

Abstract
Gene synthesis is hampered by two obstacles:
improper assembly of oligonucleotides
oligonucleotide defects incurred during chemical synthesis.
To overcome the first problem, we describe the employment of a Computationally Optimised DNA Assembly (CODA) algorithm that uses the degeneracy of the genetic code to design overlapping oligonucleotides with thermodynamic properties for self-assembly into a single, linear, DNA product. To address the second problem, we describe a hierarchical assembly strategy that reduces the incorporation of defective oligonucleotides into full-length gene constructs. The CODA algorithm and these biological methods enable fast, simple and reliable assemblies of sequence-correct full-length genes.
Keywords: synthetic biology, computational biology, DNA self-assembly, gene synthesis, Computationally Optimised DNA Assembly, CODA, bioinformatics
1 Introduction
It is often desirable to obtain a synthetic DNA molecule that encodes a protein of interest and has a sequence simultaneously optimised for several arbitrarily specified sequence properties, such as its own self-assembly, codon usage (Grosjean and Fiers, 1982) GC/AT ratio (Breslauer et al., 1986; Lio, 2002), translational kinetics (Sorensen et al., 1989), codon-pair bias (Gutman and Hatfield, 1989; Hatfield, 1993; Irwin et al., 1995) and other DNA structural scales (Seeman, 1998, 1999). Since synthesis of long gene sequences is not feasible because of cumulative chemical synthesis errors, largely point deletions (Smith et al., 2003), most synthetic genes are assembled from a large number of short partially overlapping complementary oligonucleotides (Stemmer et al., 1995). These overlapping oligonucleotides are assembled into long double-stranded DNA with ligation and polymerase extension reactions, either alone or in combination, in processes known variously as assembly PCR (Casimiro et al., 1997), splicing by overlap extension (Warrens et al., 1997), polymerase chain assembly (Smith et al., 2003), recursive PCR (Prodromou and Pearl, 1992), and others (Mandecki and Bolling, 1988; Theriault et al., 1988). However, all such approaches confront two major obstacles to accurate and reliable gene synthesis. Firstly, incorrect hybridisations during assembly produce an olio of DNA products of widely differing lengths predominantly arranged in the wrong order. Secondly, oligonucleotide synthesis defects produce frameshift (nonsense) mutations and other single-point errors. For example, with a 99.5% coupling efficiency, nearly 25% of chemically synthesised oligonucleotides 50 nucleotides in length contain an error.
Methods described in this paper address both problems by combining information technology with biological methods. Information technology is used to design a DNA sequence that encodes, simultaneously, the correct amino acid sequence, its own correct self-assembly from unpurified purchased oligonucleotides, a target expression organism, and other desired sequence properties. An efficient genetic screen to eliminate translational frameshifts caused by defective oligonucleotides also is described.
2 Methods
2.1 Materials
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). PfuUltra High-Fidelity DNA Polymerase was purchased from Strategene (La Jolla, CA). The expression vector, pET-3a, was purchased from Novagen (Gibbstown, NJ). Restriction enzymes NdeI, BamHI, EcoRI, Mung Bean Nuclease, and T4 DNA ligase were obtained from New England Biolabs. Zero Blunt TOPO PCR Cloning kit was purchased from Invitrogen. pGEM-3Z vector was obtained from Promega (Madison, WI).
2.2 Sequence optimisation engine
Each possible codon assignment influences several DNA sequence properties of interest, and all properties are optimised simultaneously. We define a general sequence objective function (figure of merit) called the Usability Codon Index, which is the harmonic mean of the desired properties, , where each property fi takes values in the half-open interval (0, 1] and higher values are better. The properties used here were:
the melting temperature gap discussed below
the codon adaptation index (Sharp and Li, 1987) for preference of codons of abundant tRNA isoacceptors (Boycheva et al., 2003; Gutman and Hatfield, 1989; Hatfield, 1993); the avoidance of consecutive rare codons (Hatfield, 1993; Moore and Maranas, 2002)
codon pair statistics to minimise codon pairs with slow translational step-times (Gutman and Hatfield, 1989; Irwin et al., 1995).
Thus, each gene sequence is designed to encode its own correct self-assembly using synonymous codon substitutions for reliable expression in any in vivo or in vitro system of choice.
