäättelä 2005; Maiuri et al. 2007). Whether a completely caspase-free type II cell death does exist still remains to be shown.
Surprisingly, we recently found (T. Kurz et al., in preparation) that apoptosis induced by iron depletion following exposure to the lipophilic iron-chelator SIH is preceded by lysosomal destabilization, indicating that lysosomal damage can be induced by iron overload followed by oxidative stress as well as by iron depletion. Any oxidative stress that might occur simultaneously with iron starvation due to general iron chelation should clearly not induce lysosomal labilization. Therefore, the destabilizing effect on lysosomes of iron starvation must be a consequence of other apoptogenic stimuli, which are yet to be identified. The finding nevertheless gives additional support to the emerging view that lysosomal destabilization is a much more common phenomenon in apoptosis than so far recognized.
Many forms of apoptosis are regulated by activation of the p53 gene. Exactly how the p53 protein triggers the apoptotic cascade is a topic of intense research (Levine et al. 2006). If lysosomal destabilization is a general phenomenon in apoptosis, it may be assumed that this phenomenon should be found early in the process of p53 activation. A mutated p53 protein exists in a thermo-labile form that is inactive at 37°C but structurally stable and active at 32°C (Michalovitz et al. 1990). When cells that stably over-express the thermo-labile form of p53 are grown at 37°C, they remain normal, but when transferred to 32°C they undergo apoptosis within 8–10 h. Following transfer to the permissive temperature of 32°C, these cells start to show signs of early lysosomal destabilization within a few hours, long before any apoptotic signs are found (Yuan et al. 2002).
Even if lysosomes seem to be more deeply involved in apoptosis than is generally recognized, many questions remain to be answered. First among these is whether lysosomes are regularly destabilized early in forms of apoptosis that are not dependent on oxidative stress, and if so, how. Lysosomal rupture as a result of oxidative stress is well understood, but why are iron starvation, serum starvation (Brunk and Svensson 1999), and p53 activation followed by lysosomal destabilization? The recently discovered protein LAPF (for lysosome-associated apoptosis-inducing protein containing PH and FYVE domains), a member of a widespread new family of Phafins (proteins containing the above domains), was shown to induce apoptosis by attaching itself to lysosomal membranes, causing lysosomal permeabilization and activation of the lysosomal–mitochondrial pathway (Chen et al. 2005). Perhaps this protein acts as a link between the p53 protein and lysosomal labilization?
Figure 7 summarizes the role of lysosomes in apoptosis and emphasizes the crosstalk between lysosomes and mitochondria that seems to occur following lysosomal rupture. This crosstalk seems to result in an amplifying loop with further lysosomal rupture and ensuing mitochondrial damage.
Fig. 7.

Lysosomal–mitochondrial interplay in apoptosis. Activation of the external apoptotic pathway, at least sometimes, results in lysosomal destabilization. This may be a result of direct influence on lysosomes by caspase 8 or by a coupled production of ceramide that is converted to the lysosomotropic detergent sphingosine by ceramidase. It may also be an effect of caspase 8-mediated activation of Bax that attaches itself to, and forms pores in, the lysosomal and outer mitochondrial membranes. Hydrogen peroxide, in combination with lysosomal redox-active iron, produces hydroxyl radicals by the Fenton reaction leading to peroxidation of the lysosomal membrane and subsequent rupture. The lysosomal membrane can be indirectly destabilized by the p53 protein in a manner that is still unknown, and by the newly discovered LAPF protein (these events are perhaps linked). Synthetic lysosomal detergents, such as MSDH, and aldehydes, e.g., 3-aminopropanal, labilize lysosomes. Released lysosomal enzymes can inflict further damage on lysosomes directly or via activation of phospholipases. The internal apoptotic pathway is initiated by damage to mitochondria (e.g., by activated Bid or Bax, phospholipases or, perhaps, lysosomal enzymes), which results in release of cytochrome c and the start of the caspase cascade with ensuing apoptosis
Lipofuscin and postmitotic ageing
Ageing, a progressive decline in an organism’s adaptability and consequent increased morbidity and mortality, largely depends on alterations occurring in long-lived postmitotic cells, such as neurons, cardiac myocytes, and retinal pigment epithelial (RPE) cells. These cells are very rarely (or never) replaced through the division and differentiation of stem cells, thus allowing biological waste materials (such as lipofuscin, irreversibly damaged mitochondria, and aberrant proteins) to accumulate and gradually substitute normal structures, leading to functional decay and cell death (Terman et al. 2007). Other ageing effects, some of which are mainly of cosmetic interest, while others are more serious, include wrinkling of the skin, cataract, and stiffening of blood vessels. These effects are thought to be a consequence of alterations in extra-cellular matrix proteins, which undergo polymerization and loose their elasticity due to glycosylation with accompanying Maillard reactions (binding of carbonyls of reducing sugars to protein-free amino groups) and ensuing Amadori reorganization (the formation of new carbonyls within the complex allowing the binding of additional amino groups and polymerization of proteins to stiff, plastic-like structures) (DeGroot 2004). These reactions, resulting in the formation of advanced glycation endproducts (AGEs) have pronounced similarities with formation of lipofuscin inside lysosomes (see below). The difference is that no sugars are involved in the case of lipofuscin formation when aldehydes, formed by degradation of oxidized lipids, act as linkers between protein residues.
It is now generally accepted that the ageing of long-lived postmitotic cells is induced by endogenously formed (mainly within mitochondria) reactive oxygen species (ROS) causing irreversible damage (mainly to mitochondria) with increased oxidative stress and apoptotic cell death (Barja 2004; Harman 1956; Ku et al. 1993; Lane 2005; Terman et al. 2006). However, if the degradation of damaged structures were to be perfect, no biological ‘garbage’ would accumulate. Therefore, one has to assume that damaged structures accumulate with age because they are not perfectly turned over by the cellular degradation systems, first of all lysosomes, which are responsible for the digestion of a variety of mainly long-lived macromolecules and all cellular organelles.
Although it is rapid and effective, lysosomal (autophagic) degradation is not completely successful. Even under normal conditions, some iron-catalyzed peroxidation occurs intralysosomally (as pointed out, lysosomes are rich in redox-active iron), resulting in oxidative modification of autophagocytosed material and yielding lipofuscin (age pigment), a non-degradable, yellowish-brown, autofluorescent, polymeric compound slowly accumulating within ageing postmitotic cells at a rate that is inversely correlated to the longevity across species (Brunk and Terman 2002).
As pointed out, accumulation of lipofuscin is mainly seen in long-lived postmitotic cells, but a disease-related pigment with similar physico-chemical properties, named ceroid, is also found in cells with a moderate replacement rate as a consequence of reparative autophagy, e.g., in liver cells damaged by ionizing radiation or inflammation (Ghadially 1975). Ceroid also forms due to increased sensitivity to oxidation, such as in vitamin E deficiency (Fattoretti et al. 2002) or disturbed lysosomal degradation, e.g., in lysosomal storage diseases (Wenger et al. 2003). Due to the variable composition of lysosomal pigment in ageing and different diseases, as well as in different tissues, a distinction between lipofuscin and ceroid is reasonable only from an etiological viewpoint. Since ceroid forms more quickly than lipofuscin, the latter is perhaps just a more restructured and advanced polymer—compare with the Amadori reorganization of glucosylated proteins.
The fatal lysosomal storage disease, juvenile neuronal lipofuscinosis, is caused by a mutation of a lysosomal membrane protein, which seems to be of importance for the fusion between lysosomes and autophagosomes. As a result, the influx of degrading enzymes to autophagosomes will be slow (Cao et al. 2006). A delayed degradation allows more time for peroxidation and formation of lipofuscin/ceroid. This scenario can be reproduced in cell cultures exposed to inhibitors of lysosomal proteases (delaying degradation) or to moderate oxidative stress (enhancing oxidation) (reviewed in Brunk and Terman 2002; Terman and Brunk 1998).
Lysosomes receive a wide variety of autophagocytosed subcellular structures, most importantly, mitochondria, which are rich in lipidaceous membrane components and iron-containing proteins, such as cytochrome c. The origin of lipofuscin/ceroid from mitochondrial components is proven by the presence of the ATP-synthase subunit c in age pigment or ceroid granules (Elleder et al. 1997). In professional scavengers, such as RPE cells and macrophages (foam cells) in atheroma, a large portion of lipofuscin (or ceroid) originates from endocytosed material (Lee et al. 1998; Nilsson et al. 2003). Depending on the nature of autophagocytosed/endocytosed material, the composition of lipofuscin varies amongst different types of postmitotic cells, and no chemical formula can be given for this complex substance, mainly composed of cross-linked protein and lipid residues. It should be pointed out that all forms of lipofuscin contain considerable amounts of redox-active iron, which sensitizes lipofuscin-loaded cells to oxidative stress (Brun and Brunk 1973; Brunk and Terman 2002; Jolly et al. 1995).
As already noted, lipofuscin accumulation apparently further compromises autophagic degradative capacity, prolonging the half-lives of long-lived proteins and organelles and creating a situation where cells are forced to exercise their functions with less than perfect tools (Fig. 8). Consistent with this idea, the capacity for autophagic degradation is found to be diminished in aged lipofuscin-loaded cells (Cuervo and Dice 2000; Terman 1995; Terman et al. 1999b), which suggests some serious consequences. For example, delayed degradation of mitochondria would result in increased damage by self-produced ROS, additionally contributing to lipofuscinogenesis and, perhaps, inducing apoptotic cell death (see above).
Fig. 8.