2.3 Computational infrastructure
The computational optimisation framework uses a multi-queue branch-and-bound algorithm (Lathrop et al., 2001). Allegro Common LISP was licensed from Franz, Inc. DNA secondary structure and melting temperatures were estimated using the Mfold software package (Zuker, 1989; Zuker et al., 1999) based on nearest-neighbour thermodynamic parameters (SantaLucia, 1998) Codon usage was estimated using the codon adaptation index (Sharp and Li, 1987) and codon pair statistics (Boycheva et al., 2003; Gutman and Hatfield, 1989; Moore and Maranas, 2002). Computations were performed on a 64-node cluster of 3 GHz Xeon dual processors.
2.4 Parallel system architecture
The system exploits both coarse- and fine-grained parallelism simultaneously on the same cluster. At a coarse-grain level, the design of a gene is divided into sub-designs for each intermediate DNA fragment. The sub-designs are run in parallel and recombined into an initial gene design which is then further refined. The coarse-grain level executes a general multi-queue branch-and-bound optimisation engine, written in LISP, consuming one cluster node for the design of each intermediate DNA fragment (Figure 1). At a fine-grain level, each step of sequence design requires estimating the melting temperatures of a large number of possible hybridisations. These are run in parallel and later recombined to yield an estimate of the melting temperature gap for that particular step of the design. The fine-grain level executes a series of PERL programs and Linux shell scripts that divide a large DNA melting run into individual Mfold thermodynamic calculations to be executed in parallel on the cluster.
Figure 1.
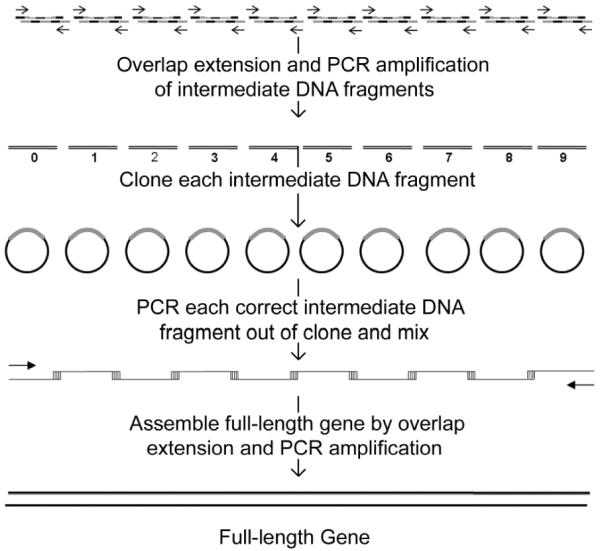
Gene Assembly Schematics. Self-assembled overlapping oligonucleotides are extended by overlap extension and PCR to generate intermediate DNA fragments. The intermediate DNA fragments are cloned to be sequenced and PCR amplified from the selected vectors. Intermediate DNA fragments with correct DNA sequences are mixed together and extended by overlap extension and PCR amplification to generate the full-length gene
2.5 Assembly of intermediate DNA fragments
For each intermediate DNA fragment, the constituent oligonucleotide set was added to a primer extension reaction at a final concentration of 0.1 μM along with an excess (1 μM) of leader and trailer primer oligonucleotide (the most 5'- and 3'-distant oligonucleotides). Each oligonucleotide set was extended into an intermediate DNA fragment with 2.5 U of PfuUltra High-Fidelity DNA polymerase (Stratagene), 300 μM dNTPs (Roche Diagnostics, Indianapolis, IN), and 1X PfuUltra reaction buffer. These primer extension and PCR amplification reactions were performed in a MJ Research PTC-225 thermal cycler using the following calculated-control protocol: 1 min denaturation step at 95°C, followed by 35 cycles of 10 s at 95°C, 40 s at 60°C, and 2 min at 72°C, and a final step of 2 min at 72°C.
2.6 Topo cloning and sequencing of intermediate DNA fragments
For each intermediate DNA fragment, 1 μL of the PCR product of the assembly reaction was used for cloning into a pCRII-Blunt-TOPO vector (Invitrogen (Carlsbad, CA)) according to the manufacturer's protocol. Reactions were incubated for 5 min at room temperature and 1 μL of each reaction was used for transformation of One Shot electrocompetent cells according to the manufacture's protocol. Following transformation, plasmid DNA was isolated for DNA sequencing.