The mitochondrial–lysosomal axis theory of ageing. This hypothesis tries to explain the relationship between lipofuscin accumulation, decreased autophagy, increased ROS production, and mitochondrial damage in senescent long-lived postmitotic cells. Mitochondria are damaged by self-produced hydrogen peroxide, at least if redox-active iron is present. In young cells, damaged mitochondria are rapidly autophagocytosed and degraded because enough lysosomal enzymes are distributed throughout the lysosomal compartment. Small amounts of hydrogen peroxide that normally diffuse into lysosomes lead to cross-linking of intralysosomal material and consequent gradual accumulation of lipofuscin in long-lived postmitotic cells that can neither be degraded nor exocytosed. By purely random reasons, senescent cells have an increased amount of lysosomal enzymes directed towards the plentiful lipofuscin-rich lysosomes. These enzymes are lost for effective autophagic degradation while the lipofusicin remains non-degradable. The consequence for the senescent postmitotic cell is further decreased autophagy, a gradual accumulation of damaged mitochondria, increased hydrogen peroxide production, and apoptotic cell death
Recently, an elegant study on Caenorhabditis elegans added new evidences for the hypothesis that lipofuscin accumulation is causally related to ageing and decline of postmitotic cells. These tiny nematodes are transparent, which allows a direct measurement of lipofuscin by spectrofluorimetry in vivo. It was found that mutant nematodes that lived for longer or shorter than the wild type, accumulated lipofuscin at a slower or quicker pace, respectively. It was also found that caloric restricted worms lived longer and accumulated lipofuscin more slowly than animals fed ad libitum. Finally, when wild type siblings that aged differently, as evaluated by changes in their motility capacity, were compared, it was found that the still mobile and youthful ones at day 11 of their life contained only 25% of the lipofuscin that was found in severely motility-impaired individuals of the same age. This implies that lipofuscin accumulation reflects biological rather than chronological age (Gerstbrein et al. 2005).
Besides the intralysosomal ‘waste’ material lipofuscin, ageing postmitotic cells accumulate extralysosomal ‘garbage’, such as damaged dysfunctional mitochondria and indigestible protein aggregates (aggresomes) that for some reason are not autophagocytosed (reviewed in Terman and Brunk 2005b, 2006). Aged mitochondria are enlarged and show considerably lowered fusion and fission activity. Their autophagy may be prevented by their size, since autophagy of large structures is apparently energy consuming, and autophagosomes seem to have an upper limit in volume (Navratil et al. 2008; Terman et al. 2007).
The accumulation of aberrant proteins within ageing postmitotic cells is a consequence of both ROS-induced damage and incomplete degradation of altered protein molecules. Although damaged proteins may partially preserve their functions, their enzymatic activity per unit mass declines (Rattan 1996; Stadtman et al. 1992). Aberrant proteins often prove to aggregate. Lewy bodies and neurofibrillary tangles (composed of α-synuclein or the hyperphosphorylated protein tau, respectively) are characteristic examples of such aggregates (Bennett 2005; Hardy and Selkoe 2002).
The importance of autophagy for removal of protein aggregates and delaying ageing was demonstrated in a recent study on Drosophila (Simonsen et al. 2008). During normal ageing of the fly, the expression of the Atg8a gene, which is important for autophagy, decreases in neurons resulting in the accumulation of aggregates of ubiquitinated protein. When the expression of Atg8a was upregulated, the aggregates disappeared and the flies showed an increased resistance to oxidative stress as well as a more than 50% prolonged lifespan.
There is accumulating evidence that progressive biological garbage accumulation underlies the development of several late-onset diseases, commonly called age-related diseases. Age-related macular degeneration is associated with a pronounced lipofuscin loading of RPE cells, which prevents phagocytosis of obsolete photoreceptor outer segments resulting in the degeneration of photoreceptors (Nilsson et al. 2003). It is possible that intralysosomal accumulation of oxidized LDL within iron-rich lysosomes of vessel macrophages may induce foam cell formation and apoptotic cell death due to lysosomal destabilization, contributing to atherosclerosis (Li et al. 2001, 2006). Moreover, accumulation of intracellular ‘waste’ material such as lipofuscin and defective mitochondria within cardiac myocytes results in progressive decline of cardiac function leading to heart failure (Terman and Brunk 2005b; Terman et al. 2008).
Formation of indigestible protein aggregates is involved in a number of age-related neurodegenerative diseases. Alzheimer’s disease is characterized by the intraneuronal aggregation of protein tau forming neurofibrillary tangles (see above) and extraneuronal β-amyloid plaque formation (Hardy and Selkoe 2002). There is evidence that β-amyloid plaques may form as a result of neuronal death following intralysosomal accumulation of amyloid β-protein (Gouras et al. 2005; Yu et al. 2005; Zheng et al. 2006). In Parkinson’s disease dopaminergic neurons of substantia nigra accumulate α-synuclein aggregates (Yu et al. 2005), while formation of huntingtin aggresomes within striatal neurons is a characteristic of Huntington’s disease (Lee and Kim 2006).
Conclusion and perspectives
There is increasing recognition that lysosomes are central to a great number of cellular functions including not only the removal of exhausted organelles but also the metabolic cycling iron and the initiation—at least in some cases—of apoptotic cell death. Lysosomal dysfunction—such as occurs in ageing and certain lysosomal storage diseases—can be a major factor limiting function and longevity of the organism. Appreciation of the central role of lysosomes in ageing, and certain disease states, may lead to the development of new therapeutic agents aimed at preserving or correcting abnormal lysosomal function.
Acknowledgments
We are grateful to Stephen Hampson for excellent linguistic advice.
Abbreviations
- AO
Acridine orange
- ATG
Autophagy-related genes
- CMA
Chaperone-mediated autophagy
- DFO
Desferrioxamine
- LDL
Low density lipoprotein
- LIP
Labile iron pool
- LMP
Lysosomal membrane permeability
- MMP
Mitochondrial membrane permeability
- MP
Mannose-6-phosphate
- MPR
Mannose-6-phosphate receptor
- MPT
Mitochondrial permeability transition
- MSDH
o-Methylserine dodecylamide hydrochloride
- ROS
Reactive oxygen species
- RPE
Retinal pigment epithelial
- SIH
Salicylaldehyde isonicotinoyl hydrazone
- SSM
Sulfide-silver method
- Tf
Transferrin
- TfR
Transferrin receptor
- TGN
trans-Golgi network
References
- Antunes F, Cadenas E, Brunk UT (2001) Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J 356:549–555 [DOI] [PMC free article] [PubMed]
- Arosio P, Levi S (2002) Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med 33:457–463 [DOI] [PubMed]
- Autelli R, Crepaldi S, De Stefanis D, Parola M, Bonelli G, Baccino FM (2005) Intracellular free iron and acidic pathways mediate TNF-induced death of rat hepatoma cells. Apoptosis 10:777–786 [DOI] [PubMed]
- Baird SK, Kurz T, Brunk UT (2006) Metallothionein protects against oxidative stress-induced lysosomal destabilization. Biochem J 394:275–283 [DOI] [PMC free article] [PubMed]
- Barbouti A, Amorgianiotis C, Kolettas E, Kanavaros P, Galaris D (2007) Hydrogen peroxide inhibits caspase-dependent apoptosis by inactivating procaspase-9 in an iron-dependent manner. Free Radic Biol Med 43:1377–1387 [DOI] [PubMed]
- Barja G (2004) Free radicals and aging. Trends Neurosci 27:595–600 [DOI] [PubMed]
- Bartrons R, Caro J (2007) Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr 39:223–229 [DOI] [PubMed]
- Baumgartner HK, Gerasimenko JV, Thorne C, Ashurst LH, Barrow SL, Chvanov MA, Gillies S, Criddle DN, Tepikin AV, Petersen OH et al (2007) Caspase-8-mediated apoptosis induced by oxidative stress is independent of the intrinsic pathway and dependent on cathepsins. Am J Physiol Gastrointest Liver Physiol 293:G296–G307 [DOI] [PubMed]
- Bennett MC (2005) The role of alpha-synuclein in neurodegenerative diseases. Pharmacol Ther 105:311–331 [DOI] [PubMed]
- Berg D, Youdim MB (2006) Role of iron in neurodegenerative disorders. Top Magn Reson Imaging 17:5–17 [DOI] [PubMed]
- Bergamini E (2006) Autophagy: a cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol Aspects Med 27:403–410 [DOI] [PubMed]
- Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A (2003) Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem 278:31401–31411 [DOI] [PubMed]
- Billings J (1874) Everybody’s friend, or: Josh Billing’s encyclopedia and proverbial philosophy of wit and humor. American Publishing Company, Hartford
- Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, Ojcius DM, Jaattela M, Kroemer G (2003) Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med 197:1323–1334 [DOI] [PMC free article] [PubMed]
- Brun A, Brunk U (1970) Histochemical indications for lysosomal localization of heavy metals in normal rat brain and liver. J Histochem Cytochem 18:820–827 [DOI] [PubMed]
- Brun A, Brunk U (1973) Heavy metal localization and age related accumulation in the rat nervous system. A histochemical and atomic absorption spectrophotometric study. Histochemie 34:333–342 [DOI] [PubMed]
- Brunk U (1973) Distribution and shifts of ingested marker particles in residual bodies and other lysosomes. Studies on in vitro cultivated human glia cells in phase II and 3. Exp Cell Res 79:15–27 [DOI] [PubMed]
- Brunk U, Ericsson JL (1972a) Electron microscopical studies on rat brain neurons. Localization of acid phosphatase and mode of formation of lipofuscin bodies. J Ultrastruct Res 38:1–15 [DOI] [PubMed]
- Brunk UT, Ericsson JL (1972b) Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lysosomal membranes. Histochem J 4:479–491 [DOI] [PubMed]