2.7 Preparation and use of CODA blue vector for frameshift screening
pGEM-3Z plasmid DNA was digested with EcoRI and treated with Mung Bean Nuclease (New England Biolabs (Beverly, MA)) according to the manufacturer's protocol. The linearised plasmid was extracted and purified from an agarosegel with a Qiagen MinElute gel extraction kit and religated with T4 DNA ligase. The four bases (AATT) deleted from this plasmid result in a + 1 frameshift in the α-complementing subunit of β-galactosidase. Intermediate DNA fragments were cloned into the BamHI site of the CODA Blue vector and transformed in E. coli JM109 and plated on LB agar plates containing 100 μg/ml ampicillin, 80 μg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) and 0.5 mM Isopropyl-β-D-thiogalactopyranoside (IPTG). Blue colonies were chosen for plasmid DNA isolation and DNA sequencing.
2.8 Construction of the full-length Integrase (IN) gene
The 5'- and 3'-most distant oligonucleotides were used to PCR amplify each sequence verified intermediate DNA fragment with PfuUltra High-Fidelity DNA polymerase (Stratagene). Next, 1 μl of each intermediate DNA fragment PCR product was combined with an IN gene leader (1 μM) containing an NdeI site and a trailer (1 μM) containing a BamHI site. The full-length IN gene was isolated by primer extension and PCR amplification with 2.5 U of PfuUltra High-Fidelity DNA polymerase (Stratagene), 300 μM dNTPs, and 1X PfuUltra buffer. These primer extension and PCR amplification reactions were performed in a MJ Research PTC-225 thermal cycler using the following calculated-control protocol: 5 min denaturation step at 95°C, followed by 35 cycles of 30 s at 95°C, 30 s at 68°C, and 5 min at 72°C, and a final step of 5 min at 72°C. The PCR products were subjected to electrophoresis in 1% agarose gel. Full-length IN gene was inserted into the NdeI and BamHI sites of the expression vector pET-3a (Novagen) and full-length IN sequence was verified by DNA sequencing and protein expression.
2.9 Probabilities
Sequencing of Intermediate DNA Fragments without Frameshift Screening
Let Pe be the per-base error rate, L the length of a clone in bases, N the number of clones sequenced, Pc the probability that any given clone is correct, and Po the probability that at least one of the N sequenced clones is correct. In these cases, Pc = (1 - Pe)L and Po = (1 - (1 - Pc)N). If the gene consists of M intermediate fragments, each of length L, and N clones are sequenced for each fragment, then the probability that every one of the M fragments has at least one correct clone is .
Sequencing of Intermediate DNA Fragments after Frameshift Screening
Let Pd be the per-base point deletion rate, Pi the per-base point insertion rate, and Pm the per-base point mutation rate. Assume Pe = Pd + Pi + Pm and 1 >> Pd, Pi, Pm so that product terms may be neglected. Let P[d=j] be the probability of exactly j deletions in any given clone, P[i=j] of exactly j insertions, and P[m=j] of exactly j mutations. Then , where (Lj) = L!/j!(L - j)! is the number of ways of choosing j of L objects. Similar expressions are obtained for P[i=j] and P[m=j]. Let Pb be the probability that an insert in the CODA Blue plasmid turns the clone blue. A clone turns blue when the number of point insertions minus the number of point deletions is an integer multiple of three. Thus, Pb = ∑((j-k mod 3) = 0)P[i=j]P[d=k]. The probability that any single clone is correct, given that it turns blue, is P[c|b] = P[d=0]P[i=0]P[m=0]/Pb = Pc/Pb. Probabilities corresponding to Po and Pg above, but given blue clones, may be obtained by substituting P[c|b] for Pc. Thus, P[o|b] = (1 - (1 - P[c|b])N) and .
3 Results
In this paper, we describe the assembly of a 1,640 bp synthetic gene of the yeast, Saccharomyces cerevisiae, transposable element Ty3 Integrase (IN) protein optimised for expression in E. coli. Figure 1 illustrates the assembly process. The gene was divided into ten intermediate DNA fragments, each of which was subdivided into six or eight short overlapping and abutting oligonucleotides. DNA sequences for each oligonucleotide were designed computationally and purchased commercially. Self-assembled intermediate DNA fragments were isolated by polymerase extension and cloned. Each intermediate DNA fragment was amplified from selected clones and combined in another primer extension and PCR amplification reaction to generate the full-length IN gene.