- Brunk UT, Eaton JW (2007) Peroxide hormesis? A commentary on “Hydrogen peroxide inhibits caspase-dependent apoptosis by inactivating procaspase-9 in an iron-dependent manner”. Free Radic Biol Med 43:1372–1373 [DOI] [PubMed]
- Brunk UT, Svensson I (1999) Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep 4:3–11 [DOI] [PubMed]
- Brunk UT, Terman A (2002) Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med 33:611–619 [DOI] [PubMed]
- Brunk UT, Dalen H, Roberg K, Hellquist HB (1997) Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med 23:616–626 [DOI] [PubMed]
- Brunk UT, Neuzil J, Eaton JW (2001) Lysosomal involvement in apoptosis. Redox Rep 6:91–97 [DOI] [PubMed]
- Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL (2006) Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem 281:20483–20493 [DOI] [PubMed]
- Castino R, Bellio N, Nicotra G, Follo C, Trincheri NF, Isidoro C (2007) Cathepsin D-Bax death pathway in oxidative stressed neuroblastoma cells. Free Radic Biol Med 42:1305–1316 [DOI] [PubMed]
- Chen W, Li N, Chen T, Han Y, Li C, Wang Y, He W, Zhang L, Wan T, Cao X (2005) The lysosome-associated apoptosis-inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain-containing proteins, induces caspase-independent apoptosis via the lysosomal–mitochondrial pathway. J Biol Chem 280:40985–40995 [DOI] [PubMed]
- Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM, Salvesen GS, Turk B (2004) Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 279:3578–3587 [DOI] [PubMed]
- Cuervo AM, Dice JF (2000) When lysosomes get old. Exp Gerontol 35:119–131 [DOI] [PubMed]
- Cuervo AM, Palmer A, Rivett AJ, Knecht E (1995) Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem 227:792–800 [DOI] [PubMed]
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:131–140 [DOI] [PubMed]
- de Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J (2006) Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. Embo J 25:5396–5404 [DOI] [PMC free article] [PubMed]
- de Duve C (1959) Lysosomes, a new group of cytoplasmic particles. In: Hayashi T (ed) Subcellular particles. The Ronald Press Co., New York, pp 128–159
- de Duve C (2005) The lysosome turns fifty. Nat Cell Biol 7:847–849 [DOI] [PubMed]
- de Duve C, Wattiaux R (1966) Functions of lysosomes. Annu Rev Physiol 28:435–492 [DOI] [PubMed]
- de Milito A, Fais S (2005) Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol 1:779–786 [DOI] [PubMed]
- DeGroot J (2004) The AGE of the matrix: chemistry, consequence and cure. Curr Opin Pharmacol 4:301–305 [DOI] [PubMed]
- Doulias PT, Christoforidis S, Brunk UT, Galaris D (2003) Endosomal and lysosomal effects of desferrioxamine: protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest. Free Radic Biol Med 35:719–728 [DOI] [PubMed]
- Dunn WA Jr (1990) Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol 110:1935–1945 [DOI] [PMC free article] [PubMed]
- Eaton JW, Qian M (2002) Molecular bases of cellular iron toxicity. Free Radic Biol Med 32:833–840 [DOI] [PubMed]
- Elleder M, Sokolova J, Hrebicek M (1997) Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol 93:379–390 [DOI] [PubMed]
- Fattoretti P, Bertoni-Freddari C, Casoli T, Di Stefano G, Solazzi M, Corvi E (2002) Morphometry of age pigment (lipofuscin) and of ceroid pigment deposits associated with vitamin E deficiency. Arch Gerontol Geriatr 34:263–268 [DOI] [PubMed]
- Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E (2006) Mitochondria in hematopoiesis and hematological diseases. Oncogene 25:4757–4767 [DOI] [PubMed]
- Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB (2002) PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ Res 90:1012–1019 [DOI] [PubMed]
- Garner B, Li W, Roberg K, Brunk UT (1997) On the cytoprotective role of ferritin in macrophages and its ability to enhance lysosomal stability. Free Radic Res 27:487–500 [DOI] [PubMed]
- Garner B, Roberg K, Brunk UT (1998) Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radic Res 29:103–114 [DOI] [PubMed]
- Gerstbrein B, Stamatas G, Kollias N, Driscoll M (2005) In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditis elegans. Aging Cell 4:127–137 [DOI] [PubMed]
- Ghadially FN (1975) Ultrastructural pathology of the cell and matrix second edit. Butterworths, London
- Glaumann H, Ericsson JL, Marzella L (1981) Mechanisms of intralysosomal degradation with special reference to autophagocytosis and heterophagocytosis of cell organelles. Int Rev Cytol 73:149–182 [DOI] [PubMed]
- Gouras GK, Almeida CG, Takahashi RH (2005) Intraneuronal abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging 26:1235–1244 [DOI] [PubMed]
- Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene 23:2881–2890 [DOI] [PubMed]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356 [DOI] [PubMed]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 211:298–300 [DOI] [PubMed]
- Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W, Trauzold A, Hethke A, Schutze S (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 11:550–563 [DOI] [PubMed]
- Jolly RD, Douglas BV, Davey PM, Roiri JE (1995) Lipofuscin in bovine muscle and brain: a model for studying age pigment. Gerontology 41:283–295 [DOI] [PubMed]
- Kågedal K, Zhao M, Svensson I, Brunk UT (2001) Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J 359:335–343 [DOI] [PMC free article] [PubMed]
- Kaushik S, Cuervo AM (2006) Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med 27:444–454 [DOI] [PMC free article] [PubMed]
- Kidane TZ, Sauble E, Linder MC (2006) Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol 291:445–455 [DOI] [PubMed]
- Kiselyov K, Jennigs JJ Jr, Rbaibi Y, Chu CT (2007) Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy 3:259–262 [DOI] [PMC free article] [PubMed]
- Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937 [DOI] [PubMed]
- Koorts AM, Viljoen M (2007) Ferritin and ferritin isoforms I: Structure–function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113:30–54 [DOI] [PubMed]
- Kroemer G, Jäättelä M (2005) Lysosomes and autophagy in cell death control. Nat Rev Cancer 5:886–897 [DOI] [PubMed]
- Kruszewski M (2003) Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res 531:81–92 [DOI] [PubMed]
- Ku HH, Brunk UT, Sohal RS (1993) Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic Biol Med 15:621–627 [DOI] [PubMed]
- Kurz T, Leake A, Von Zglinicki T, Brunk UT (2004) Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem J 378:1039–1045 [DOI] [PMC free article] [PubMed]
- Kurz T, Gustafsson B, Brunk UT (2006) Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J 273:3106–3117 [DOI] [PubMed]
- Kurz T, Terman A, Brunk UT (2007) Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 462:220–230 [DOI] [PubMed]
- Lane N (2005) Power, sex, suicide: mitochondria and the meaning of life. Oxford University Press, Oxford, pp 289–301
- Lee ST, Kim M (2006) Aging and neurodegeneration. Molecular mechanisms of neuronal loss in Huntington’s disease. Mech Ageing Dev 127:432–435 [DOI] [PubMed]
- Lee FY, Lee TS, Pan CC, Huang AL, Chau LY (1998) Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis 138:281–288 [DOI] [PubMed]
- Levine AJ, Hu W, Feng Z (2006) The P53 pathway: what questions remain to be explored? Cell Death Differ 13:1027–1036 [DOI] [PubMed]
- Li W, Dalen H, Eaton JW, Yuan XM (2001) Apoptotic death of inflammatory cells in human atheroma. Arterioscler Thromb Vasc Biol 21:1124–1130 [DOI] [PubMed]
- Li W, Yuan XM, Ivanova S, Tracey KJ, Eaton JW, Brunk UT (2003) 3-Aminopropanal, formed during cerebral ischaemia, is a potent lysosomotropic neurotoxin. Biochem J 371:429–436 [DOI] [PMC free article] [PubMed]
- Li W, Ostblom M, Xu LH, Hellsten A, Leanderson P, Liedberg B, Brunk UT, Eaton JW, Yuan XM (2006) Cytocidal effects of atheromatous plaque components: the death zone revisited. Faseb J 20:2281–2290 [DOI] [PubMed]
- Liu X, Jin W, Theil EC (2003) Opening protein pores with chaotropes enhances Fe reduction and chelation of Fe from the ferritin biomineral. Proc Natl Acad Sci USA 100:3653–3658 [DOI] [PMC free article] [PubMed]
- Lorenzo K, Ton P, Clark JL, Coulibaly S, Mach L (2000) Invasive properties of murine squamous carcinoma cells: secretion of matrix-degrading cathepsins is attributable to a deficiency in the mannose 6-phosphate/insulin-like growth factor II receptor. Cancer Res 60:4070–4076 [PubMed]
- Luzio JP, Pryor PR, Bright NA (2007) Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8:622–632 [DOI] [PubMed]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752 [DOI] [PubMed]
- Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ (1993) Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol 265:C1015–C1029 [DOI] [PubMed]
- Michalovitz D, Halevy O, Oren M (1990) Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 62:671–680 [DOI] [PubMed]
- Miyajima H (2003) Aceruloplasminemia, an iron metabolic disorder. Neuropathology 23:345–350 [DOI] [PubMed]
- Miyawaki H (1965) Histochemistry and electron microscopy of iron-containing granules, lysosomes, and lipofuscin in mouse mammary glands. J Natl Cancer Inst 34:601–623 [PubMed]