3.1 Computationally Optimised DNA Assembly (CODA)
Given the amino acid sequence of the Ty3 IN protein, the CODA algorithm described in Methods generated a list of linearly overlapping and abutting oligonucleotides usually 45-50 nucleotides (nts) long, optimised for self-assembly and expression in E. coli. Correct self-assembly is achieved by a designed melting temperature gap, defined as [the minimum melting temperature of any correct hybridisation] minus [the maximum melting temperature of any incorrect hybridisation]. Correct hybridisations occur only at the overlap regions between adjacent segments when they are aligned to hybridise linearly across the entire extent of the overlap. Incorrect hybridisations occur as:
hairpins, where an oligonucleotide folds back and hybridises to itself
dimers, where an oligonucleotide is partially self-complementary
inter-oligonucleotide mismatches, where one oligonucleotide is partially complementary to another
shifted correct matches, where a misaligned overlap region is partially complementary to another region within the same overlap.
While previous approaches have removed local DNA structure such as hairpins and palindromes, the key extension here is to examine all possible incorrect hybridisations exhaustively.
A melting temperature gap
Different oligonucleotides possess different melting temperatures depending on their base composition and degree of base pairing complementarity (Breslauer et al., 1986; Mathews et al., 1999; SantaLucia, 1998). The CODA algorithm takes advantage of the degeneracy of the genetic code to assign codons so that correct hybridisations of the designed overlapping oligonucleotides have a high melting temperature and incorrect hybridisations have a low melting temperature. Thus, there exists a temperature gap within which, with high thermodynamic probability, correct hybridisations are annealed and incorrect hybridisations are melted. The graphs in Figure 2 show the computational melting temperatures for correct and incorrect hybridisations without (Figure 2(a)) and with (Figure 2(b)) melting temperature gap optimisations (CODA). In Figure 2(a), codons simply were chosen to be the most preferred codons in E. coli highly expressed genes, i.e., codons yielding a Codon Adaptation Index of 1.0. Not surprisingly, the melting temperature distributions of correct and incorrect hybridisations intersect when no CODA optimisation is performed. On the other hand, the melting temperature distributions of CODA designed sequences show a melting temperature gap of 18oC between correct hybridisations and incorrect hybridisations (Figure 2(b)).
Figure 2.

Hybridisation melting temperatures and assembly of the integrase (IN) gene of the S. cerevisiae transposable element Ty3. Computational melting temperatures without (A) and with (B) CODA melting temperature optimisations. Solid and dashed lines represent correct hybridisations of oligonucleotides and intermediate DNA fragments, respectively. Dot-dash and dotted lines represent corresponding incorrect hybridisations. Lanes 1-10, IN intermediate DNA fragments 1 through 10 assembled from oligonucleotides without (C) and with (D) CODA melting temperature optimisations. Lanes 11 and 13, molecular weight markers. Lane 12, corresponding assembly of full-length IN gene (1,640 bp)
Assembly of intermediate DNA fragments and full-length IN gene
The full-length IN gene was designed as ten separate intermediate DNA fragments overlapping by approximately 50 nts. Each intermediate DNA fragment was designed as a set of 6-8 oligonucleotides overlapping one another by 20-30 nts (fragment 0: 196 bp, 8 oligos; fragment 1: 224 bp, 8 oligos; fragment 2: 224 bp, 8 oligos; fragment 3: 223 bp, 8 oligos; fragment 4: 227 bp, 8 oligos; fragment 5: 223 bp, 8 oligos; fragment 6: 224 bp, 8 oligos; fragment 7: 172 bp, 6 oligos; fragment 8: 175 bp, 6 oligos; fragment 9: 174 bp, 8 oligos).
The electrophoretograms of Figure 2(c) and (d) show the results of gene assemblies from oligonucleotides with the melting temperature distributions of Figure 2(a) and (b), respectively. Intermediate DNA fragments assembled from oligonucleotides optimised only for codon usage, but not for self-assembly (Figure 2(a)), show a broad range of products both larger and smaller than expected (Figure 2(c), lanes 1-10). However, all intermediate DNA fragments assembled from CODA designed oligonucleotides (Figure 2(b)) resulted in single products of the expected length (Figure 2(d), lanes 1-10). In addition, a single full-length (1,640 bp) gene product was obtained from the assembly of ten CODA designed intermediate DNA fragments (Figure 2(d), lane 12), whereas no apparent full-length product was obtained from the assembly of fragments not CODA designed for self-assembly (Figure 2(c), lane 12).
3.2 Codon usage optimisation
In addition to optimising genes for self-assembly, the CODA algorithm also optimises genes for expression in an in vivo or in vitro system of choice. The Ty3 IN gene was optimised to be expressed in E. coli. The data plotted in Figure 3(a) compare codon usage in the CODA designed gene with genomic codon usage of highly expressed genes in E. coli. These data show that the E. coli codon usage of the CODA designed IN gene is comparable to the codon usage of highly expressed E. coli genes. Indeed, the codon optimised synthetic IN gene expressed from a pET-3a vector in E. coli strain BL-21 (DE3) produced more than 15% of the total cell protein (Figure 3(b)).
Figure 3.

Codon usage in the CODA designed Ty3 IN gene vs. codon usage in E. coli highly expressed genes. A. X (solid regression line), CODA designed IN gene. O (dashed regression line), E. coli highly expressed genes. Each codon x-coordinate is usage per 1,000 in E. coli average genomic DNA; y-coordinate is in the CODA gene (X, solid line) or in highly expressed genes (O, dashed line). B. 10% SDS coomassie stained gel. Protein Marker (Lane 1). Protein products from uninduced (Lane 2) and induced (Lane 3) cells
3.3 Errors in oligonucleotide synthesis
The chemical synthesis of oligonucleotides is never 100% efficient. Each base addition in the synthesis process is commonly 98-99.5% efficient, resulting in the formation of failed sequences with single-base deletions and other defects. Our prior sequencing results of several CODA synthetic genes, assembled from hundreds of commercially prepared, partially overlapping oligonucleotides, revealed an error rate per base in non-overlapping (single stranded) regions nearly twice as high as the error rate per base in overlapping (double stranded) regions. We reasoned that this difference was due to the selection for perfect complementarity in overlapping complementary regions. Therefore, to achieve a lower error rate in the gene assembly process, we have chosen to assemble genes with completely overlapping (abutting) oligonucleotides.
To obtain an estimate of the error rate per bp, we sequenced 8,300 bp from four independent clones of each of the ten integrase intermediate DNA fragments. The sequencing results show that, on average, we encounter an error every 218 bp (0.46%). Of these, we observe: a bp deletion every 553 bp (0.18%); a bp insertion every 638 bp (0.16%); and a bp substitution every 692 bp (0.12%).
To reduce this error rate, we developed a simple genetic frameshift screen using the CODA Blue vector (Section 2). This vector was altered from pGEM-3Z (Promega) by introducing a + 1 frameshift, such that it is not able to restore β-galactosidase activity in an E. coli JM109 strain with a lacZ gene missing the α-fragment sequence. Therefore, cells containing intact CODA Blue plasmid grow as white colonies on LB agar plates containing IPTG and X-gal. The intermediate DNA fragments of IN gene were engineered to be 3N + 1 base pairs long and to contain flanking BamHI sites. When these BamHI-(3N + 1)-BamHI intermediate DNA gene fragments are cloned into the BamHI site of the CODA Blue vector, a - 1 frameshift is created that restores the correct reading frame of the α-fragment coding sequence. Now the plasmid produces active, in frame, α-fragment that restores β-galactosidase activity. Thus, cells containing the CODA Blue plasmid with a BamHI-(3N + 1)-BamHI insert grow as easily identifiable blue colonies.
We cloned intermediate DNA gene fragments into the CODA Blue vector and picked blue colonies. This eliminated all point deletion or insertion errors except where the fragment length is altered by a multiple of three nucleotides. The sequence data from 8,300 bp, as four blue colonies from each of the ten intermediate DNA fragments, demonstrate that the frameshift screen nearly halves the total error rate per bp. The sequencing results show that, on average, we encounter an error every 417 bp (0.24%). Of these, we observe: a bp deletion every 750 bp (0.13%); a bp insertion every 4,165bp (0.024%); and a bp substitution every 1,190 bp (0.084%). The data in Table 1 show that, as expected, all deletions and insertions that escape the frameshift screen, in fact, are multiples of three.
Table 1.