- Montcourrier P, Silver I, Farnoud R, Bird I, Rochefort H (1997) Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin Exp Metastasis 15:382–392 [DOI] [PubMed]
- Navratil M, Terman A, Arriaga EA (2008) Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp Cell Res 314:164–172 [DOI] [PubMed]
- Nilsson E, Ghassemifar R, Brunk UT (1997) Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J 29:857–865 [DOI] [PubMed]
- Nilsson SE, Sundelin SP, Wihlmark U, Brunk UT (2003) Aging of cultured retinal pigment epithelial cells: oxidative reactions, lipofuscin formation and blue light damage. Doc Ophthalmol 106:13–16 [DOI] [PubMed]
- Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M (2004) Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 200:425–435 [DOI] [PMC free article] [PubMed]
- Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA 75:3327–3331 [DOI] [PMC free article] [PubMed]
- Pandolfo M (2006) Iron and Friedreich ataxia. J Neural Transm Suppl 70:143–146 [DOI] [PubMed]
- Persson HL, Nilsson KJ, Brunk UT (2001) Novel cellular defenses against iron and oxidation: ferritin and autophagocytosis preserve lysosomal stability in airway epithelium. Redox Rep 6:57–63 [DOI] [PubMed]
- Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT (2003) Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med 34:1295–1305 [DOI] [PubMed]
- Pisoni RL, Acker TL, Lisowski KM, Lemons RM, Thoene JG (1990) A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J Cell Biol 110:327–335 [DOI] [PMC free article] [PubMed]
- Podgorski I, Sloane BF (2003) Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp 70:263–276 [DOI] [PubMed]
- Radisky DC, Kaplan J (1998) Iron in cytosolic ferritin can be recycled through lysosomal degradation in human fibroblasts. Biochem J 336:201–205 [DOI] [PMC free article] [PubMed]
- Rattan SI (1996) Synthesis, modifications, and turnover of proteins during aging. Exp Gerontol 31:33–47 [DOI] [PubMed]
- Richardson DR (2002) Therapeutic potential of iron chelators in cancer therapy. Adv Exp Med Biol 509:231–249 [DOI] [PubMed]
- Roberts S, Bomford A (1988) Ferritin iron kinetics and protein turnover in K562 cells. J Biol Chem 263:19181–19187 [PubMed]
- Rochefort H, Capony F, Garcia M (1990) Cathepsin D: a protease involved in breast cancer metastasis. Cancer Metastasis Rev 9:321–331 [DOI] [PubMed]
- Rupar CA, Albo S, Whitehall JD (1992) Rat liver lysosome membranes are enriched in alpha-tocopherol. Biochem Cell Biol 70:486–488 [DOI] [PubMed]
- Sakaida I, Kyle ME, Farber JL (1990) Autophagic degradation of protein generates a pool of ferric iron required for the killing of cultured hepatocytes by an oxidative stress. Mol Pharmacol 37:435–442 [PubMed]
- Schafer FQ, Buettner GR (2000) Acidic pH amplifies iron-mediated lipid peroxidation in cells. Free Radic Biol Med 28:1175–1181 [DOI] [PubMed]
- Schraufstätter I, Hyslop PA, Jackson JH, Cochrane CG (1988) Oxidant-induced DNA damage of target cells. J Clin Invest 82:1040–1050 [DOI] [PMC free article] [PubMed]
- Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P, Zhong H (2001) The metabolism of tumours: 70 years later. Novartis Found Symp 240:251–260 [PubMed]
- Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, Pablo RD, Tacchetti C, Rubinsztein DC, Ballabio A (2008) A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17:119–129 [DOI] [PubMed]
- Settembre C, Fraldi A, Rubinsztein DC, Ballabio A (2008) Lysosomal storage diseases as disorders of autophagy. Autophagy 4:113–114 [DOI] [PubMed]
- Seymour CA, Peters TJ (1978) Organelle pathology in primary and secondary haemochromatosis with special reference to lysosomal changes. Br J Haematol 40:239–253 [DOI] [PubMed]
- Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995 [DOI] [PMC free article] [PubMed]
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4:176–184 [DOI] [PubMed]
- Sipe JC, Lee P, Beutler E (2002) Brain iron metabolism and neurodegenerative disorders. Dev Neurosci 24:188–196 [DOI] [PubMed]
- Stadtman ER, Starke-Reed PE, Oliver CN, Carney JM, Floyd RA (1992) Protein modification in aging. Exs 62:64–72 [DOI] [PubMed]
- Suzuki K, Ohsumi Y (2007) Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 581:2156–2161 [DOI] [PubMed]
- Takagi H, Shi D, Ha Y, Allewell NM, Theil EC (1998) Localized unfolding at the junction of three ferritin subunits. A mechanism for iron release? J Biol Chem 273:18685–18688 [DOI] [PubMed]
- Tang D, Lahti JM, Grenet J, Kidd VJ (1999) Cycloheximide-induced T-cell death is mediated by a Fas-associated death domain-dependent mechanism. J Biol Chem 274:7245–7252 [DOI] [PubMed]
- Tenopoulou M, Doulias PT, Barbouti A, Brunk U, Galaris D (2005) Role of compartmentalized redox-active iron in hydrogen peroxide-induced DNA damage and apoptosis. Biochem J 387:703–710 [DOI] [PMC free article] [PubMed]
- Terman A (1995) The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology 41:319–326 [DOI] [PubMed]
- Terman A, Brunk UT (1998) Lipofuscin: mechanisms of formation and increase with age. Apmis 106:265–276 [DOI] [PubMed]
- Terman A, Brunk UT (2004) Lipofuscin. Int J Biochem Cell Biol 36:1400–1404 [DOI] [PubMed]
- Terman A, Brunk UT (2005a) The aging myocardium: roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ 14:107–114 [DOI] [PubMed]
- Terman A, Brunk UT (2005b) Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 68:355–365 [DOI] [PubMed]
- Terman A, Brunk UT (2006) Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal 8:197–204 [DOI] [PubMed]
- Terman A, Abrahamsson N, Brunk UT (1999a) Ceroid/lipofuscin-loaded human fibroblasts show increased susceptibility to oxidative stress. Exp Gerontol 34:755–770 [DOI] [PubMed]
- Terman A, Dalen H, Brunk UT (1999b) Ceroid/lipofuscin-loaded human fibroblasts show decreased survival time and diminished autophagocytosis during amino acid starvation. Exp Gerontol 34:943–957 [DOI] [PubMed]
- Terman A, Gustafsson B, Brunk UT (2006) The lysosomal–mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 163:29–37 [DOI] [PubMed]
- Terman A, Gustafsson B, Brunk UT (2007) Autophagy, organelles and ageing. J Pathol 211:134–143 [DOI] [PubMed]
- Terman A, Kurz T, Gustafsson B, Brunk UT (2008) The involvement of lysosomes in myocardial aging and disease. Curr Cardiol Rev (in press) [DOI] [PMC free article] [PubMed]
- Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresic K, Turk V (2002) Apoptotic pathways: involvement of lysosomal proteases. Biol Chem 383:1035–1044 [DOI] [PubMed]
- Vaisman B, Fibach E, Konijn AM (1997) Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood 90:831–838 [PubMed]
- Viarengo A, Burlando B, Ceratto N, Panfoli I (2000) Antioxidant role of metallothioneins: a comparative overview. Cell Mol Biol 46:407–417 [PubMed]
- Victor BC, Sloane BF (2007) Cysteine cathepsin non-inhibitory binding partners: modulating intracellular trafficking and function. Biol Chem 388:1131–1140 [DOI] [PubMed]
- Walton JH, Lewis HA (1916) The partition coefficients of hydrogen peroxide between water and certain organic solvents. J Am Chem Soc 38:633–638 [DOI]
- Wenger DA, Coppola S, Liu SL (2003) Insights into the diagnosis and treatment of lysosomal storage diseases. Arch Neurol 60:322–328 [DOI] [PubMed]
- Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ (2007) Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem 282:28960–28970 [DOI] [PubMed]
- Yeung BH, Huang DC, Sinicrope FA (2006) PS-341 (bortezomib) induces lysosomal cathepsin B release and a caspase-2-dependent mitochondrial permeabilization and apoptosis in human pancreatic cancer cells. J Biol Chem 281:11923–11932 [DOI] [PubMed]
- Yorimitsu T, Klionsky DJ (2005) Autophagy: molecular machinery for self-eating. Cell Death Differ 12:1542–1552 [DOI] [PMC free article] [PubMed]
- Yu Z, Li W, Brunk UT (2003a) 3-Aminopropanal is a lysosomotropic aldehyde that causes oxidative stress and apoptosis by rupturing lysosomes. Apmis 111:643–652 [DOI] [PubMed]
- Yu Z, Persson HL, Eaton JW, Brunk UT (2003b) Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med 34:1243–1252 [DOI] [PubMed]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO et al (2005) Macroautophagy—a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171:87–98 [DOI] [PMC free article] [PubMed]
- Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L, Brunk UT (2002) Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci USA 99:6286–6291 [DOI] [PMC free article] [PubMed]
- Zdolsek JM, Roberg K, Brunk UT (1993) Visualization of iron in cultured macrophages: a cytochemical light and electron microscopic study using autometallography. Free Radic Biol Med 15:1–11 [DOI] [PubMed]
- Zhang AS, Sheftel AD, Ponka P (2005) Intracellular kinetics of iron in reticulocytes: evidence for endosome involvement in iron targeting to mitochondria. Blood 105:368–375 [DOI] [PubMed]
- Zhao M, Antunes F, Eaton JW, Brunk UT (2003) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem 270:3778–3786 [DOI] [PubMed]
- Zheng L, Roberg K, Jerhammar F, Marcusson J, Terman A (2006) Autophagy of amyloid beta-protein in differentiated neuroblastoma cells exposed to oxidative stress. Neurosci Lett 394:184–189 [DOI] [PubMed]