Observed and expected errors per intermediate DNA fragment. d, i, and s are the number of deletions, insertions, and substitutions in each intermediate DNA fragment. Integrase and Integrase blue are as in (a). # gives the observed number (and % the percentage) of fragments with each indicated condition (40 fragments of each were sequenced). Exp(.) is the expected number of fragments, based on the probabilistic formulae developed in the text with Pd = 0.18%, Pi = 0.16% and Ps = 0.12% taken from (a). L = 225, M = 10
| Source | Total #. bps sequenced | Deletions #. (Pd%) | Insertions #. (Pi %) | Substitutions #. (Ps %) | # errors total no. (Perr %) |
|---|---|---|---|---|---|
| Integrase | 8300 | 15 (0.18%) | 13 (0.16%) | 10 (0.12%) | 38 (0.46%) |
| Integrase blue | 8300 | 11 (0.13%) | 2 (0.024%) | 7 (0.084%) | 20 (0.24%) |
4 Discussion
In this report, we described the use of a CODA algorithm for the design of overlapping oligonucleotides that self-assemble into a single linear DNA product. We have employed this algorithm to construct synthetic genes that can be optimised for high-level protein expression in any standard in vivo or in vitro production system. The data in Figure 2(b) demonstrate that the CODA algorithm achieves a predicted melting temperature gap between the highest melting incorrect hybridisation and the lowest melting correct hybridisation for linear self-assembly of the overlapping oligonucleotides. Thus, at an annealing temperature for correct hybridisations, but far above that for all incorrect hybridisations, self-assembly into a single linear DNA product is of high thermodynamic probability (Figure 2(d)). In addition to optimising for self-assembly, the CODA algorithm also uses the degeneracy of the genetic code to maximise the use of abundant tRNA isoacceptors for frequently used codons (Figure 3(a)) (Boycheva et al., 2003; Gutman and Hatfield, 1989; Hatfield, 1993), to avoid consecutive rare codons (Hatfield, 1993; Moore and Maranas, 2002), and to use codon pair statistics (Gutman and Hatfield, 1989; Hatfield, 1993; Irwin et al., 1995) to minimise unfavourable codon pairs for high level protein expression (Figure 3(b)).
While our results demonstrate that it is possible to design oligonucleotides that cleanly self-assemble into a single DNA product, it is not possible to chemically synthesise these oligonucleotides without errors. The sequencing results of intermediate DNA fragments that were not screened by CODA Blue vector show that the cumulative error of all bp substitutions, insertions and deletions is 0.46%. This means that a sequence error is encountered about every 200 bp. Therefore, if all 74 of the overlapping ~45 bp oligonucleotides for the Ty3 integrase gene were combined at once to produce a full-length gene, it would be littered with mutations. This motivates the hierarchical assembly strategy described in this report (Figure 1). The probability of incorporating a defective oligonucleotide into a self-assembled DNA molecule is a function of the error rate per bp and the length of the molecule. Furthermore, Table 2 shows that if intermediate DNA fragments 200-250 bp in length are assembled and screened with the CODA Blue frameshift vector, then the probability of obtaining a correct sequence is high (~90 to 95%) if only two CODA Blue inserts for each intermediate DNA fragment are sequenced. Thus, building the gene in a hierarchical fashion from verified correct intermediate DNA fragments enables the rapid assembly of a sequence-correct, full-length gene. The efficacy of the CODA Blue frameshift screen is illustrated in Figure 4. For example, if four clones for each intermediate DNA fragment are sequenced, these data predict a greater than 90% chance of obtaining at least one correct sequence for each of the ten integrase intermediate DNA fragments for the assembly of a correct full-length gene. In the absence of this screen this chance drops to less than 20%.
Table 2.
Probabilities for obtaining error-free intermediate DNA fragments. The error per base values are from Table 1. M = 10
| Intermediate DNA fragment clone length (L) | Probability of one given correct individual clone (Pc) | Probability of one correct individual clone if two clones are sequenced (Po, N = 2) | Probability of one correct individual clone if four clones are sequenced (Po, N = 4) | Probability of one correct individual clone if six clones are sequenced (Po, N = 6) | |
|---|---|---|---|---|---|
| Without CODA | 200 | 0.398 | 0.637 | 0.868 | 0.952 |
| Blue Screen | 250 | 0.316 | 0.532 | 0.781 | 0.897 |
| 300 | 0.251 | 0.439 | 0.685 | 0.823 | |
| With CODA | 200 | 0.747 | 0.936 | 0.9959 | 0.9997 |
| Blue Screen | 250 | 0.672 | 0.893 | 0.988 | 0.9987 |
| 300 | 0.593 | 0.834 | 0.9730 | 0.9955 |
Figure 4.

Probability of Obtaining at Least One Correct Sequence for each of the Intermediate DNA Fragments. Pg (light bars) is the probability that every one of the M intermediate DNA fragments has at least one clone with the correct sequence. P[g|b] (dark bars) is the probability that every one of the M intermediate DNA fragments has at least one clone with the correct sequence given that only pCODAblue screened blue clones are considered
The hierarchical assembly described here is facilitated by oligonucleotides that are CODA designed for self-assembly into a single product (Figure 2(d)). Otherwise, even with the CODA Blue frameshift screen, many clones of widely varying lengths would need to be screened and sequenced to obtain one of the correct length and sequence (Figure 2(c)). This addresses the problem of how to assemble and PCR amplify a full-length DNA molecule from the mixture of randomly hybridised oligonucleotides (Stemmer et al., 1995) while avoiding mutations incorporated with defective oligonucleotides into the full-length gene, even when a stringent biological selection such as that described by Smith et al. (2003) is not possible.
Acknowledgments
Acknowledgements
We are appreciative of the expert technical assistance of Ms. Elaine Ito. We are especially grateful to IGB Director Pierre Baldi, Deans Susan Bryant, Thomas Cesario, Debra Richardson, and Ronald Stern, and Vice Chancellor William Parker, for their generous financial support of this project. This work was supported in part by grants from the University of California Biological Research and Education Program (to RHL), the National Science Foundation (44630121784 to RHL and GWH) and the National Institutes of Health (GM68903 to GWH). CDW is the recipient of a Biomedical Informatics Training (BIT) Program Predoctoral Fellowship (NIH T15 LM-07443).
Biographical notes
Liza S.Z. Larsen, PhD, is the Director of Scientific Operations at CODA Genomics, Inc., Laguna Hills CA. She received her PhD Degree in Molecular and Computational Biology from the University of California, Irvine, in affiliation with the Department of Microbiology and Molecular Genetics and the UCI Institute for Genomics and Bioinformatics in 2006. Her research interests are in the areas of synthetic gene construction, translation engineering, bioinformatics, and virology.
Christopher D. Wassman is a PhD student in the Department Computer Science and is affiliated with the Institute for Genomics and Bioinformatics of the University of California, Irvine. His research interests are in the areas of synthetic gene construction, site-directed mutagenesis, computational biology and biophysics.
G. Wesley Hatfield, PhD, is a Professor Emeritus and the Associate Director of the Institute for Genomics and Bioinformatics at the University of California, Irvine. He holds a PhD Degree from Purdue University and a BA Degree from the University of California at Santa Barbara. His primary areas of scientific expertise include molecular biology, biochemistry, microbial physiology, functional genomics, and bioinformatics. His recent academic interests include mathematical modelling of metabolic systems in bacteria and yeasts, and the application and development of genomic and bioinformatics methods to elucidate the effects of chromosome structure and DNA topology on gene expression.
Richard H. Lathrop, PhD, is a Professor in the School of Information and Computer Science at the University of California, Irvine. He received a BA in Mathematics from Reed College, and a Masters in Computer Science, the Graduate Degree of Electrical Engineer, and a PhD in Artificial Intelligence from the Massachusetts Institute of Technology. His research interests are in the areas of protein structure prediction, protein-DNA interactions, rational drug design and discovery, and DNA self-assembly and synthetic gene construction.
Footnotes
Reference to this paper should be made as follows: Larsen, L.S.Z., Wassman, C.D., Hatfield, G.W. and Lathrop, R.H. (2008) `Computationally Optimised DNA Assembly of synthetic genes', Int. J. Bioinformatics Research and Applications, Vol. 4, No. 3, pp.324-336.
References
- Boycheva S, Chkodrov G, Ivanov I. `Codon pairs in the genome of Escherichia coli'. Bioinformatics. 2003;Vol. 19:987–998. doi: 10.1093/bioinformatics/btg082. [DOI] [PubMed] [Google Scholar]
- Breslauer KJ, Frank R, Blocker H, Marky LA. `Predicting DNA duplex stability from the base sequence'. Proc. Natl. Acad. Sci. U.S.A. 1986;Vol. 83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro DR, Wright PE, Dyson HJ. `PCR-based gene synthesis and protein NMR spectroscopy'. Structure. 1997;Vol. 5:1407–1412. doi: 10.1016/s0969-2126(97)00291-8. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Fiers W. `Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes'. Gene. 1982;Vol. 18:199–209. doi: 10.1016/0378-1119(82)90157-3. [DOI] [PubMed] [Google Scholar]
- Gutman GA, Hatfield GW. `Nonrandom utilization of codon pairs in Escherichia coli'. Proc. Natl. Acad. Sci. U.S.A. 1989;Vol. 86:3699–3703. doi: 10.1073/pnas.86.10.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield DL, Lee BJ, Pirtle RM. Transfer RNA in Protein Synthesis. CRC Press; Boca Raton, FL: 1993. `Codon pair utilization bias in bacteria, yeast and mammals'; pp. 157–190. [Google Scholar]
- Irwin B, Heck JD, Hatfield GW. `Codon pair utilization biases influence translational elongation step times'. J. Biol. Chem. 1995;Vol. 270:22801–22806. doi: 10.1074/jbc.270.39.22801. [DOI] [PubMed] [Google Scholar]
- Lathrop RH, Sazhin A, Sun Y, Steffin N, Irani SS. `A multi-queue branch-and-bound algorithm for anytime optimal search with biological applications'. Genome Inform. Ser. Workshop Genome Inform. 2001;Vol. 12:73–82. [PubMed] [Google Scholar]
- Lio P. `Investigating the relationship between genome structure, composition, and ecology in prokaryotes'. Mol. Biol. E. 2002;Vol. 19:789–800. doi: 10.1093/oxfordjournals.molbev.a004136. [DOI] [PubMed] [Google Scholar]
- Mandecki W, Bolling TJ. `FokI method of gene synthesis'. Gene. 1988;Vol. 68:101–107. doi: 10.1016/0378-1119(88)90603-8. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. `Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure'. J. Mol. Biol. 1999;Vol. 288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Moore GL, Maranas CD. `eCodonOpt: a systematic computational framework for optimizing codon usage in directed evolution experiments'. Nucleic Acids Res. 2002;Vol. 30:2407–2416. doi: 10.1093/nar/30.11.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Pearl LH. `Recursive PCR: a novel technique for total gene synthesis'. Protein Eng. 1992;Vol. 5:827–829. doi: 10.1093/protein/5.8.827. [DOI] [PubMed] [Google Scholar]
- SantaLucia J., Jr. `A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics'. Proc. Natl. Acad. Sci. U.S.A. 1998;Vol. 95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman NC. `DNA nanotechnology: novel DNA constructions'. Annu. Rev. Biophys.Biomol. Struct. 1998;Vol. 27:225–248. doi: 10.1146/annurev.biophys.27.1.225. [DOI] [PubMed] [Google Scholar]
- Seeman NC. `DNA engineering and its application to nanotechnology'. Trends Biotechnol. 1999;Vol. 17:437–443. doi: 10.1016/s0167-7799(99)01360-8. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. `The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications'. Nucleic Acids Res. 1987;Vol. 15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HO, Hutchison CA, III, Pfannkoch C, Venter JC. `Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides'. Proc. Natl. Acad. Sci. U.S.A. 2003;Vol. 100:15440–15445. doi: 10.1073/pnas.2237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MA, Kurland CG, Pedersen S. `Codon usage determines translation rate in Escherichia coli'. J. Mol. Biol. 1989;Vol. 207:365–377. doi: 10.1016/0022-2836(89)90260-x. [DOI] [PubMed] [Google Scholar]
- Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. `Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides'. Gene. 1995;Vol. 164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- Theriault NY, Carter JB, Pulaski SP. `Optimization of ligation reaction conditions in gene synthesis'. Biotechniques. 1988;Vol. 6:470–474. [PubMed] [Google Scholar]
- Warrens AN, Jones MD, Lechler RI. `Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest'. Gene. 1997;Vol. 186:29–35. doi: 10.1016/s0378-1119(96)00674-9. [DOI] [PubMed] [Google Scholar]
- Zuker M. `On finding all suboptimal foldings of an RNA molecule'. Science. 1989;Vol. 244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- Zuker M. `Mfold web server for nucleic acid folding and hybridization prediction'. Nucleic Acids Res. 2003;Vol. 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M, Mathews DH, Turner DH. RNA Biochemistry and Biotechnology. Kluwer Academic Publishers; Dordrecht: 1999. `Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide'; pp. 11–43. [Google Scholar]